Abstract
The present study characterized the interactions of microbial populations in activated sludge systems during the operational period after an increase in the wastewater flow rate and consequential ammonia accumulation using a 16S rRNA gene sequencing-based network analysis. Two hundred microbial populations accounting for 81.8% of the total microbiome were identified. Based on a co-occurrence analysis, Nitrosomonas-type ammonia oxidizers had one of the largest number of interactions with diverse bacteria, including a bulking-associated Thiothrix organism. These results suggest that an increased flow rate has an impact on constituents by changing ammonia concentrations and also that Nitrosomonas- and Thiothrix-centric responses are critical for ammonia removal and microbial community recovery.
Activated sludge systems, a representative biological treatment technology, are widely used to remediate municipal and industrial wastewaters. Activated sludge consists of a number of microorganisms with metabolic activities to remove carbon and nitrogen in wastewaters (15, 27, 28, 32). By the early 2000s, the predominant and functionally important microorganisms in these systems were identified using 16S rRNA-targeted molecular approaches, such as clone libraries and fluorescence in situ hybridization (42, 44). In the past several years, comprehensive surveys on the microbiomes of full-scale systems have been conducted by employing high-throughput DNA sequencing technology (9, 11, 13, 18, 20, 23, 25, 39, 46). These studies identified common microbial constituents in these systems and reported the effects of seasonal/regional variations on microbiome assemblages. However, limited information is currently available on the effects of changes in operating conditions (e.g., the organic loading rate, wastewater type, and aeration rate) on microbiomes in full-scale activated sludge systems. Without an understanding of sludge microbiome dynamics under disturbed conditions, the development of strategies to maintain treatment efficiency and stability may be difficult. In the present study, we investigated temporal changes in an activated sludge microbiome after an increase in the wastewater flow rate through 16S rRNA gene iTag sequencing with a special focus on the microorganisms associated with ammonia oxidation and filamentous sludge bulking.
Activated sludge samples were collected from two full-scale conventional activated sludge tanks in different flow lines treating municipal sewage wastewater in Japan (named AB1 and AB2 with a working volume of 9,000 m3 each). Samples were collected at six time points in duplicate after increasing the wastewater flow rate and stored at −20°C prior to DNA extraction. BOD, ammonia, nitrite, and nitrate in the sludge water were analyzed by standard methods for wastewater analyses (19). DNA was extracted from sludge samples by a direct lysis protocol that includes bead beating, phenol-chloroform extraction, and ethanol precipitation (30, 31, 37). 16S rRNA genes were amplified with the primer set Univ515F/Univ806R and Q5 DNA polymerase (New England Biolabs Japan, Tokyo, Japan) (1, 38). MiSeq sequencing was performed using v2 chemistry (Illumina, San Diego, CA, USA). Raw paired-end 16S rRNA gene reads were assembled and screened with mothur 1.35.1 using sequence length (≥200 nt) and quality score (≥30) cut-offs (40). Screened sequence data were grouped into OTUs with the UCLUST algorithm using a 97% sequence identity cut-off (8). Representative sequences for each OTU were aligned using PyNAST (4), chimeric sequences were removed using ChimeraSlayer (12), and phylogeny was assigned using blast retained on Greengenes database ver. 13_8 (29). Chao1 and coverage values were calculated by QIIME 1.9.1 (5). Weighted UniFrac distances were used for a principal coordinate analysis (PCoA) (26). Spearman’s rank correlation coefficients rs were calculated using PAST software (14). A co-occurrence network analysis was performed with significant interactions (rs>0.609 and P value<0.001), and the network was visualized by Cytoscape version 3.2.1 (43). A circular bar plot was illustrated using ggplot2 (45) and R studio ver. 1.1.45 (https://www.rstudio.com/) under R ver. 3.5.0 (17). To investigate the relationships between the relative abundances of OTUs and physicochemical parameters, a redundancy analysis (RDA) was performed using CANOCO software version 5 (Microcomputer Power, Ithaca, NY, USA) (24).
The operational performance of two activated sludge systems treating municipal sewage wastewater is shown in Fig. 1 and S1. AB1 and AB2 tanks achieved high BOD removal efficiency (>95%) during the period monitored. These tanks were continuously operated with a sewage wastewater flow rate of ca. 26,000 m3 d−1 before Nov. 14 and after Nov. 22. Between Nov. 15 and 21, the flow rate increased to more than 31,810 m3 d−1 (up to 37,150 and 37,320 m3 d−1 in the AB1 and AB2 tanks, respectively) due to periodic maintenance of the final sedimentation tank. Although the flow rate decreased to the levels of standard operational conditions after Nov. 22, the concentrations of nitrogen constituents increased and fluctuated, i.e., ammonia concentrations increased to 1.2–6.4 mg L−1 between Dec. 8 and Jan. 18 and decreased to <0.9 mg L−1 on Jan. 25, nitrite concentrations were 2.1–7.0 mg L−1 in late November and remained at low levels of <2.1 mg L−1 by Jan. 25, and nitrate accumulated to concentrations of 11–20 mg L−1 between Dec. 7 and Jan. 5 and then returned to the level of standard operational conditions on Jan. 25 (<8.1 mg L−1; Fig. 1B).

To elucidate the effects of changes in the wastewater flow rate on the composition of the activated sludge microbiome, 16S rRNA gene iTag libraries were constructed for sludge samples collected from the AB1 and AB2 tanks between Dec. 7 and Jan. 25 (Tables S1 and S2). A total of 2,030,063 reads of 16S rRNA genes were obtained and assigned to 37,151 OTUs. High Good’s coverage values (>96%) suggested that the OTUs obtained adequately estimated the diversity of the activated sludge microbiome. Bacterial taxa commonly associated with activated sludge (11, 13, 20, 25, 46), Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Deltaproteobacteria, and Bacteroidetes, were detected in all samples and accounted for >72.2% of the total population (Fig. 1C). A comparison of microbiome compositions between samples using a weighted UniFrac-based 3D principal coordinate analysis clearly showed that compositions shifted with time (Fig. S2). These results suggest that particular microbial populations may have been affected by changes in the wastewater flow rate and resulting accumulation of nitrogen constituents in AB1 and AB2.
To analyze the interactions between major microbial constituents, we focused on the top 200 abundant OTUs accounting for 81.8% of the total microbiome dataset across 24 sludge samples from AB1 and AB2. We found two OTUs (AB23287 and AB38882) related to Nitrosomonas-type ammonia oxidizing bacteria (AOB) with a relatively low abundance by Jan. 18 (<0.1% of the total population) and marked increases on Jan. 25 up to the 71st–81st percentile, respectively (0.5% of the total microbiome) (Fig. 2, S3A, and S3B, Table S2). The abundance-based network analysis indicated the complex co-occurrence patterns of dominant organisms, including a total of 2,509 (1,252 positive and 1,257 negative) significant interactions in activated sludge samples (Fig. S4A, Table S3). Two Nitrosomonas-related OTUs had a large number of degrees (edges) within the network (Fig. 3A and 3B). Nitrosomonas-related OTUs shared 26 positive and 27 negative interactions between OTUs belonging to the cultured phyla Proteobacteria, Bacteroidetes, Spirochaetes, Verrucomicrobia, Planctomycetes, and Gemmatimonadetes and uncultivated phyla TM7 (“Ca. Saccharibacteria”), OD1 (“Ca. Parcubacteria”), and SAR406 (“Ca. Marinimicrobia”). Previous studies revealed that nitrifying bacteria forge beneficial interactions with heterotrophic bacteria belonging to the phyla Proteobacteria (particularly the classes Alphaproteobacteria and Gammaproteobacteria), Bacteroidetes, and Chloroflexi through metabolite exchange (22, 35). Another proteomicbased study demonstrated that the addition of an enrichment culture of heterotrophic bacteria to a pure culture of Nitrosomonas induced the up-regulation of the ammonia oxidation pathway (41). Metabolites, such as siderophore (21), acyl-homoserine lactone (3), and amino acids (6, 7), have been shown to increase the growth of Nitrosomonas strains. For example, Nitrosomonas-type AOB may be stimulated in situ by siderophore production by positively interacting with Myxococcales-related organisms (OTUs AB38105 and AB35792; Fig. 2 and 3), a feature often observed for members of myxobacteria (10). Based on RDA, the proliferation of Nitrosomonas- and Myxococcales-type OTUs positively correlated with mixed liquor dissolved oxygen (MLDO) and the solid retention time (SRT), but negatively correlated with reactor ammonia concentrations (Fig. S5). These results suggest that high O2 and low ammonia are favorable for the growth of the above OTUs even though both compounds are substrates for Nitrosomonas. In addition, arrows of relatively abundant (>0.4% of the total activated sludge microbiome) OTUs having a negative correlation with two Nitrosomonas-related OTUs displayed reversed associations with the above parameters. In brief, metabolically diverse bacteria may play a supporting role for the recovery of Nitrosomonas-type AOB in the activated sludge system.
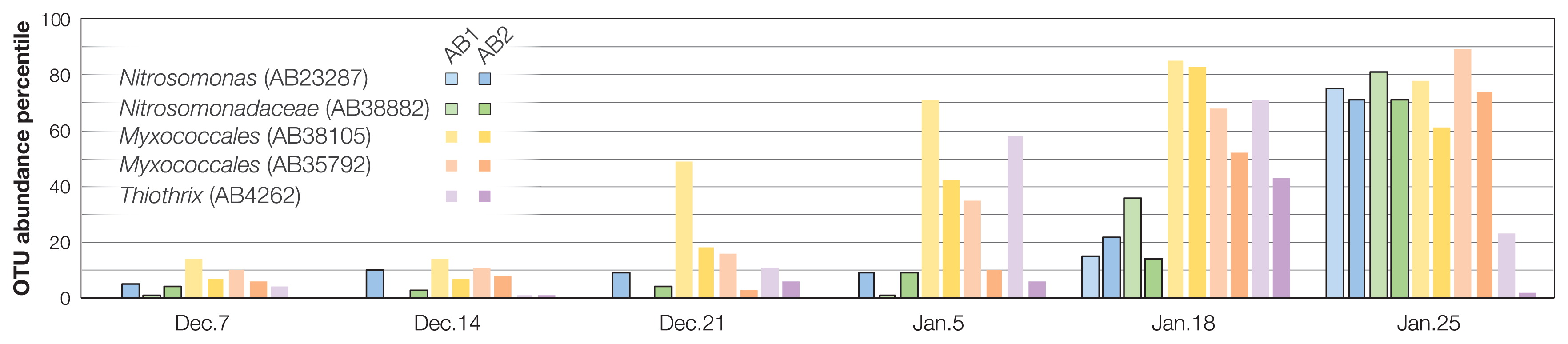

The relative abundance of an OTU AB4262 related to Thiothrix, which has been identified as a causative agent for filamentous sludge bulking (34), increased up to the 71st percentile (0.32% abundance) of the total microbiome on Jan. 18, and then decreased to less than the 23rd percentile (0.07% abundance) on Jan. 25 (Fig. 2 and S3C, Table S2). Although severe sludge bulking was not observed during the operation period in the AB1 and AB2 tanks (i.e., sludge volume indexes (SVI) showed stable values, 112–168; Fig. S1), a previous study reported that >0.19% of the Thiothrix population caused increases in suspended solids, which are likely associated with bulking events, and ammonia ion concentrations may have been one of the influential factors for Thiothrix proliferation in a full-scale nitrification and denitrification wastewater treatment plant in the U.S. (2). Thiothrix-related OTU AB4262 had a positive interaction with Nitrosomonas-type OTU AB38882 (Fig. S4B), even though its proliferation in the AB1 tank was earlier than that in the AB2 tank on Jan. 5 (Fig. 2). These results suggest that an increase in the wastewater flow rate and resulting ammonia accumulation trigger a co-increase in Thiothrix and Nitrosomonas populations. Although a Thiothrix-related population in an industrial wastewater treatment plant with bulking issues was shown to be suppressed by the addition of raw feed wastewater and the subsequent proliferation of a number of bacteria (36), interactions between Thiothrix and other bacterial constituents in activated sludge microbiomes remain unclear.
In conclusion, high-throughput microbiome profiling combined with a co-occurrence network analysis revealed complex interactions among diverse microorganisms in activated sludge tanks during the period after an increase in the wastewater flow rate. Nitrosomonas- and Thiothrix-related organisms may be affected by an increase in the flow rate. Although the reason for the proliferation of Nitrosomonas and Thiothrix organisms cannot currently be fully explained, further studies using metagenomics, metatranscriptomics, and metabolomics (16, 33) will provide more comprehensive information for elucidating the key factors shaping interactions between Nitrosomonas, Thiothrix, and other microorganisms in the activated sludge microbiome.
The sequence reported in the present study was deposited in the DDBJ database under DDBJ/EMBL/GenBank accession number DRA007026.
Acknowledgements
This study was supported by The Japan Society for the Promotion of Science with Grants-in-Aid for Scientific Research (No. 18H01576 and 18H03367).
Reference
- 1. Aoyagi, T., S.
Hanada, H.
Itoh, Y.
Sato, A.
Ogata, M.W.
Friedrich, Y.
Kikuchi, and T.
Hori. 2015. Ultra-high-sensitivity stable-isotope probing of rRNA by high-throughput sequencing of isopycnic centrifugation gradients. Environ Microbiol Rep. 7:282-287.
- 2. Asvapathanagul, P., B.H.
Olson, P.B.
Gedalanga, A.
Hashemi, Z.
Huang, and J.
La. 2015. Identification and quantification of Thiothrix eikelboomii using qPCR for early detection of bulking incidents in a full-scale water reclamation plant. Appl Microbiol Biotechnol. 99:4045-4057.
- 3. Burton, E.O., H.W.
Read, M.C.
Pellitteri, and W.J.
Hickey. 2005. Identification of acyl-homoserine lactone signal molecules produced by Nitrosomonas europaea strain Schmidt. Appl Environ Microbiol. 71:4906-4909.
- 4. Caporaso, J.G., K.
Bittinger, F.D.
Bushman, T.Z.
DeSantis, G.L.
Andersen, and R.
Knight. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 26:266-267.
- 5. Caporaso, J.G., J.
Kuczynski, J.
Stombaugh, et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 7:335-336.
- 6. Clark, C., and E.L.
Schmidt. 1967. Growth response of Nitrosomonas europaea to amino acids. J Bacteriol. 93:1302-1308.
- 7. Clark, C., and E.L.
Schmidt. 1967. Uptake and utilization of amino acids by resting cells of Nitrosomonas europaea. J Bacteriol. 93:1309-1315.
- 8. Edgar, R.C., B.J.
Haas, J.C.
Clemente, C.
Quince, and R.
Knight. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 27:2194-2200.
- 9. Ferrera, I., and O.
Sanchez. 2016. Insights into microbial diversity in wastewater treatment systems: How far have we come?
Biotechnol Adv. 34:790-802.
- 10. Gaitatzis, N., B.
Kunze, and R.
Muller. 2005. Novel insights into siderophore formation in myxobacteria. Chembiochem. 6:365-374.
- 11. Griffin, J.S., and G.F.
Wells. 2017. Regional synchrony in full-scale activated sludge bioreactors due to deterministic microbial community assembly. ISME J. 11:500-511.
- 12. Haas, B.J., D.
Gevers, A.M.
Earl, et al. 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21:494-504.
- 13. Hai, R., Y.
Wang, X.
Wang, Y.
Li, and Z.
Du. 2014. Bacterial community dynamics and taxa-time relationships within two activated sludge bioreactors. PLoS One. 9:e90175.
- 14. Hammer, Ø., D.A.T.
Harper, and P.D.
Ryan. 2001. PAST: paleontological statistics software package for education and data analysis. Palaeontol Electronica. 4:1-9.
- 15. Hanada, A., T.
Kurogi, N.M.
Giang, T.
Yamada, Y.
Kamimoto, Y.
Kiso, and A.
Hiraishi. 2014. Bacteria of the candidate phylum TM7 are prevalent in acidophilic nitrifying sequencing-batch reactors. Microbes Environ. 29:353-362.
- 16. Hiraoka, S., C.C.
Yang, and W.
Iwasaki. 2016. Metagenomics and bioinformatics in microbial ecology: Current status and beyond. Microbes Environ. 31:204-212.
- 17. Ihaka, R., and R.
Gentleman. 1996. R: a language for data analysis and graphics. J Comput Graph Stat. 5:299-314.
- 18. Isazadeh, S., S.
Jauffur, and D.
Frigon. 2016. Bacterial community assembly in activated sludge: mapping beta diversity across environmental variables. MicrobiologyOpen. 5:1050-1060.
- 19. Japan Sewage Works Association. 2015. Gesui Shikenhou (Standard Methods for Wastewater Analysis). Japan Sewage Works Association, Tokyo. (in Japanese).
- 20. Ju, F., and T.
Zhang. 2015. Bacterial assembly and temporal dynamics in activated sludge of a full-scale municipal wastewater treatment plant. ISME J. 9:683-695.
- 21. Keluskar, R., A.
Nerurkar, and A.
Desai. 2013. Mutualism between autotrophic ammonia-oxidizing bacteria (AOB) and heterotrophs present in an ammonia-oxidizing colony. Arch Microbiol. 195:737-747.
- 22. Kindaichi, T., T.
Ito, and S.
Okabe. 2004. Ecophysiological interaction between nitrifying bacteria and heterotrophic bacteria in autotrophic nitrifying biofilms as determined by microautoradiography-fluorescence in situ hybridization. Appl Environ Microbiol. 70:1641-1650.
- 23. Lee, S.H., H.J.
Kang, and H.D.
Park. 2015. Influence of influent wastewater communities on temporal variation of activated sludge communities. Water Res. 73:132-144.
- 24. Lepš, J., and P.
Šmilauer. 2003. Multivariate Analysis of Ecological Data Using CANOCO. Cambridge University Press, New York.
- 25. Liu, T., S.
Liu, M.
Zheng, Q.
Chen, and J.
Ni. 2016. Performance assessment of full-scale wastewater treatment plants based on seasonal variability of microbial communities via high-throughput sequencing. PLoS One. 11:e0152998.
- 26. Lozupone, C., M.E.
Lladser, D.
Knights, J.
Stombaugh, and R.
Knight. 2011. UniFrac: an effective distance metric for microbial community comparison. ISME J. 5:169-172.
- 27. Lv, X.M., M.F.
Shao, C.L.
Li, J.
Li, X.L.
Gao, and F.Y.
Sun. 2014. A comparative study of the bacterial community in denitrifying and traditional enhanced biological phosphorus removal processes. Microbes Environ. 29:261-268.
- 28. Matsunaga, K., K.
Kubota, and H.
Harada. 2014. Molecular diversity of eukaryotes in municipal wastewater treatment processes as revealed by 18S rRNA gene analysis. Microbes Environ. 29:401-407.
- 29. McDonald, D., M.N.
Price, J.
Goodrich, E.P.
Nawrocki, T.Z.
DeSantis, A.
Probst, G.L.
Andersen, R.
Knight, and P.
Hugenholtz. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6:610-618.
- 30. Narihiro, T., N.K.
Kim, R.
Mei, M.K.
Nobu, and W.T.
Liu. 2015. Microbial community analysis of anaerobic reactors treating soft drink wastewater. PLoS One. 10:e0119131.
- 31. Narihiro, T., M.K.
Nobu, N.K.
Kim, Y.
Kamagata, and W.T.
Liu. 2015. The nexus of syntrophy-associated microbiota in anaerobic digestion revealed by long-term enrichment and community survey. Environ Microbiol. 17:1707-1720.
- 32. Narihiro, T.
2016. Microbes in the water infrastructure: underpinning our society. Microbes Environ. 31:89-92.
- 33. Narihiro, T., and Y.
Kamagata. 2017. Genomics and metagenomics in microbial ecology: recent advances and challenges. Microbes Environ. 32:1-4.
- 34. Nielsen, P.H., M.A.
de Muro, and J.L.
Nielsen. 2000. Studies on the in situ physiology of Thiothrix spp. present in activated sludge. Environ Microbiol. 2:389-398.
- 35. Okabe, S., T.
Kindaichi, and T.
Ito. 2005. Fate of 14C-labeled microbial products derived from nitrifying bacteria in autotrophic nitrifying biofilms. Appl Environ Microbiol. 71:3987-3994.
- 36. Onetto, C.A., K.L.
Eales, and P.R.
Grbin. 2017. Remediation of Thiothrix spp. associated bulking problems by raw wastewater feeding: A full-scale experience. Syst Appl Microbiol. 40:396-399.
- 37. Sato, Y., T.
Hori, R.R.
Navarro, H.
Habe, and A.
Ogata. 2015. Effect of a microbiota activator on accumulated ammonium and microbial community structure in a pilot-scale membrane bioreactor. J Gen Appl Microbiol. 61:132-138.
- 38. Sato, Y., T.
Hori, R.R.
Navarro, R.
Naganawa, H.
Habe, and A.
Ogata. 2016. Effects of organic-loading-rate reduction on sludge biomass and microbial community in a deteriorated pilot-scale membrane bioreactor. Microbes Environ. 31:361-364.
- 39. Saunders, A.M., M.
Albertsen, J.
Vollertsen, and P.H.
Nielsen. 2016. The activated sludge ecosystem contains a core community of abundant organisms. ISME J. 10:11-20.
- 40. Schloss, P.D., S.L.
Westcott, T.
Ryabin, et al. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 75:7537-7541.
- 41. Sedlacek, C.J., S.
Nielsen, K.D.
Greis, W.D.
Haffey, N.P.
Revsbech, T.
Ticak, H.J.
Laanbroek, and A.
Bollmann. 2016. Effects of bacterial community members on the proteome of the ammonia-oxidizing bacterium Nitrosomonas sp. strain Is79. Appl Environ Microbiol. 82:4776-4788.
- 42. Seviour, R.J., T.
Mino, and M.
Onuki. 2003. The microbiology of biological phosphorus removal in activated sludge systems. FEMS Microbiol Rev. 27:99-127.
- 43. Shannon, P., A.
Markiel, O.
Ozier, N.S.
Baliga, J.T.
Wang, D.
Ramage, N.
Amin, B.
Schwikowski, and T.
Ideker. 2003. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13:2498-2504.
- 44. Wagner, M., and A.
Loy. 2002. Bacterial community composition and function in sewage treatment systems. Curr Opin Biotechnol. 13:218-227.
- 45. Wickham, H.
2009. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag, New York.
- 46. Zhang, T., M.F.
Shao, and L.
Ye. 2012. 454 pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 6:1137-1147.
