Abstract
Nitrite reductase is a key enzyme for denitrification. There are two types of nitrite reductases: copper-containing NirK and cytochrome cd1-containing NirS. Most denitrifiers possess either nirK or nirS, although a few strains been reported to possess both genes. We herein report the presence of nirK and nirS in the soil-denitrifying bacterium Bradyrhizobium sp. strain TSA1T. Both nirK and nirS were identified and actively transcribed under denitrification conditions. Based on physiological, chemotaxonomic, and genomic properties, strain TSA1T (=JCM 18858T=KCTC 62391T) represents a novel species within the genus Bradyrhizobium, for which we propose the name Bradyrhizobium nitroreducens sp. nov.
Denitrification is a microbial respiratory process in which nitrogen oxides, such as nitrate and nitrite, are reduced in a stepwise manner to gaseous nitrogen products (NO, N2O, and N2) (36). Each step in denitrification is catalyzed by distinct enzymes: nitrate reductase (Nar), nitrite reductase (Nir), nitric oxide reductase (Nor), and nitrous oxide reductase (Nos). There are two types of nitrite reductases: the copper-containing NirK type and cytochrome cd1-containing NirS type (36). The genes encoding these enzymes (nirK and nirS) have been frequently used as markers to identify denitrifiers (2). Although a few strains have been reported to harbor both nirK and nirS (3, 6, 22), most denitrifiers possess either nirK or nirS.
We previously isolated several Bradyrhizobium sp. strains that possess nirS from rice paddy soil in Japan (8). We then isolated additional nirS-harboring Bradyrhizobium sp. denitrifiers from other soils (15). The presence of nirS in Bradyrhizobium spp. is unique because most of the other Bradyrhizobium strains possess nirK (1, 21, 22). In addition, these nirS sequences form distinct clusters from other nirS sequences (8). Most of the nirK and nirS sequences (e.g., 16–42% and 15–35%, respectively) obtained by culture-independent analyses were found to be closely related to the nirK and nirS of the Bradyrhizobium sp. denitrifier (Fig. S1) (32, 33), suggesting an important role for Bradyrhizobium spp. in denitrification in the environment. Based on our preliminary PCR examination, we detected nirK in some nirS-positive Bradyrhizobium strains. In order to further clarify whether these strains possess both nirK and nirS, these genes need to be identified by whole genome sequencing.
We herein report the presence of both nirK and nirS in Bradyrhizobium sp. denitrifiers based on a genome analysis and transcription experiments. We further analyzed the physiological traits, such as nitrogen fixation and nodulation abilities, of the representative strain TSA1T. The results of these analyses indicated that nirK- and nirS-positive Bradyrhizobium strains were taxonomically distinct from any of the previously established species of the genus Bradyrhizobium. Thus, we propose Bradyrhizobium nitroreducens sp. nov. for strain TSA1T. Strain TSA1T was selected as the representative of nirK- and nirS-positive Bradyrhizobium based on a phylogenetic analysis targeting the 16S rRNA gene and internal transcribed spacer (ITS) sequences (8).
Strain TSA1T was originally isolated from rice paddy soil collected at the Institute for Sustainable Agro-Ecosystems, the Graduate School of Agricultural and Life Sciences, The University of Tokyo (Nishitokyo, Tokyo, Japan; 35°44′ N, 139°32′ E) (8). The culture of strain TSA1T was maintained on 100-fold diluted nutrient broth (DNB) (BD Difco™; Becton, Dickinson and Company, Franklin, Lakes, NJ, USA) supplemented with 3 mM nitrate and 4.4 mM succinate (DNBNS medium) or 1.5% agar plates of DNBNS medium (DNBNS agar medium) under an anoxic incubation at 30°C as described previously (8). Flagellar motility was assayed on semi-solid DNBNS medium containing 0.075% agar (11). In fatty acid and quinone analyses, cells were aerobically grown on R2A agar (BD Difco™) and R2A liquid medium, respectively, and harvested at the exponential phase of growth. Cellular fatty acids were analyzed by gas chromatography, as previously described (11). Quinones were analyzed by reverse-phase HPLC as described elsewhere (13). Growth at different temperatures, pH, and antibiotic supplement conditions was examined in R2A liquid medium under an aerobic incubation at 30°C. In the carbon utilization test, modified yeast extract (YE) medium containing a minimal amount of yeast extract (10 mg L−1) and 10 g L−1 carbon source was used as described previously (28). The growth rates of the strain were measured in R2A broth medium and yeast extract mannitol (YEM) broth medium (27) at 30 and 37°C. Cell morphology was observed using the Philips CM12 transmission electron microscope (TEM) after cells were negatively stained with 1% of aqueous potassium phosphotungstic acid (pH 7.0).
Potential nitrogen fixation ability was examined by measuring acetylene reduction activity as described previously (11). Denitrification ability was analyzed using the acetylene block method as described previously (23). N2O-reducing ability was examined using 15N-labeled N2O (15N, 99 atom. %; Cambridge Isotope Laboratories) and gas chromatography/mass spectrometry (GC/MS) as described previously (9). A nodulation assay was performed using siratro as the host plant as described elsewhere (20). Bradyrhizobium diazoefficiens USDA110T was used as the positive control for these assays.
The genomic DNA of strain TSA1T was extracted for genome sequencing as previously described (14). The library was prepared using the TruSeq DNA sample prep kit (Illumina, San Diego, CA, USA) and sequenced using the Illumina HiSeq 2000 platform with 101-bp paired-end sequencing chemistry. Sequence reads were assembled using Velvet v. 12.0.8 (35) followed by gene annotation performed with the NCBI Prokaryotic Genome Annotation Pipeline (25). A genome sequence and annotation summary is shown in Table S1. Average nucleotide identity (ANI) values were calculated using JSpecies (19) in order to examine the relatedness of strain TSA1T and its close relatives. We also calculated the percentage of conserved proteins (POCP) as described by Qin et al. (17).
A phylogenetic analysis was performed using the 16S rRNA gene, ITS, nirK, and nirS sequences in order to examine sequence relatedness among Bradyrhizobium and other denitrifying strains. Multilocus sequencing typing (MLST) was also performed using concatenated sequences of glnII, recA, rpoB, and dnaK (7). Phylogenetic trees were generated using the neighbor-joining and maximum-likelihood methods with a bootstrap analysis (n=1,000) using MEGA6 software (24).
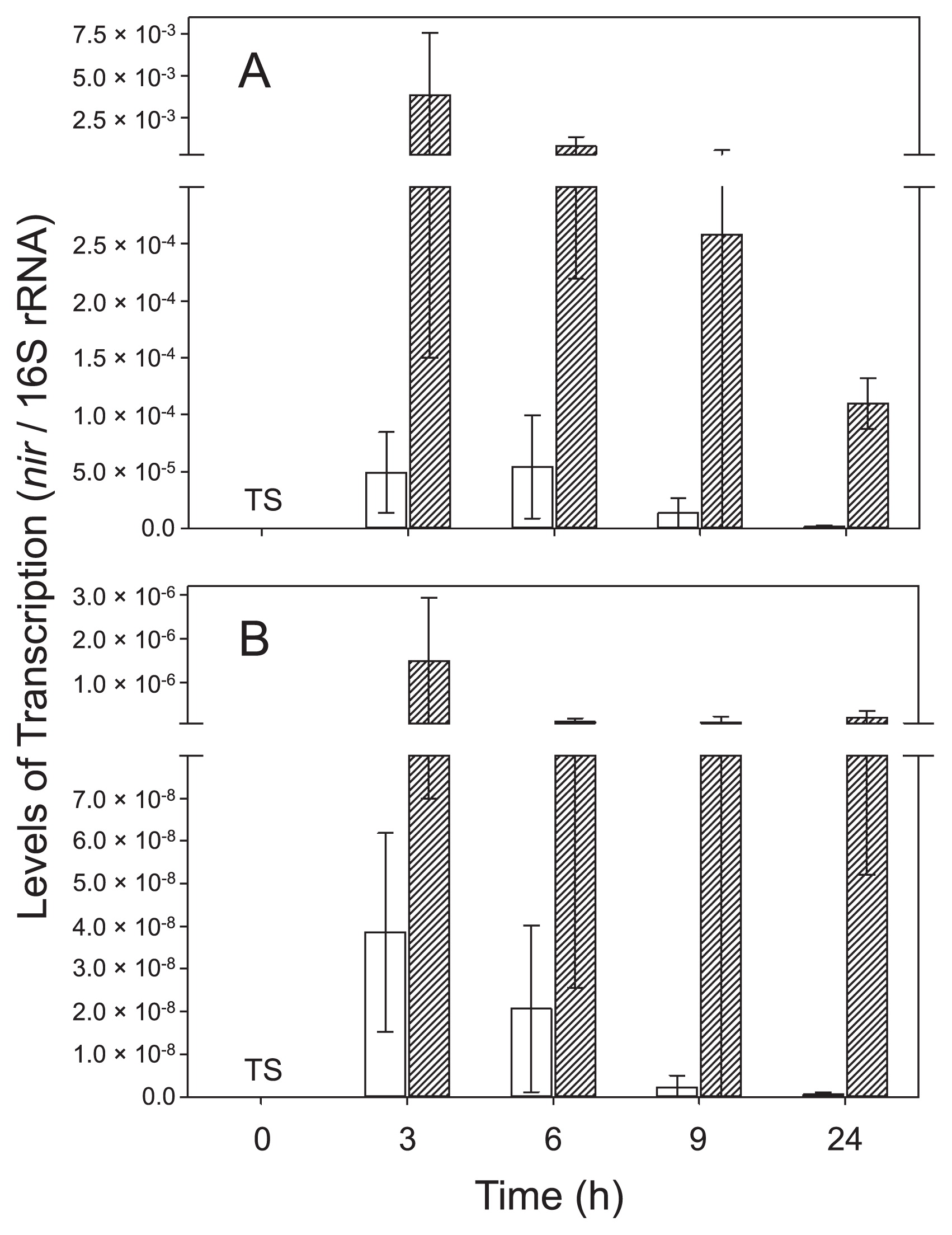
In order to examine whether nirK and nirS are transcribed under denitrification conditions, we performed reverse transcription quantitative PCR (RT-qPCR) targeting these gene transcripts as well as 16S rRNA. RNA was isolated from TSA1T cells grown in R2A broth supplemented with 10 mM acetate and 5 mM nitrate under oxic and anoxic conditions for 3, 6, 12, and 24 h. RNA was also isolated from preculture cells grown in the same medium but without nitrate under oxic conditions (=0-h samples). The absence of genomic DNA in RNA samples was verified by PCR targeting the 16S rRNA gene as described previously (7). The primers nirSCd3aF and nirSR3cd were used for the quantification of nirS (12), while we designed the new primers BRnirK_F (5′-TTCGT CTATCACTGCGCC-3′) and BRnirK_R (5′-CAGCTTCTT CATCACCTCTTC-3′) for the quantification of nirK because currently available primers had several base mismatches to the nirK of strain TSA1T. RNA was reverse transcribed using the PrimeScript RT reagent kit (Takara Bio, Otsu, Shiga, Japan) with random hexamers as described previously (10). The reaction mixture for RT-qPCR contained 1× SYBR Premix ExTaq ROXplus (Takara Bio), 0.2 μM of each primer, and 100 ng cDNA. RT-qPCR was performed using the StepOnePlus Real-Time PCR System (v. 2.3; Applied Biosystems, Foster City, CA, USA) under the following conditions: 95°C for 30 s, followed by 40 cycles of 95°C for 5 s and 60°C for 30 s. Transcription levels were normalized using the quantity of 16S rRNA.
The morphological, cultural, physiological, and biochemical characteristics of the nirK and nirS-harboring Bradyrhizobium strains represented by strain TSA1T are summarized in Table 1. Cells were rod-shaped, measuring 0.4–0.6 μm in width and 1.6–2.2 μm in length, and were motile with a single polar flagellum (Fig. S4). The colonies that formed on R2A agar medium were smooth, circular, white, and convex. Cells of strain TSA1T grew in a temperature range of 25–37°C (optimum, 30°C) in R2A and YEM media. The doubling times of TSA1T in R2A and YEM media at 30°C were 10.6 and 11.2 h, respectively, similar to other Bradyrhizobium strains (34). Cells also grew under a wide range of pH conditions (pH 4.5–10.0) similar to B. ottawaense O99T, but dissimilar to the other Bradyrhizobium strains. Unlike B. ottawaense O99T, strain TSA1T did not grow in media containing >1% NaCl (w/v). Strain TSA1T showed resistance to 100 μg mL−1 ampicillin, 20 μg mL−1 tetracycline, and 300 μg mL−1 polymyxin. Resistance to these antibiotics made TSA1T distinct from other closely related Bradyrhizobium strains. Growth was inhibited by 100 μg mL−1 erythromycin and 50 μg mL−1 kanamycin. Cells grew using d- and l-arabinose, d-fructose, d-glucose, maltose, d-mannitol, d-mannose, l-rhamnose, gluconate, d-sorbitol, and d-xylose. Growth was not observed in YE medium supplemented with cellobiose, inositol, d-lactose, and d-sucrose.
Table 1
Phenotypic, genetic, and chemotaxonomic characteristics that separate strain TSA1
T from closely related phylogenetic neighbors. Strains: 1, TSA1
T; 2,
B. yuanmingense CCBAU 10071
T; 3,
B. liaoningense 2281
T; 4,
B. diazoefficiens USDA110
T; 5,
B. ottawaense O99
T; 6,
B. oligotropha S58
T.
| Characteristic |
1 |
2 |
3 |
4 |
5 |
6 |
| Denitrification |
+ |
ND |
ND |
+ |
− |
+ |
| nir type |
nirK, nirS |
nirK |
nirKa |
nirK |
nirK |
nirK, nirS |
| Nitrogen fixation |
+ |
+ |
+ |
+ |
+ |
+ |
| Nodulation |
− |
+ |
+ |
+ |
+ |
+ |
| Major FA |
16:0, 18:1 ω7c |
ND |
ND |
16:0, 18:1 ω6c/ω7c |
16:0, 18:1 ω6c/ω7c |
18:1 |
| Major quinone |
Q10 |
ND |
ND |
Q10b |
ND |
Q10 |
| DNA G+C content (mol %) |
64.3 |
62–64 |
60–64 |
64 |
62.6 |
65.1 |
| Carbon source utilization |
| d-Arabinose |
+ |
W |
W |
+ |
ND |
ND |
| l-Arabinose |
+ |
+ |
+ |
W |
+ |
+ |
| Cellobiose |
− |
− |
− |
− |
ND |
− |
| d-Fructose |
+ |
W |
− |
W |
ND |
ND |
| d-Glucose |
+ |
W |
− |
W |
+ |
+ |
| Inositol |
− |
− |
− |
− |
ND |
− |
| d-Lactose |
− |
− |
− |
+ |
− |
− |
| Maltose |
+ |
W |
− |
− |
+ |
− |
| d-Mannitol |
+ |
W |
W |
W |
+ |
+ |
| d-Mannose |
+ |
W |
+ |
W |
+ |
+ |
| l-Rhamnose |
+ |
W |
W |
W |
+ |
+ |
| Sodium gluconate |
+ |
− |
W |
− |
+ |
+ |
| d-Sorbitol |
W |
− |
− |
W |
ND |
− |
| d-Sucrose |
− |
− |
− |
W |
ND |
− |
| d-Xylose |
+ |
+ |
+ |
W |
+ |
+ |
| Growth in/at |
| 37°C |
+ |
+ |
− |
− |
+ |
+ |
| pH 4.5 |
+ |
W |
W |
+ |
+ |
ND |
| pH 8.0 |
+ |
+ |
+ |
− |
+ |
ND |
| pH 10.0 |
+ |
− |
− |
− |
+ |
ND |
| 1% NaCl |
− |
− |
− |
− |
+ |
− |
| Resistance (μg mL−1) |
| Polymyxin (300) |
+ |
+ |
ND |
− |
ND |
ND |
| Erythromycin (100) |
− |
− |
− |
− |
+ |
ND |
| Tetracycline (20) |
+ |
+ |
W |
− |
+ |
ND |
| Ampicillin (10) |
+ |
− |
− |
W |
− |
ND |
|
| Reference |
This study |
(4, 31) |
(4, 30) |
(4) |
(34) |
(16, 18) |
a Based on the genome of strain CCNWSX0360 (GenBank accession LUKO01000171)
b Assessed in this study.
+, growth; −, no growth; w, weakly positive; ND, not determined.
Strain TSA1T exhibited the ability to grow under anoxic conditions with nitrate as the terminal electron acceptor as well as under aerobic conditions. They also reduced exogenous N2O to N2. The presence of all of the functional genes for denitrification, including napA, nirK, nirS, norB, and nosZ, on an 8.1-Mbp contig of the genome (Contig01) of strain TSA1T (Table S2) supported this result.
Based on RT-qPCR results, nirK and nirS of strain TSA1T were both actively transcribed under anoxic conditions in the presence of nitrate (i.e., denitrification conditions), although the number of nirK transcripts was always higher than that of nirS transcripts (Fig. 1). The transcription of nirK and nirS was the most intensive under denitrification conditions at the first time point (3 h), and decreased thereafter. The transcription of nirK and nirS was suppressed when oxygen was present and nitrate was absent. Although the addition of nitrate increased the transcription levels of nirK and nirS under oxic conditions, transcription levels were still significantly lower than those under anoxic conditions (P<0.05 by ANOVA). These results suggest that nirK and nirS in strain TSA1T are regulated in response to the availability of oxygen and nitrate. The occurrence of both nirK and nirS in B. oligotrophicum S58T (formally known as Agromonas oligotropha) (16) was recently reported (22). The findings reported by Sanchez and Minamisawa (22) suggest that NirK-type and NirS-type nitrite reductases are functionally redundant in strain S58T; however, the functional redundancy of nitrite reductases in strain TSA1T remains unknown.
Genes related to nitrogen fixation, such as fixR, nifA, sufBCDSE, nifDKENX, nifHV, and fixABC, were identified on the genome of strain TSA1T. These gene clusters are highly conserved among symbiotic nitrogen-fixing bradyrhizobia (16). The nitrogen-fixing ability of strain TSA1T was confirmed in an acetylene reduction assay. Strain TSA1T lacks the common nodulation gene cluster nodABC on its genome, and did not nodulate siratro (data not shown). In addition, the photosynthetic gene cluster (pufBALM) was not detected on the genome of strain TSA1T. These results suggest that strain TSA1T is a nitrogen-fixing, non-phototrophic, free-living Bradyrhizobium.
Strain TSA1T was the most closely related to B. diazoefficiens USDA110T based on the 16S rRNA gene sequence analysis, with a similarity value of 99.7% (Fig. S2); however, strain TSA1T was more closely related to B. liaoningense CCNWSX0360 and B. yuanmingense CCBAU10071T based on an analysis of ITS sequences (Fig. 2A). The MLST analysis based on four housekeeping genes suggested that strain TSA1T was also closely related to B. ottawaense O99T (Fig. S3). The nirK sequence of strain TSA1T was also closely related to those of B. ottawaense, B. diazoefficiens, B. liaoningense, and B. yuanmingense (Fig. 2B). While B. oligotrophicum S58T also possesses the nirK and nirS homologs, this strain was relatively distantly related to strain TSA1T based on the 16S rRNA gene, ITS, and nirK sequences. In contrast, nirS sequences from strain TSA1T and B. oligotrophicum S58T were similar to each other with 87% sequence similarity (Fig. 2C). These phylogenetic analyses suggest that strain TSA1T has a common evolutionary history with B. diazoefficiens, B. liaoningense, B. yuanmingense, and B. ottawaense, and nirS was horizontally acquired from elsewhere by strain TSA1T and B. oligotrophicum S58T. We attempted to provide evidence of horizontal gene transfer (e.g., integrons, phages, insertion sequences, and genomic islands) around nirS, but were unsuccessful.
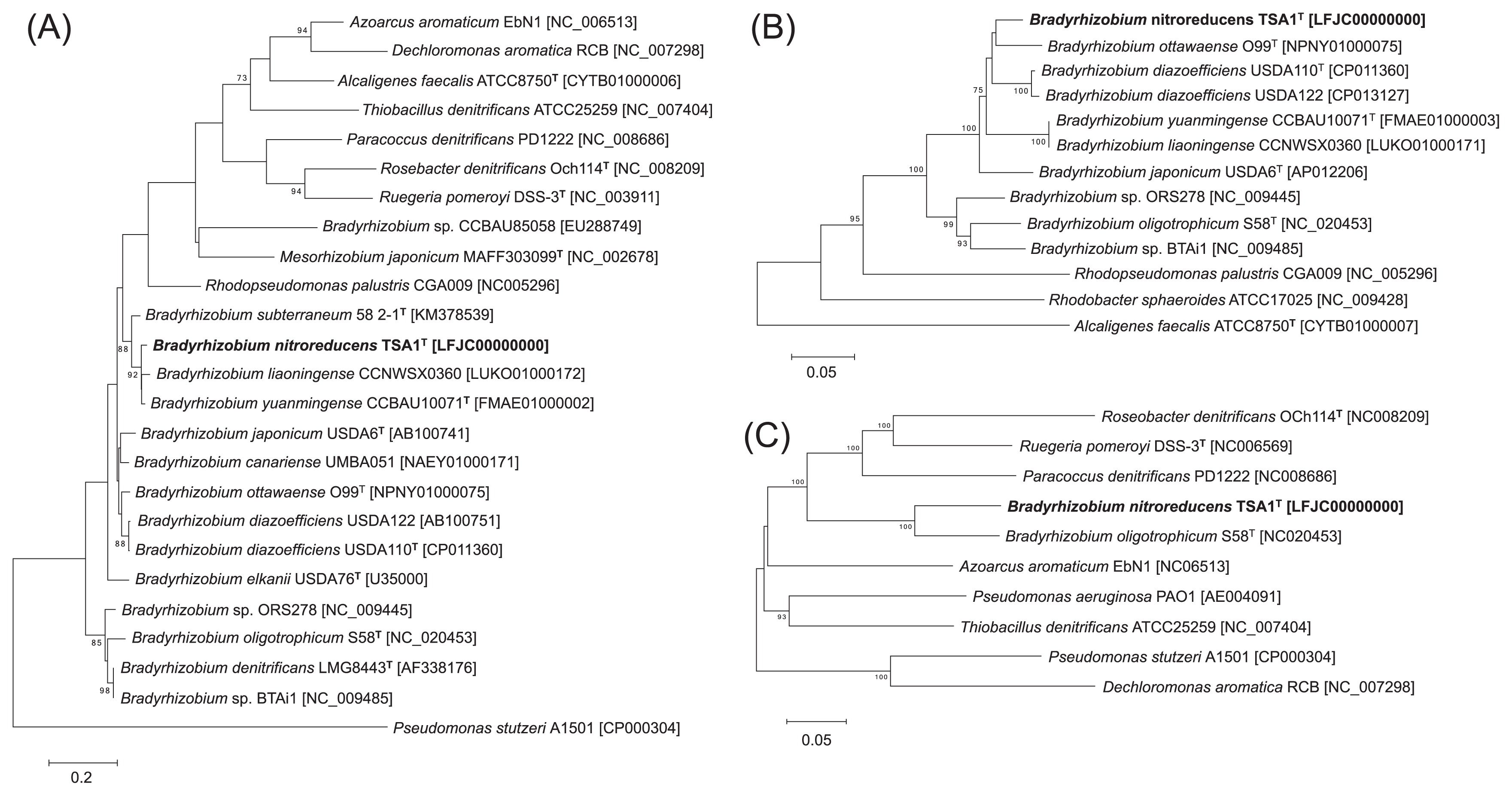
The average nucleotide identity (ANI) values between strain TSA1T and closely related Bradyrhizobium species (selected based on the 16S rRNA and ITS phylogenies) ranged between 61 and 86% (Table S3). These ANI values were smaller than 95%, which is recommended as the species boundary (5), indicating that strain TSA1T needs to be classified as a novel species within the genus Bradyrhizobium. The POCP value between strain TSA1T and most of the other closely related Bradyrhizobium species ranged between 54 and 69% (Table S4). The POCP value of 50% was proposed as the cut-off for the genus (17), supporting the affiliation of strain TSA1T to the genus Bradyrhizobium. The POCP value between strain TSA1T and B. oligotrophicum S58T was smaller than the genus cut-off value of 50% (37.08%, Table S4). However, B. oligotrophicum S58T also had low POCP values with other Bradyrhizobium species, suggesting that B. oligotrophicum S58T had different gene contents from other Bradyrhizobium species, which is consistent with the phylogenetic analysis based on the four housekeeping genes (Fig. S3).
The G+C content of strain TSA1T was 64.3% based on draft genome sequencing. This value is similar to those of other closely related Bradyrhizobium spp. (Table 1). Strain TSA1T had Q-10 as the only ubiquinone, similar to other Bradyrhizobium spp. (29). The fatty acid profile of TSA1T contained 75.8% C18:1 ω7c, 15.0% C16:0, 5.9% C18:0, 2.0% C12:0, and 1.3% C14:0, which is similar to other Bradyrhizobium spp. (26). These results support the affiliation of strain TSA1T to the genus Bradyrhizobium.
In conclusion, the group of Bradyrhizobium sp. strains represented by strain TSA1T is unique, in that they possess both Cu-type and cytochrome cd1-type nitrite reductase genes (nirK- and nirS). Although nirK and nirS were also found on the genome of B. oligotrophicum S58T, strain TSA1T is phylogenetically distantly related to strain S58T. Since a large proportion of nirK and nirS clone libraries was related to the nitrite reductase genes of Bradyrhizobium sp. TSA1T (32, 33), these bacteria may play an important role in denitrification in soil. Based on the 16S rRNA gene sequence analysis in combination with physiological, chemotaxonomic, and genomic properties, strain TSA1T (=JCM 18858T=KCTC 62391T) represents a novel species within the genus Bradyrhizobium, for which we propose the name Bradyrhizobium nitroreducens sp. nov.
Description of Bradyrhizobium nitroreducens sp. nov.
Bradyrhizobium nitroreducens (ni.tro.re.du’cens. Gr. n. nitron nitrate, nitrite; L. adj. from pres. part. of verb reduco reduce, bring back to a condition; N.L. adj. nitroreducens reducing nitrate and nitrite).
Cells are Gram-negative rods measuring 0.4–0.6 μm in width and 1.6–2.2 μm in length. Motile by means of a polar flagellum. Colonies formed on R2A agar medium are smooth, circular, white, and convex. Facultative anaerobic. Cells grow with oxygen, nitrate, or N2O as the terminal electron acceptor. Denitrification positive. Cells possess both nirK and nirS as the nitrite reductase gene. Nitrogen fixation positive. Cells do not form nodules to siratro. Cells have the ability to grow at 25–37°C (optimum 30°C) and pH 4.5–10. No growth occurs in the presence of 1% NaCl in R2A medium. Cells are resistant to ampicillin (100 μg mL−1), tetracycline (20 μg mL−1), and polymyxin (300 μg mL−1), but sensitive to erythromycin (100 μg mL−1) and kanamycin (50 μg mL−1). Usable carbon and energy sources are d- and l-arabinose, d-fructose, d-glucose, maltose, d-mannitol, d-mannose, l-rhamnose, gluconate, d-sorbitol, and d-xylose. Cellobiose, inositol, d-lactose, and d-sucrose are not used. The major cellular fatty acids (>10% of total fatty acids) are C16:0 and C18:1ω7c. The predominant quinone is ubiquinone-10. The type strain TSA1T (=JCM 18858T=KCTC 62391T) was isolated from rice paddy soil in Tokyo, Japan. The DNA G+C content of the type strain is 64.3% as calculated from draft genome sequencing.
The GenBank/EMBL/DDBJ accession numbers for the 16S rRNA gene sequence and the genome sequence of strain TSA1T are AB542368 and LFJC00000000, respectively.
Acknowledgements
This work was supported, in part, by the Programme for Promotion of Basic and Applied Research for Innovations in Bio-oriented Industry (BRAIN) and Science and Technology Research Promotion Program for Agriculture, Forestry, Fisheries and Food Industry, and by the Minnesota’s Discovery, Research and InnoVation Economy (MnDRIVE) initiative of the University of Minnesota.
References
- 1. Bedmar, E.J., E.F. Robles, and M.J. Delgado. 2005. The complete denitrification pathway of the symbiotic, nitrogen-fixing bacterium Bradyrhizobium japonicum. Biochem Soc Trans. 33:141-144.
- 2. Braker, G., J. Zhou, L. Wu, A.H. Devol, and J.M. Tiedje. 2000. Nitrite reductase genes (nirK and nirS) as functional markers to investigate diversity of denitrifying bacteria in pacific northwest marine sediment communities. Appl Environ Microbiol. 66:2096-2104.
- 3. Campbell, M.A., G. Nyerges, J.A. Kozlowski, A.T. Poret-Peterson, L.Y. Stein, and M.G. Klotz. 2011. Model of the molecular basis for hydroxylamine oxidation and nitrous oxide production in methanotrophic bacteria. FEMS Microbiol Lett. 322:82-89.
- 4. Delamuta, J.R.M., R.A. Ribeiro, E. Ormeño-Orrillo, I.S. Melo, E. Martínez-Romero, and M. Hungria. 2013. Polyphasic evidence supporting the reclassification of Bradyrhizobium japonicum group ia strains as Bradyrhizobium diazoefficiens sp. nov. Int J Syst Evol Microbiol. 63:3342-3351.
- 5. Goris, J., K.T. Konstantinidis, J.A. Klappenbach, T. Coenye, P. Vandamme, and J.M. Tiedje. 2007. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 57:81-91.
- 6. Graf, D.R.H., C.M. Jones, and S. Hallin. 2014. Intergenomic comparisons highlight modularity of the denitrification pathway and underpin the importance of community structure for N2O emissions. PLoS One. 9:e114118.
- 7. Grönemeyer, J.L., T. Hurek, W. Bünger, and B. Reinhold-Hurek. 2016. Bradyrhizobium vignae sp. nov., a nitrogen-fixing symbiont isolated from effective nodules of Vigna and Arachis. Int J Syst Evol Microbiol. 66:62-69.
- 8. Ishii, S., N. Ashida, S. Otsuka, and K. Senoo. 2011. Isolation of oligotrophic denitrifiers carrying previously uncharacterized functional gene sequences. Appl Environ Microbiol. 77:338-342.
- 9. Ishii, S., H. Ohno, M. Tsuboi, S. Otsuka, and K. Senoo. 2011. Identification and isolation of active N2O reducers in rice paddy soil. ISME J. 5:1936-1945.
- 10. Ishii, S., K. Joikai, S. Otsuka, K. Senoo, and S. Okabe. 2016. Denitrification and nitrate-dependent Fe(II) oxidation in various Pseudogulbenkiania strains. Microbes Environ. 31:293-298.
- 11. Ishii, S., N. Ashida, H. Ohno, T. Segawa, S. Yabe, S. Otsuka, A. Yokota, and K. Senoo. 2017. Noviherbaspirillum denitrificans sp. nov., a denitrifying bacterium isolated from rice paddy soil and Noviherbaspirillum autotrophicum sp. nov., a denitrifying, facultatively autotrophic bacterium isolated from rice paddy soil and proposal to reclassify Herbaspirillum massiliense as Noviherbaspirillum massiliense comb. nov. Int J Syst Evol Microbiol. 67:1841-1848.
- 12. Kandeler, E., K. Deiglmayr, D. Tscherko, D. Bru, and L. Philippot. 2006. Abundance of narG, nirS, nirK, and nosZ genes of denitrifying bacteria during primary successions of a glacier foreland. Appl Environ Microbiol. 72:5957-5962.
- 13. Komagata, K., and K.-I. Suzuki. 1988. Lipid and cell-wall analysis in bacterial systematics, p.161-207. In R.R. Colwell, and R. Grigorova (ed.), Methods in Microbiology. vol. 19. Academic Press, New York.
- 14. Narihiro, T., and Y. Kamagata. 2017. Genomics and metagenomics in microbial ecology: recent advances and challenges. Microbes Environ. 32:1-4.
- 15. Nishizawa, T., K. Tago, Y. Uei, S. Ishii, K. Isobe, S. Otsuka, and K. Senoo. 2012. Advantages of functional single-cell isolation method over standard agar plate dilution method as a tool for studying denitrifying bacteria in rice paddy soil. AMB Express. 2:50.
- 16. Okubo, T., S. Fukushima, M. Itakura, et al. 2013. Genome analysis suggests that the soil oligotrophic bacterium Agromonas oligotrophica (Bradyrhizobium oligotrophicum) is a nitrogen-fixing symbiont of Aeschynomene indica. Appl Environ Microbiol. 79:2542-2551.
- 17. Qin, Q.-L., B.-B. Xie, X.-Y. Zhang, X.-L. Chen, B.-C. Zhou, J. Zhou, A. Oren, and Y.-Z. Zhang. 2014. A proposed genus boundary for the prokaryotes based on genomic insights. J Bacteriol. 196:2210-2215.
- 18. Ramírez-Bahena, M.-H., R. Chahboune, A. Peix, and E. Velázquez. 2013. Reclassification of Agromonas oligotrophica into the genus Bradyrhizobium as Bradyrhizobium oligotrophicum comb. nov. Int J Syst Evol Microbiol. 63:1013-1016.
- 19. Richter, M., and R. Rosselló-Móra. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA. 106:19126-19131.
- 20. Saito, A., H. Mitsui, R. Hattori, K. Minamisawa, and T. Hattori. 1998. Slow-growing and oligotrophic soil bacteria phylogenetically close to Bradyrhizobium japonicum. FEMS Microbiol Ecol. 25:277-286.
- 21. Sameshima-Saito, R., K. Chiba, and K. Minamisawa. 2006. Correlation of denitrifying capability with the existence of nap, nir, nor and nos genes in diverse strains of soybean bradyrhizobia. Microbes Environ. 21:174-184.
- 22. Sánchez, C., and K. Minamisawa. 2018. Redundant roles of Bradyrhizobium oligotrophicum Cu-type (nirK) and cd1-type (nirS) nitrite reductase genes under denitrifying conditions. FEMS Microbiol Lett. 365:fny015.
- 23. Tago, K., S. Ishii, T. Nishizawa, S. Otsuka, and K. Senoo. 2011. Phylogenetic and functional diversity of denitrifying bacteria isolated from various rice paddy and rice-soybean rotation fields. Microbes Environ. 26:30-35.
- 24. Tamura, K., G. Stecher, D. Peterson, A. Filipski, and S. Kumar. 2013. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30:2725-2729.
- 25. Tatusova, T., M. DiCuccio, A. Badretdin, et al. 2016. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44:6614-6624.
- 26. Tighe, S.W., P. de Lajudie, K. Dipietro, K. Lindström, G. Nick, and B.D. Jarvis. 2000. Analysis of cellular fatty acids and phenotypic relationships of Agrobacterium, Bradyrhizobium, Mesorhizobium, Rhizobium and Sinorhizobium species using the Sherlock Microbial Identification System. Int J Syst Evol Microbiol. 50:787-801.
- 27. Vincent, J.M. 1970. A manual for the practical study of the root-nodule bacteria, International Biological Programme Handbook No. 15. Blackwell Scientific Publications, Oxford.
- 28. Wagner, S.C., H.D. Skipper, and P.G. Hartel. 1995. Medium to study carbon utilization by bradyrhizobium strains. Can J Microbiol. 41:633-636.
- 29. Wang, J.Y., R. Wang, Y.M. Zhang, H.C. Liu, W.F. Chen, E.T. Wang, X.H. Sui, and W.X. Chen. 2013. Bradyrhizobium daqingense sp. nov., isolated from soybean nodules. Int J Syst Evol Microbiol. 63:616-624.
- 30. Xu, L.M., C. Ge, Z. Cui, J. Li, and H. Fan. 1995. Bradyrhizobium liaoningense sp. nov., isolated from the root nodules of soybeans. Int J Syst Evol Microbiol. 45:706-711.
- 31. Yao, Z.Y., F.L. Kan, E.T. Wang, G.H. Wei, and W.X. Chen. 2002. Characterization of rhizobia that nodulate legume species of the genus Lespedeza and description of Bradyrhizobium yuanmingense sp. nov. Int J Syst Evol Microbiol. 52:2219-2230.
- 32. Yoshida, M., S. Ishii, S. Otsuka, and K. Senoo. 2009. Temporal shifts in diversity and quantity of nirS and nirK in a rice paddy field soil. Soil Biol Biochem. 41:2044-2051.
- 33. Yoshida, M., S. Ishii, D. Fujii, S. Otsuka, and K. Senoo. 2012. Identification of active denitrifiers in rice paddy soil by DNA- and RNA-based analyses. Microbes Environ. 27:456-461.
- 34. Yu, X., S. Cloutier, J.T. Tambong, and E.S.P. Bromfield. 2014. Bradyrhizobium ottawaense sp. nov., a symbiotic nitrogen fixing bacterium from root nodules of soybeans in Canada. Int J Syst Evol Microbiol. 64:3202-3207.
- 35. Zerbino, D.R., and E. Birney. 2008. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821-829.
- 36. Zumft, W.G. 1997. Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev. 61:533-616.
