Abstract
Hydrogen sulfide (H2S) is emitted from industrial activities, and several chemotrophs possessing Sox enzymes are used for its removal. Oral malodor is a common issue in the dental field and major malodorous components are volatile sulfur compounds (VSCs), including H2S and methyl mercaptan. Paracoccus pantotrophus is an aerobic, neutrophilic facultatively autotrophic bacterium that possesses sulfur-oxidizing (Sox) enzymes in order to use sulfur compounds as an energy source. In the present study, we cloned the Sox enzymes of P. pantotrophus GB17 and evaluated their VSC-degrading activities for the prevention of oral malodor. Six genes, soxX, soxY, soxZ, soxA, soxB, and soxCD, were amplified from P. pantotrophus GB17. Each fragment was cloned into a vector for the expression of 6×His-tagged fusion proteins in Escherichia coli. Recombinant Sox (rSox) proteins were purified from whole-cell extracts of E. coli using nickel affinity chromatography. The enzyme mixture was investigated for the degradation of VSCs using gas chromatography. Each of the rSox enzymes was purified to apparent homogeneity, as confirmed by SDS-PAGE. The rSox enzyme mixture degraded H2S in dose- and time-dependent manners. All rSox enzymes were necessary for degrading H2S. The H2S-degrading activities of rSox enzymes were stable at 25–80°C, and the optimum pH was 7.0. The amount of H2S produced by periodontopathic bacteria or oral bacteria collected from human subjects decreased after an incubation with rSox enzymes. These results suggest that the combination of rSox enzymes from P. pantotrophus GB17 is useful for the prevention of oral malodor.
Hydrogen sulfide (H2S) is emitted from industrial activities, particularly from biogas and protein-rich industrial waste. H2S exhibits high toxicity and is very corrosive to internal combustion engines. Many commercial chemical technologies are currently used to remove H2S in industry (41). However, these chemical H2S removal processes are expensive due to high chemical requirements as well as energy and disposal costs, and mostly exert short-term effects only. Biological treatment methods for the removal of H2S are desirable, and several studies have investigated this issue (10, 32). The oxidation of H2S is mediated by various aerobic lithotrophic and anaerobic phototrophic bacteria (35).
In the dental field, oral malodor, the presence of unpleasant or foul-smelling breath, is a common issue worldwide and may have significant social or psychological effects on those affected by it (6, 15, 21). Oral malodor is a mixture of malodorous components, including volatile sulfur compounds (VSCs), such as H2S, methyl mercaptan (CH3SH), and dimethyl sulfide (6, 15). VSCs are produced in saliva, gingival crevices, on the tongue surface, and in other areas via the putrefactive activities of microorganisms on sulfur-containing amino acids, such as cysteine and methionine (3, 6). H2S and CH3SH are primarily responsible for oral malodor and comprise approximately 90% of the VSC content in mouth air (43).
VSCs also exert adverse effects on oral tissues. Previous studies demonstrated that H2S damages gingival epithelial cells (42), increases the permeability of the oral mucosa in vitro (24), and causes apoptosis in human gingival fibroblasts (2). H2S has predominantly been detected in pockets (27) associated with periodontal bacteria, including Fusobacterium nucleatum, one of the most active oral bacteria to produce H2S from L-cysteine (26). The presence of CH3SH is also known to be involved in the induction or progression of periodontal disease. Exposure to CH3SH inhibits cell migration in periodontal ligament cells (18) as well as epithelial cell growth and proliferation (38). Porphyromonas gingivalis, a black-pigmented anaerobic bacterium and major pathogen in adult periodontitis, produces large amounts of CH3SH in human serum (26) from L-methionine.
Several attempts have been made to prevent and reduce oral malodor (6, 40, 45). Mechanical prophylaxis using a toothbrush and tongue scraper represents a basic method to remove bacterial cells and substrates from the oral cavity; however, organisms still grow and accumulate. Mouth rinses containing antimicrobial agents, such as chlorhexidine and cetylpyridinium chloride, have been used to decrease bacterial numbers, leading to a reduction in oral malodor (9, 47). Additionally, zinc ions have been used to reduce VSCs through their oxidizing effects on the thiol groups in VSC precursors (1, 16). However, the effects of specifically formulated mouth rinses on oral malodor generally remain unclear (40). Therefore, new methods to control VSCs in the oral cavity need to be established for the prevention of oral malodor.
The purpose of this study was to examine the applicability of the sulfur-oxidizing (Sox) enzyme system to the control of oral malodor. We selected the sulfur-oxidizing bacterium, Paracoccus pantotrophus, which is an aerobic, Gram-negative, neutrophilic facultatively autotrophic bacterium that grows using thiosulfate or molecular hydrogen as an energy source and heterotrophically using various carbon sources (13). The Sox enzyme system in P. pantotrophus has already been examined. The sox gene region of P. pantotrophus comprises 12 open reading frames and seven genes, soxXYZABCD, which encode proteins that are essential for sulfur oxidation in vitro (34). The Sox proteins of P. pantotrophus are located in the periplasm (12), and four proteins, SoxXA, SoxYZ, SoxB, and SoxCD, are known to be required for the H2S-, sulfur-, thiosulfate-, and sulfite-dependent reduction of horse cytochrome c (34). We report the cloning of sox genes from P. pantotrophus GB17—soxX, soxY, soxZ, soxA, soxB, and soxCD—and the characteristics of recombinant Sox (rSox) enzymes. We investigated the rSox enzymatic degradation of VSCs produced by periodontopathic bacteria and oral bacteria.
Materials and Methods
Bacterial strains and culture conditions
All strains were cultivated at 37°C. P. pantotrophus GB17 (NBRC 102493) was obtained from NBRC (Kisarazu, Japan) and used throughout this study. Seed cultures were grown aerobically in brain heart infusion (BHI; Becton, Dickinson and Company, Sparks, MD, USA) broth supplemented with 4 mM magnesium sulfate. Escherichia coli XL II was grown aerobically in Luria Bertani (LB; Difco Laboratories, Detroit, MI, USA) medium. P. gingivalis W83 was grown anaerobically in GAM broth (Nissui Medical, Tokyo, Japan) supplemented with hemin (5 μg mL−1) and menadione (1 μg mL−1). F. nucleatum ATCC10953 was grown anaerobically in BHI broth supplemented with 5 mg mL−1 yeast extract and 0.3 mg mL−1 cysteine-HCl. Ampicillin (100 μg mL−1) was added when appropriate.
DNA manipulation
Standard DNA recombinant procedures, such as DNA isolation, restriction endonuclease digestion, ligation, the transformation of competent E. coli cells, and agarose gel electrophoresis, were performed as described by Sambrook et al. (37). Chromosomal DNA was isolated from P. pantotrophus GB17 cells using the Dr. GenTLE (from yeast) High Recovery DNA extraction kit (Takara Bio, Otsu, Japan).
DNA amplification
We used Tks Gflex DNA polymerase (Takara Bio) to improve the fidelity of the PCR assay for soxX, soxY, soxZ, soxA, soxB, and soxCD genes. The reaction mixture (50 μL in total) contained 25 μL of 2× Gflex PCR buffer (containing 2 mM of Mg2+ and 400 μM dNTPs; Takara Bio), 1 μL of Tks Gflex DNA polymerase (Takara Bio), 0.01 nM of each primer, and 1 μL of a DNA template, and the volume was adjusted with nuclease-free water (Roche Diagnostics, Indianapolis, IN, USA). The reaction was performed for 30 cycles under the following conditions: initial denaturation at 94°C for 1 min, denaturation at 98°C for 10 s, annealing at 48°C for 15 s, and extension at 68°C for 2.5 min.
Cloning of sox genes from P. pantotrophus GB17
Six sox genes (soxX, soxY, soxZ, soxA, soxB, and soxCD) were amplified by PCR from the 5′ terminus of the sox gene (13 kbp): the soxX fragment (474 bp), soxY fragment (423 bp), soxZ fragment (330 bp), soxA fragment (873 bp), soxB fragment (1695 bp), and soxCD fragment (2431 bp). These short double-stranded DNAs were compatible with a BamHI site on the 5′-end and a HindIII site on the 3′-end. Primers were designed to create BamHI and HindIII restriction sites (underlined) within the PCR product (Table 1). The pQE30 vector (Qiagen, Tokyo, Japan) was used for the construction of histidine-tagged recombinant proteins. Each DNA fragment, containing a sox gene, was digested with BamHI and HindIII and ligated into the pQE30 vector for the expression of 6×His-tagged fusion proteins. In order to obtain the Sox products of P. pantotrophus GB17, E. coli XL II competent cells were transformed with each resulting plasmid (soxX, soxY, soxZ, soxA, soxB, and soxCD). Positive colonies were selected and re-plated on tryptic soy (TS; Becton, Dickinson, and Company) agar containing ampicillin (100 μg mL−1). The nucleotide sequences of the inserted fragments were confirmed by PCR in order to verify that the fragments were correct and did not contain nucleotide substitutions or deletions. A BLAST nucleotide sequence analysis was also performed for DNA sequence identification.
Table 1
Oligonucleotide primers used in this study
| Fragment |
Primer |
Sequence (5′ to 3′)a |
| rSoxX |
rSoxX-Forward |
AGGGATCCATGAGCAGCCATCTATGG |
| rSoxX-Reverse |
CTAAGCTTGTCGAGCCTGTAGAGATC |
| rSoxY |
rSoxY-Forward |
AGGGATCCATGATCCTTTCAAGACGC |
| rSoxY-Reverse |
GCAAGCTTAATCTCCTGTTACTGGAC |
| rSoxZ |
rSoxZ-Forward |
AGGGATCCATGGCAGATGATGCAAAG |
| rSoxZ-Reverse |
TGAAGCTTGATGTTGCGGCGCTTAGG |
| rSoxA |
rSoxA-Forward |
GAGGATCCATGCCGCGCTTTACCAAG |
| rSoxA-Reverse |
GGAAGCTTGAAGCATTGCCCTTTCGA |
| rSoxB |
rSoxB-Forward |
ACGGATCCATGATTACCCGACGTGAG |
| rSoxB-Reverse |
CAAAGCTTAACGCTCCTTCGTGATTG |
| rSoxCD |
rSoxCD-Forward |
GTGGATCCATGAAAGACGAGCTCACC |
| rSoxCD-Reverse |
AGAAGCTTCTGTCTCATGCGTCACTT |
a Nucleotides underlined in each primer sequence show the position of the restriction endonuclease site incorporated to facilitate cloning.
Transformants were grown in LB medium with ampicillin (100 μg mL−1) at 37°C until optical density at 550 nm (OD550) reached 0.5. Isopropyl-β-D(–)-thiogalactopyranoside was added to the culture at a final concentration of 1 mM, and cultures were grown for an additional 4 h. Cells were harvested by centrifugation (5,000×g, 4°C, 15 min) and lysed using an ultrasonic sonifier equipped with a microtip (Model W-220-F; Heat Systems Ultrasonics, Plainview, NY, USA) on ice. The cell extract was obtained by centrifugation (10,000×g, 4°C, 15 min) and subjected to Ni-NTA resin (Qiagen) affinity column chromatography. Purification procedures followed the manufacturer’s instructions, and the purities of recombinant Sox proteins were analyzed using SDS-PAGE. Eluted proteins were refolded by sequential dialysis against a urea-decreasing phosphate buffer at 4°C (48). The amounts of proteins were assessed using the Lowry method with bovine serum albumin as the standard (19).
SDS-PAGE and Western blotting
SDS-PAGE was performed using 15% polyacrylamide gels according to the method of Laemmli (17). After electrophoresis, the gel was stained with Coomassie brilliant blue R-250. The low molecular weight electrophoresis calibration kit (Amersham Pharmacia Biotech, Uppsala, Sweden) was used for molecular mass markers. In Western blotting, proteins subjected to SDS-PAGE were transferred electrophoretically to a nitrocellulose membrane according to the method of Burnette (8). After blocking with 2% skimmed milk in Tris-buffered saline (20 mM Tris, 150 mM NaCl, pH 7.2) containing 0.1% Triton X-100 (TBS-T), the membrane was reacted with a HRP-conjugated mouse anti-6×His monoclonal antibody (Wako Pure Chemical, Osaka, Japan). The membrane was washed three times with TBS-T, and fluorescence detection was performed using a chemiluminescence detection system (Clarity Western ECL blotting, Bio-Rad Laboratories, Hercules, CA, USA).
Enzyme assay
We investigated the ability of a rSox enzyme mixture reconstituted from rSoxX, rSoxY, rSoxZ, rSoxA, rSoxB, and rSoxCD to degrade H2S generated from sodium hydrogen sulfide (NaHS). The assay mixture (1 mL) for the assessment of H2S-degrading activity contained various amounts of P. pantotrophus GB17 cells or rSox enzymes and 20 nmol of NaHS to generate H2S in 10 mM phosphate buffer (pH 7.0). All reactions were performed in sterile 15-mL polypropylene tubes sealed with a silicone plug. NaHS was added to start the reaction. After an incubation at 37°C for 2 h, a sample (2.5 mL) of the vapor above the assay mixture in the tube was removed using a gas-tight syringe and analyzed by gas chromatography (model GC-2014; Shimadzu, Kyoto, Japan) using a glass column packed with 25% β,β′-oxydipropionitrile on a 60–80-mesh Chromosorb W AW-DMCS-ST device (Shimadzu) fit with a flame photometric detector at 70°C. The concentration of VSCs was measured using standard H2S and CH3SH gas prepared with a Permeater PD-1B (Gastec, Ayase, Japan).
The assay mixture (1 mL) used to assess rSox enzyme activity to degrade VSCs produced by periodontopathic bacteria was prepared following the method of Yoshimura et al. (49). Briefly, bacterial strains were grown at 37°C until an OD550 of approximately 0.6 was reached. Cells were harvested and washed three times with buffered salt solution (40 mM potassium phosphate buffer, 50 mM NaCl, pH 7.7). Cells were suspended in buffered salt solution to an OD550 of 0.3 for F. nucleatum ATCC10953 and OD550 of 1.0 for P. gingivalis W83. In order to measure H2S, a reaction mixture was prepared consisting of 100 μL of a F. nucleatum ATCC10953 or P. gingivalis W83 cell suspension and 0.5 nmol each of rSoxX, rSoxY, rSoxZ, rSoxA, rSoxB, and rSoxCD. Buffered salt solution was then added to the tube, which was sealed with a silicone plug. The reaction was initiated by adding 30 μL of 33 mM L-cysteine. In order to measure CH3SH, 30 μL of 33 mM L-methionine was added instead of L-cysteine. Assay mixtures were maintained at 37°C and, after a 2-h incubation, the reactions were stopped by adding 500 μL of 3 M phosphoric acid. A sample (2.5 mL) of the vapor above the assay mixture was analyzed by gas chromatography 10 min later, as described above.
The assay mixture used to assess the degrading activity of H2S produced by oral bacteria was prepared as described by Tonzetich and Johnson (44) with modifications. Paraffin-stimulated whole saliva was collected from three healthy and non-smoking participants (28–58 years of age), which was approved by the Ethics Committee of Kagoshima University (authorization number 572). The bacterial sediment was collected by centrifugation (10,000×g, 4°C, 15 min) and washed three times with buffered salt solution. The bacterial sediment was disrupted by ultrasonication on ice and suspended in buffered salt solution to an OD550 of 1.0 in order to measure extracellular H2S produced from L-cysteine by oral bacteria. The reaction mixture (3 mL) consisted of a 2-mL bacterial suspension and 0.5 nmol each of rSoxX, rSoxY, rSoxZ, rSoxA, rSoxB, and rSoxCD in buffered salt solution. Subsequent procedures were as described above.
In order to evaluate heat stability, rSox enzymes were mixed and heated at 25 to 100°C for 30 min and then subjected to the H2S-degrading assay. The effects of pH on rSox enzyme activity were examined using 10 mM phosphate buffer with pH 4 to 9.
Statistical analysis
Data were averaged for three independent experiments. Statistical analyses were performed using the Student’s t-test or a one-way analysis of variance (ANOVA) followed by Dunnett’s test. P-values<0.05 were considered to indicate significance.
Results
Purification of recombinant Sox proteins
Fig. 1A shows purified recombinant proteins. rSoxX migrated at 23 kDa; rSoxY at 18 kDa; rSoxZ at 21, 22, and 43 kDa; rSoxA at 43 kDa; and rSoxB at 68 kDa (Fig. 1A). In the Western blot analysis, the anti-6×His antibody reacted with the rSoxX, rSoxY, rSoxZ, rSoxA, and rSoxB proteins (Fig. 1B). rSoxCD was separated into two bands (51 and 54 kDa) by SDS-PAGE, and the 54-kDa band reacted with the anti-6×His antibody. Since 6×His was attached to rSoxC, the 51-kDa band appeared to be rSoxD and the 54-kDa band rSoxC. rSoxZ showed three bands and the antibody to 6×His reacted with all of these components; therefore, the 43-kDa band appeared to be a dimer, and the 21-kDa band lacked some portion of rSoxZ (the 22-kDa band).
P. pantotrophus GB17 activity to degrade H2S
Whole cells of P. pantotrophus GB17 were examined for the degradation of H2S generated from NaHS. P. pantotrophus GB17 cells showed H2S-degrading activity, and H2S concentrations decreased in a cell number-dependent manner (Fig. 2). Approximately 50% of H2S was degraded when a bacterial suspension containing 1.0×107 cells was used.
rSox enzyme activity to degrade H2S
A combination of the six rSox proteins (rSoxX, rSoxY, rSoxZ, rSoxA, rSoxB, and rSoxCD) was investigated for its ability to degrade H2S generated from NaHS. After being incubated, the rSox enzyme mixture degraded H2S in a dose-dependent manner (Fig. 3A). The concentration of H2S significantly decreased and reached a stable level after 0.125 nmol of proteins was used. In time-course experiments, the ratio of the H2S concentration (sample/control) decreased to approximately 45% in the 2-h incubation and was stable thereafter (Fig. 3B). In the thermal stability study, the amount of H2S in the assay mixture increased gradually as the temperature was raised (Fig. 4A). However, the amount of H2S was still significantly lower than that with the control, even after heating at 80°C. After heating at 100°C, no significant difference was observed in the amount of H2S from that with the control. Regarding the effects of pH on rSox enzyme activity, the percentage of the H2S concentration (sample/control) was the lowest at pH 7.0, and higher values were observed at other pH values (Fig. 4B).
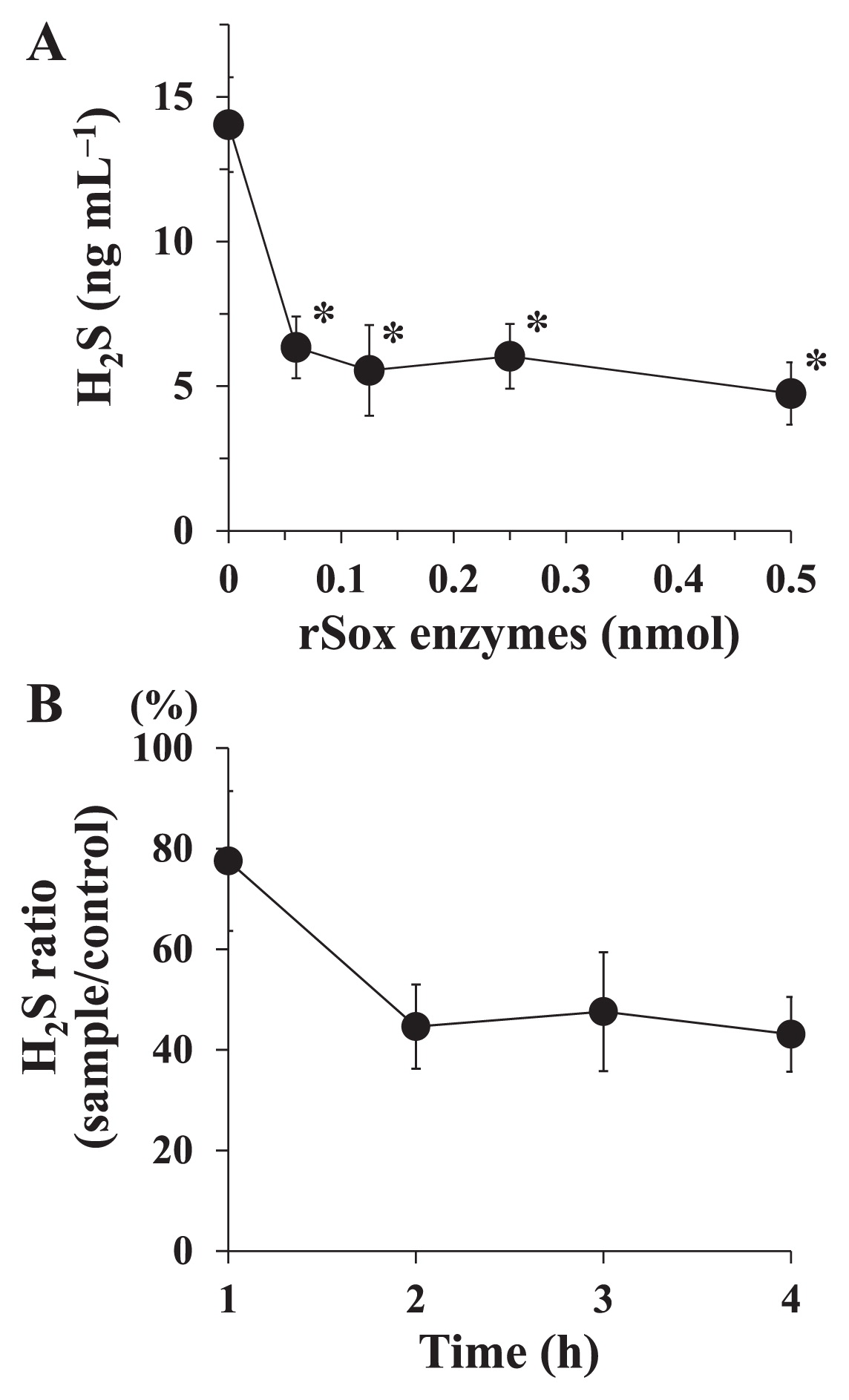
Table 2 shows the effects of the components of the rSox enzyme mixture on the degradation of H2S. In this experiment, the rSoxX and rSoxA or rSoxY and rSoxZ pair was omitted from the assay because these pairs of enzymes work together in the Sox system (12, 34). When a mixture of rSox enzymes reconstituted from all rSox enzymes was used, H2S levels were significantly lower than those with the control (no rSox enzymes). In other cases where certain rSox enzymes were omitted from the assay, H2S levels were not significantly different from the control, and H2S ratios relative to control levels were approximately 70%. Bovine serum albumin, which was used as a non-sulfur-oxidizing control, showed no reducing effect on H2S.
Table 2
Effects of each rSox enzyme on the degradation of H
2S generated from NaHS
| Sox enzymes added to the assay |
H2S (ng mL−1)a |
Ratio to the control (%) |
|
| X |
A |
Y |
Z |
B |
CD |
BSAb |
| − |
− |
− |
− |
− |
− |
− |
16.9±3.8 |
|
| + |
+ |
+ |
+ |
+ |
+ |
− |
6.4±1.1* |
37.9 |
| − |
− |
+ |
+ |
+ |
+ |
− |
11.9±2.5 |
70.4 |
| + |
+ |
− |
− |
+ |
+ |
− |
11.5±2.1 |
68.0 |
| + |
+ |
+ |
+ |
− |
+ |
− |
12.0±4.1 |
71.0 |
| + |
+ |
+ |
+ |
+ |
− |
− |
12.0±1.5 |
71.0 |
| − |
− |
− |
− |
− |
− |
+ |
17.2±4.7 |
101.8 |
In order to assess the role of each rSox component, an enzyme mixture was prepared as shown in the table. The rSox enzyme mixture (0.125 nmol each) prepared was reacted with 20 nmol of NaHS at 37°C for 2 h.
a Values are the means±SDs of three independent experiments.
b BSA, bovine serum albumin (0.125 nmol) was used as a non-sulfur-oxidizing control.
* P<0.05 significantly different from the control (without rSox enzymes or BSA), as assessed by ANOVA followed by Dunnett’s test.
We investigated the ability of rSox enzymes to degrade H2S produced by F. nucleatum ATCC10953 and P. gingivalis W83. The rSox enzyme mixture significantly degraded H2S produced by the two bacterial strains (Table 3). We also assessed CH3SH degradation by the rSox enzyme mixture and found that it only slightly decreased CH3SH levels from those with the control (no enzymes). Oral bacteria were collected from human saliva and used to produce H2S. The rSox enzyme mixture significantly degraded H2S produced from the oral bacteria of all subjects; however, the degree of degradation differed among subjects (Table 4).
Table 3
rSox enzyme activity to degrade VSCs produced by periodontopathic bacteria
| Bacterial strain |
H2S (ng mL−1)a |
CH3SH (ng mL−1)a |
|
|
| rSox enzymes |
rSox enzymes |
|
|
| (−) |
(+) |
(−) |
(+) |
| F. nucleatum ATCC10953 |
26.7±2.2 |
12.1±1.9* |
ND |
ND |
| P. gingivalis W83 |
19.6±4.5 |
9.1±3.5* |
22.0±2.9 |
15.9±1.8 |
a Values are the means±SDs of three independent experiments.
* P<0.05 significantly different from the control (without rSox enzymes), as assessed by the Student’s
t-test.
Table 4
rSox enzyme activity to degrade H
2S produced by oral bacteria
| Subject |
H2S (ng mL−1)a |
Ratio (%)(rSox/Control) |
|
| Control |
rSox enzymes |
| A |
23.8±3.8 |
13.3±4.7* |
55.9 |
| B |
22.3±6.3 |
6.5±0.7* |
29.1 |
| C |
8.1±1.1 |
2.7±0.2* |
33.3 |
a Values are the means±SDs of three independent experiments.
* P<0.05 significantly different from the control (without rSox enzymes), as assessed by the Student’s
t-test.
Discussion
In the present study, we demonstrated the degradation of H2S by rSox enzymes from P. pantotrophus GB17 in view of oral malodor prevention. We produced rSox enzymes to examine their VSC-degrading activities. We attempted to construct rSoxXA, rSoxYZ, rSoxB, and rSoxCD proteins; however, only rSoxB and rSoxCD proteins were successfully obtained. Thus, we constructed each enzyme as rSoxX, rSoxY, rSoxZ, and rSoxA. We demonstrated that the combination of rSoxX, rSoxY, rSoxZ, rSoxA, rSoxB, and rSoxCD proteins degraded H2S, and this activity was almost constant after reaching the maximum effect (Fig. 3A). Regarding the use of recombinant Sox enzymes, rSoxXA expressed in E. coli was found to be functional in the reconstituted enzyme system to activate H2S-dependent cytochrome c reduction (35). This study is the first to describe a Sox system composed of each recombinant Sox protein for degrading H2S.
High cytochrome c-reducing activity by the Sox system was observed using H2S as the substrate (34), and SoxXA, SoxYZ, SoxB, and SoxCD were required to achieve optimal activity (12, 34). In the present study, we demonstrated that all of the enzymes—rSoxX, rSoxY, rSoxZ, rSoxA, rSoxB, and rSoxCD—were necessary for H2S degradation (Table 2). The central protein of the Sox enzyme system is SoxYZ, to which the sulfur substrate is covalently linked, oxidized, and finally released as sulfate by three other Sox proteins (11). SoxXA binds the sulfur substrate and covalently links it to the thiol of the single cysteine 110 residue of the SoxY subunit. SoxY forms a complex with SoxZ. The outer sulfur atom is oxidized by sulfur dehydrogenase SoxCD, yielding SoxY-cysteine-S-sulfate. Finally, SoxY-cysteine-S-sulfate is hydrolyzed by the dimanganese SoxB protein to yield sulfate and regenerate SoxY for a new reaction cycle of SoxYZ (13, 36). When SoxB or SoxYZ was omitted from the assay mixture, residual H2S-dependent activities decreased to approximately 12 or 9%, respectively, that of the mixture of SoxXA, SoxYZ, SoxB, and SoxCD (34). SoxCD was also required to achieve the maximum rate of H2S-dependent cytochrome c reduction, showing a residual rate of 19% that of the mixture of all Sox enzymes when SoxCD was omitted. The results obtained in this study are consistent with these findings.
In P. pantotrophus GB17, each sox gene encodes a protein with a different molecular mass: SoxX, 16,421 Da; SoxY, 13,830 Da; SoxZ, 11,850 Da; SoxA, 31,884 Da; SoxB, 61,897 Da; SoxC, 43,442 Da; and SoxD, 37,637 Da (12). In the present study, the estimated molecular masses of rSox enzymes by SDS-PAGE were larger than those predicted by the nucleotide sequences (Fig. 1A). All rSox proteins, except for rSoxD, had a 6×His tag; however, the differences observed were larger than the size of 6×His (approximately 1.2 kDa). Previous studies (12, 29) also showed that purified Sox proteins had different molecular masses from the predicted ones (SoxX, 16 kDa; SoxY, 12 kDa; SoxZ, 16 kDa; SoxA, 29 kDa; SoxB, 59 kDa; SoxC, 47 kDa; and SoxD, 50 kDa) under denaturing SDS-PAGE. Friedrich et al. (12) reported that the molecular mass of SoxY assessed by SDS-PAGE differed from that predicted by the nucleotide sequence due to an unknown reason. Furthermore, Quentmeier et al. (29) found that the SoxC and SoxD proteins had different molecular masses assessed by SDS-PAGE from those deduced from the nucleotide sequence of the mature protein. Mobility on SDS-PAGE gels may be affected by the amino acid composition, glycosylation, or phosphorylation of the protein. In the present study, amino acid compositions and predicted pI values were obtained using the Compute pI/Mw tool in ExPASy. None of the rSox enzymes appeared to have a unique amino acid composition, and pI values were between 4.5 and 6.6. These characteristics did not appear to affect the mobilities of the proteins on SDS-PAGE gels. A BLAST nucleotide sequence analysis showed the identities of all of the sox genes of P. pantotrophus GB17 used in this study; therefore, the reason for the differences observed in mass remains unclear.
The optimal pH for the H2S-degrading activities of rSox enzymes was 7.0, and activities were stable at 25–80°C. P. pantotrophus was originally isolated as Thiosphaera pantotropha (33), was reclassified as P. denitrificans (20), and then renamed (31). T. pantotropha GB17 obtains energy through the Sox system and grows at pH 6.5–10.5, with an optimum pH of 8.0, and at a temperature of 15–42°C, with an optimum at 37°C (33). Another study demonstrated the optimal growth conditions of P. denitrificans at pH 7.5–8.0 and 30–37°C (11). These findings suggest that the Sox system functions under these pH and temperature conditions, which appears to be reasonable. We also found that whole cells from P. pantotrophus GB17 degraded H2S, even after heating at 100°C (data not shown). Thermophilic sulfur-oxidizing bacteria have recently been isolated from geothermal fields (25, 39). Although it currently remains unclear whether these bacteria contain the Sox system, Sox enzymes may possess thermophilic properties. Ghosh et al. (14) suggested that the Sox system originated in ancient thermophilic bacteria and evolved through extensive horizontal gene transfer. Further studies are needed in order to clarify the mechanisms responsible for the thermal stabilities of rSox enzymes. The heat stabilities of rSox enzymes may be advantageous in potential clinical applications, because intra-oral temperature changes after the ingestion of various cold/hot meals and beverages (4, 22).
In the present study, we demonstrated that rSox enzymes significantly degraded H2S produced by periodontopathic bacteria. The reducing effects of rSox enzymes in reaction mixtures with P. gingivalis or F. nucleatum were weaker than those obtained in reaction mixtures with NaHS. We examined the effects of rSox enzymes on the growth of these bacterial cells and found no significant effects (data not shown). rSox enzymes also showed the potential to decrease CH3SH produced by P. gingivalis. The Sox system, reconstituted from SoxXA, SoxYZ, SoxB, and SoxCD, mediates thiosulfate-, sulfite-, sulfur-, and H2S-dependent cytochrome c reduction (12, 34); however, CH3SH-dependent reduction has not yet been demonstrated. The availability of CH3SH as a substrate for the Sox system needs to be clarified in further studies. We also showed that H2S produced by oral bacteria collected from human saliva decreased after an incubation with rSox enzymes. These results indicate the potential of rSox enzymes to reduce H2S produced in the oral cavity.
Many randomized controlled trials have reported the reducing effects of mouth rinses containing chlorhexidine, cetylpyridinium chloride, or metal ions on oral malodor, whereas several studies have reported no significant effects (7, 30, 40). Systematic reviews showed that available data are insufficient to assess precision (5) and concluded that the strength of the recommendation to use these ingredients for oral malodor reduction is graded as ‘weak’ (40). It currently remains unclear whether Sox enzyme-reducing effects on H2S are superior to existing components. Chlorhexidine and cetylpyridinium chloride act to suppress bacterial growth and may be accompanied by several adverse effects in oral tissues. The Sox system appears to be advantageous in this point because its target is H2S produced by oral bacteria, not oral bacteria or oral tissues. Furthermore, previous studies reported that H2S participates in physiological regulation in the human body (28, 46, 50). Therefore, the Sox system has potential as an effective agent for controlling body systems as a H2S regulator.
More data are needed for the final goal of clinical applications of this enzyme system. The toxicity of the Sox enzyme system as well as the stabilities of rSox enzymes against proteases present in saliva need to be examined. The maximum effect of the rSox enzyme mixture appeared 2 h after the reaction started (Fig. 3B). In the present study, we examined rSox enzyme activities using assay mixtures in a tube sealed with a silicon plug. Since many components including the type and amount of the sulfur source, the pH of oral fluids, saliva flow rates, and oxygen levels in saliva and dental plaque affect the formation of VSCs in the mouth (23), the duration needed to achieve the maximum effect of the rSox enzyme mixture in the oral cavity currently remains unclear. We expect the reducing effects of rSox enzymes to last for a long time. Although their effects lasted for 4 h in the assay used in the present study, the duration of these effects in the oral environment has yet to be established. Further studies are needed in order to clarify these issues before clinical applications are realized.
In conclusion, we demonstrated that the combination of seven rSox enzymes from P. pantotrophus GB17 exhibited H2S-reducing activity. These results suggest the potential of the rSox system to prevent oral malodor.
Acknowledgements
We thank Dr. Yuichi Oogai, Department of Oral Microbiology, Kagoshima University Graduate School of Medical and Dental Sciences for his help. This study was supported in part by a research grant from the Futokukai Foundation (A. R.).
References
- 1. Ademovski, S.E., G.R. Persson, E. Winkel, A. Tangerman, P. Lingstrom, and S. Renvert. 2013. The short-term treatment effects on the microbiota at the dorsum of the tongue in intra-oral halitosis patients—a randomized clinical trial. Clin Oral Investig. 17:463-473.
- 2. Aoyama, I., K. Yaegaki, B. Calenic, H. Ii, N. Ishkitiev, and T. Imai. 2012. The role of p53 in an apoptotic process caused by an oral malodorous compound in periodontal tissues: a review. J Breath Res. 6:017104.
- 3. Aylikci, B.U., and H. Colak. 2013. Halitosis: From diagnosis to management. J Nat Sci Biol Med. 4:14-23.
- 4. Barclay, C.W., D. Spence, and W.R. Laird. 2005. Intra-oral temperatures during function. J Oral Rehabil. 32:886-894.
- 5. Blom, T., D.E. Slot, M. Quirynen, and G.A. Van der Weijden. 2012. The effect of mouthrinses on oral malodor: a systematic review. Int J Dent Hyg. 10:209-222.
- 6. Bollen, C.M., and T. Beikler. 2012. Halitosis: the multidisciplinary approach. Int J Oral Sci. 4:55-63.
- 7. Borden, L.C., E.S. Chaves, J.P. Bowman, B.M. Fath, and G.L. Hollar. 2002. The effect of four mouthrinses on oral malodor. Compend Contin Educ Dent. 23:531-536, 538, 540 passim; quiz 548.
- 8. Burnette, W.N. 1981. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem. 112:195-203.
- 9. Carvalho, M.D., C.M. Tabchoury, J.A. Cury, S. Toledo, and G.R. Nogueira-Filho. 2004. Impact of mouthrinses on morning bad breath in healthy subjects. J Clin Periodontol. 31:85-90.
- 10. Cha, J.M., H.J. Shin, S.H. Roh, and S.I. Kim. 2007. Hydrogen sulfide removal by immobilized Thiobacillus novellas on SiO2 in a fluidized bed reactor. J Microbiol Biotechnol. 17:320-324.
- 11. Dahl, C., C. Friedrich, and A. Kletzin. 2008. Sulfur Oxidation in Prokaryotes, eLS. 10.1002/9780470015902.a0021155John Wiley & Sons, New York.
- 12. Friedrich, C.G., A. Quentmeier, F. Bardischewsky, D. Rother, R. Kraft, S. Kostka, and H. Prinz. 2000. Novel genes coding for lithotrophic sulfur oxidation of Paracoccus pantotrophus GB17. J Bacteriol. 182:4677-4687.
- 13. Friedrich, C.G., D. Rother, F. Bardischewsky, A. Quentmeier, and J. Fischer. 2001. Oxidation of reduced inorganic sulfur compounds by bacteria: emergence of a common mechanism? Appl Environ Microbiol. 67:2873-2882.
- 14. Ghosh, W., S. Mallick, and S.K. DasGupta. 2009. Origin of the Sox multienzyme complex system in ancient thermophilic bacteria and coevolution of its constituent proteins. Res Microbiol. 160:409-420.
- 15. Hughes, F.J., and R. McNab. 2008. Oral malodour—a review. Arch Oral Biol. 53 (Suppl 1):S1-7.
- 16. Kleinberg, I., and D.M. Codipilly. 2002. Cysteine challenge testing: a powerful tool for examining oral malodour processes and treatments in vivo. Int Dent J. 52 (Suppl 3):221-228.
- 17. Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680-685.
- 18. Lancero, H., J. Niu, and P.W. Johnson. 1996. Exposure of periodontal ligament cells to methyl mercaptan reduces intracellular pH and inhibits cell migration. J Dent Res. 75:1994-2002.
- 19. Lowry, O.H., N.J. Rosebrough, A.L. Farr, and R.J. Randall. 1951. Protein measurement with the Folin phenol reagent. J Biol Chem. 193:265-275.
- 20. Ludwig, W., G. Mittenhuber, and C.G. Friedrich. 1993. Transfer of Thiosphaera pantotropha to Paracoccus denitrificans. Int J Syst Bacteriol. 43:363-367.
- 21. Madhushankari, G.S., A. Yamunadevi, M. Selvamani, K.P. Mohan Kumar, and P.S. Basandi. 2015. Halitosis—an overview: part-I— classification, etiology, and pathophysiology of halitosis. J Pharm Bioallied Sci. 7:S339-343.
- 22. Michailesco, P.M., J. Marciano, A.R. Grieve, and M.J. Abadie. 1995. An in vivo recording of variations in oral temperature during meals: a pilot study. J Prosthet Dent. 73:214-218.
- 23. Morita, M., and H.L. Wang. 2001. Association between oral malodor and adult periodontitis: a review. J Clin Periodontol. 28:813-819.
- 24. Ng, W., and J. Tonzetich. 1984. Effect of hydrogen sulfide and methyl mercaptan on the permeability of oral mucosa. J Dent Res. 63:994-997.
- 25. Odintsova, E.V., H.W. Jannasch, J.A. Mamone, and T.A. Langworthy. 1996. Thermothrix azorensis sp. nov., an obligately chemolithoautotrophic, sulfur-oxidizing, thermophilic bacterium. Int J Syst Bacteriol. 46:422-428.
- 26. Persson, S., M.B. Edlund, R. Claesson, and J. Carlsson. 1990. The formation of hydrogen sulfide and methyl mercaptan by oral bacteria. Oral Microbiol Immunol. 5:195-201.
- 27. Persson, S. 1992. Hydrogen sulfide and methyl mercaptan in periodontal pockets. Oral Microbiol Immunol. 7:378-379.
- 28. Pichette, J., and J. Gagnon. 2016. Implications of hydrogen sulfide in glucose regulation: how H2S can alter glucose homeostasis through metabolic hormones. Oxid Med Cell Longev. 2016:3285074.
- 29. Quentmeier, A., R. Kraft, S. Kostka, R. Klockenkamper, and C.G. Friedrich. 2000. Characterization of a new type of sulfite dehydrogenase from Paracoccus pantotrophus GB17. Arch Microbiol. 173:117-125.
- 30. Quirynen, M., H. Zhao, C. Soers, C. Dekeyser, M. Pauwels, W. Coucke, and D. Steenberghe. 2005. The impact of periodontal therapy and the adjunctive effect of antiseptics on breath odor-related outcome variables: a double-blind randomized study. J Periodontol. 76:705-712.
- 31. Rainey, F.A., D.P. Kelly, E. Stackebrandt, J. Burghardt, A. Hiraishi, Y. Katayama, and A.P. Wood. 1999. A re-evaluation of the taxonomy of Paracoccus denitrificans and a proposal for the combination Paracoccus pantotrophus comb. nov. Int J Syst Bacteriol. 49 (Pt 2):645-651.
- 32. Ramirez, M., J.M. Gomez, G. Aroca, and D. Cantero. 2009. Removal of hydrogen sulfide by immobilized Thiobacillus thioparus in a biotrickling filter packed with polyurethane foam. Bioresour Technol. 100:4989-4995.
- 33. Robertson, L.A., and J.G. Kuenen. 1983. Thiosphaera pantotropha gen. nov. sp. nov., a facultatively anaerobic, facultatively autotrophic sulphur bacterium. J Gen Microbiol. 129:2847-2855.
- 34. Rother, D., H.J. Henrich, A. Quentmeier, F. Bardischewsky, and C.G. Friedrich. 2001. Novel genes of the sox gene cluster, mutagenesis of the flavoprotein SoxF, and evidence for a general sulfur-oxidizing system in Paracoccus pantotrophus GB17. J Bacteriol. 183:4499-4508.
- 35. Rother, D., and C.G. Friedrich. 2002. The cytochrome complex SoxXA of Paracoccus pantotrophus is produced in Escherichia coli and functional in the reconstituted sulfur-oxidizing enzyme system. Biochim Biophys Acta, Proteins and Proteomics. 1598:65-73.
- 36. Rother, D., J. Ringk, and C.G. Friedrich. 2008. Sulfur oxidation of Paracoccus pantotrophus: the sulfur-binding protein SoxYZ is the target of the periplasmic thiol-disulfide oxidoreductase SoxS. Microbiology. 154:1980-1988.
- 37. Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual, 2nd ed. Cold Spring Harbor Laboratory Press, New York.
- 38. Setoguchi, T., M. Machigashira, M. Yamamoto, Y. Yotsumoto, M. Yoshimori, Y. Izumi, and K. Yaegaki. 2002. The effects of methyl mercaptan on epithelial cell growth and proliferation. Int Dent J. 52 (Suppl 3):241-246.
- 39. Skirnisdottir, S., G.O. Hreggvidsson, O. Holst, and J.K. Kristjansson. 2001. Isolation and characterization of a mixotrophic sulfur-oxidizing Thermus scotoductus. Extremophiles. 5:45-51.
- 40. Slot, D.E., S. De Geest, F.A. van der Weijden, and M. Quirynen. 2015. Treatment of oral malodour. Medium-term efficacy of mechanical and/or chemical agents: a systematic review. J Clin Periodontol. 42 (Suppl 16):S303-316.
- 41. Syed, M., G. Sereanu, P. Falletta, and M. Béland. 2006. Removal of hydrogen sulfide from gas streams using biological processes—A review. Can Biosys Eng. 48:2.1-2.14.
- 42. Takeuchi, H., T. Setoguchi, M. Machigashira, K. Kanbara, and Y. Izumi. 2008. Hydrogen sulfide inhibits cell proliferation and induces cell cycle arrest via an elevated p21 Cip1 level in Ca9-22 cells. J Periodontal Res. 43:90-95.
- 43. Tonzetich, J. 1971. Direct gas chromatographic analysis of sulphur compounds in mouth air in man. Arch Oral Biol. 16:587-597.
- 44. Tonzetich, J., and P.W. Johnson. 1977. Chemical analysis of thiol, disulphide and total sulphur content of human saliva. Arch Oral Biol. 22:125-131.
- 45. van den Broek, A.M., L. Feenstra, and C. de Baat. 2008. A review of the current literature on management of halitosis. Oral Dis. 14:30-39.
- 46. Vandiver, M., and S.H. Snyder. 2012. Hydrogen sulfide: a gasotransmitter of clinical relevance. J Mol Med (Berl). 90:255-263.
- 47. Wigger-Alberti, W., K. Gysen, E.M. Axmann, and K.P. Wilhelm. 2010. Efficacy of a new mouthrinse formulation on the reduction of oral malodour in vivo. A randomized, double-blind, placebo-controlled, 3 week clinical study. J Breath Res. 4:017102.
- 48. Wingfield, P.T. 1995. Purification of recombinant proteins, p.6.1.1-6.2.15. In J.E. Coligan, B.M. Dunn, H.L. Ploegh, D.W. Speicher, and P.T. Wingfield (ed.), Current Protocols in Protein Science. 1. John Wiley & Sons, New York.
- 49. Yoshimura, M., Y. Nakano, Y. Yamashita, T. Oho, T. Saito, and T. Koga. 2000. Formation of methyl mercaptan from L-methionine by Porphyromonas gingivalis. Infect Immun. 68:6912-6916.
- 50. Zhou, C.F., and X.Q. Tang. 2011. Hydrogen sulfide and nervous system regulation. Chin Med J (Engl). 124:3576-3582.



