Abstract
Giant viruses of ‘Megaviridae’ have the ability to widely disperse around the globe. We herein examined ‘Megaviridae’ communities in four distinct aquatic environments (coastal and offshore seawater, brackish water, and hot spring freshwater), which are distantly located from each other (between 74 and 1,765 km), using a meta-barcoding method. We identified between 593 and 3,627 OTUs in each sample. Some OTUs were detected in all five samples tested as well as in many of the Tara Oceans metagenomes, suggesting the existence of viruses of this family in a wide range of habitats and the ability to circulate on the planet.
‘Megaviridae’, also referred to as the extended Mimiviridae, is a rapidly expanding proposed family of nucleocytoplasmic large DNA viruses (NCLDVs) (see [24, 32] and references therein; [7] for an alternative proposal). ‘Megaviridae’ includes giant viruses such as mimiviruses (20), Cafeteria roenbergensis virus (10), and Prymnesium kappa virus RF01 (17). Early marine metagenomic surveys revealed the existence of viruses of ‘Megaviridae’ in the sea (13, 25, 26). They were later found to be highly abundant (14) and active (5) across oceanic regions, at an abundance estimated to be 103 to 105 genomes mL−1 seawater. The taxon richness of this viral group appears to be very large and exceeds that of the whole domain of Bacteria (24), suggesting that a diverse array of eukaryotes are potential hosts of these mostly unidentified viruses. Isolated lineages of this viral family are still rare; they infect unicellular algae (12, 17, 27, 31–33) and aquatic heterotrophic protists, such as amoeba infecting mimiviruses (1, 9, 10, 29). Furthermore, a viral group infecting sturgeons, a group of fishes from which caviar is obtained, was recently shown to be related to ‘Megaviridae’ (8). Therefore, viruses in this group appear to have important, but vastly unknown ecological functions in the aquatic environment through the regulation of the populations of their eukaryotic hosts.
Meta-barcoding approaches using degenerate polymerase chain reaction (PCR) primers that amplify specific genes from environmental DNA have been successful in characterizing the community structures of ‘Megaviridae’ in aquatic environments (18, 21, 37). We recently proposed a set of 82 pairs of degenerate PCR primers (i.e., MEGAPRIMER) targeting the B family DNA polymerase genes (polBs) of ‘Megaviridae’ (22). These primers were designed based on 904 metagenomic ‘Megaviridae’ polBs in addition to 17 polBs from viruses with sequenced genomes. The MEGAPRIMER approach revealed 5,595 ‘Megaviridae’ non-singleton operational taxonomic units (OTUs) at a nucleotide sequence identity of 97% in a seawater sample taken at Osaka Bay, Japan. However, MEGAPRIMER was only tested for a single seawater sample in our previous study. Therefore, the effectiveness of MEGAPRIMER has not yet been sufficiently demonstrated. In the present study, we used the same primer set to investigate ‘Megaviridae’ community structures in four different aquatic environments.
Between June 2016 and October 2016, samples were collected from four geographically distant locations in Japan to cover a broad range of aquatic environments (Fig. 1 and Table 1). The sampling sites were distantly located from each other (between 74 and 1,765 km). Osaka Bay is a typical eutrophic environment surrounded by densely populated districts and forests with the input of nutrients from rivers. The Japan Sea represents a marginal sea environment that is semi-isolated from the north Pacific. A mangrove site in Ishigaki Island was selected for the potential existence of ‘Megaviridae’ (25). The Miyuki hot spring of Shirahama was also selected to examine the existence of ‘Megaviridae’ in a high temperature environment. Four liters of surface water (from a depth of between 0 and 5 m) was collected at each sampling location. Filtration and DNA extraction were performed as previously described (22). Each primer pair of MEGAPRIMER was used in a separate PCR amplification as previously described (22). In the present study, we did not select amplicons based on visualization in gel electrophoresis as previously reported (22); we mixed all 82 amplicons and an identical barcode was attached to PCR products from the same sample to distinguish sequences from different samples. One sequencing run was performed using a MiSeq platform with MiSeq V3 (2×300 bp) reagent kits (Illumina, San Diego, CA, USA) and with a spike-in of PhiX at 50% to serve as an internal control. Raw reads were processed using the ‘Megaviridae’ Amplicon Processing System (MAPS) as previously described (22). OTUs were formed using CD-HIT-EST (11) with a nucleotide sequence identity of 97%. Rarefaction curves were generated using matplotlib package version 2.0.2 (16). The most abundant 100 OTUs in each sample were selected (423 OTUs after removing redundancy) and used to build a phylogenetic tree based on their translated sequences. The phylogenetic tree was generated using FastTree version 2.1.9 (28) with a default setting (the JTT+CAT model) and visualized using Python ETE3 package version 3.0.0b35 (15). A Unifrac analysis (23) was performed with the scikit-bio package version 0.5.1 of Python and visualized using R. Metagenomic genes from published Tara Oceans data (34) downloaded from MGENES (https://www.genome.jp/mgenes/) were initially screened for homologs of the mimivirus PolB sequence with TBLASTN (2), and then analyzed with CD-HIT-EST to identify genes nearly identical to OTUs (nucleotide sequence identity>97%). Raw read data were deposited to DDBJ (accession number DRA008113), and sequence data are also available from our ftp site (ftp://ftp.genome.jp/pub/db/community/MEGAPRIMER_papers).

Table 1
Locations and sampling dates of samples used in the present study
| Sample |
Longitude |
Latitude |
Depth (m) |
Temperature (°C) |
Salinity |
Date |
Reference |
| Osaka Bay Oct. |
N 34°19′28″ |
E 135°7′15″ |
5 |
22.01 |
33.10 |
October 30, 2015 |
(22) |
| Osaka Bay Aug. |
N 34°19′28″ |
E 135°7′15″ |
5 |
25.62 |
32.51 |
August 22, 2016 |
This study |
| Japan Sea |
N 37°20′06″ |
E 134°49′85″ |
0 |
25.5 |
32.03 |
July 25, 2016 |
This study |
| Ishigaki Island |
N 24°19′19″ |
E 124°03′23″ |
0 |
29.5 |
NA* |
October 13, 2016 |
This study |
| Miyuki hot spring |
N 33°40′35″ |
E 135°20′18″ |
0 |
69.4 |
0.8 |
June 21 2016 |
This study |
* The salinity meter broke down at this sampling site.
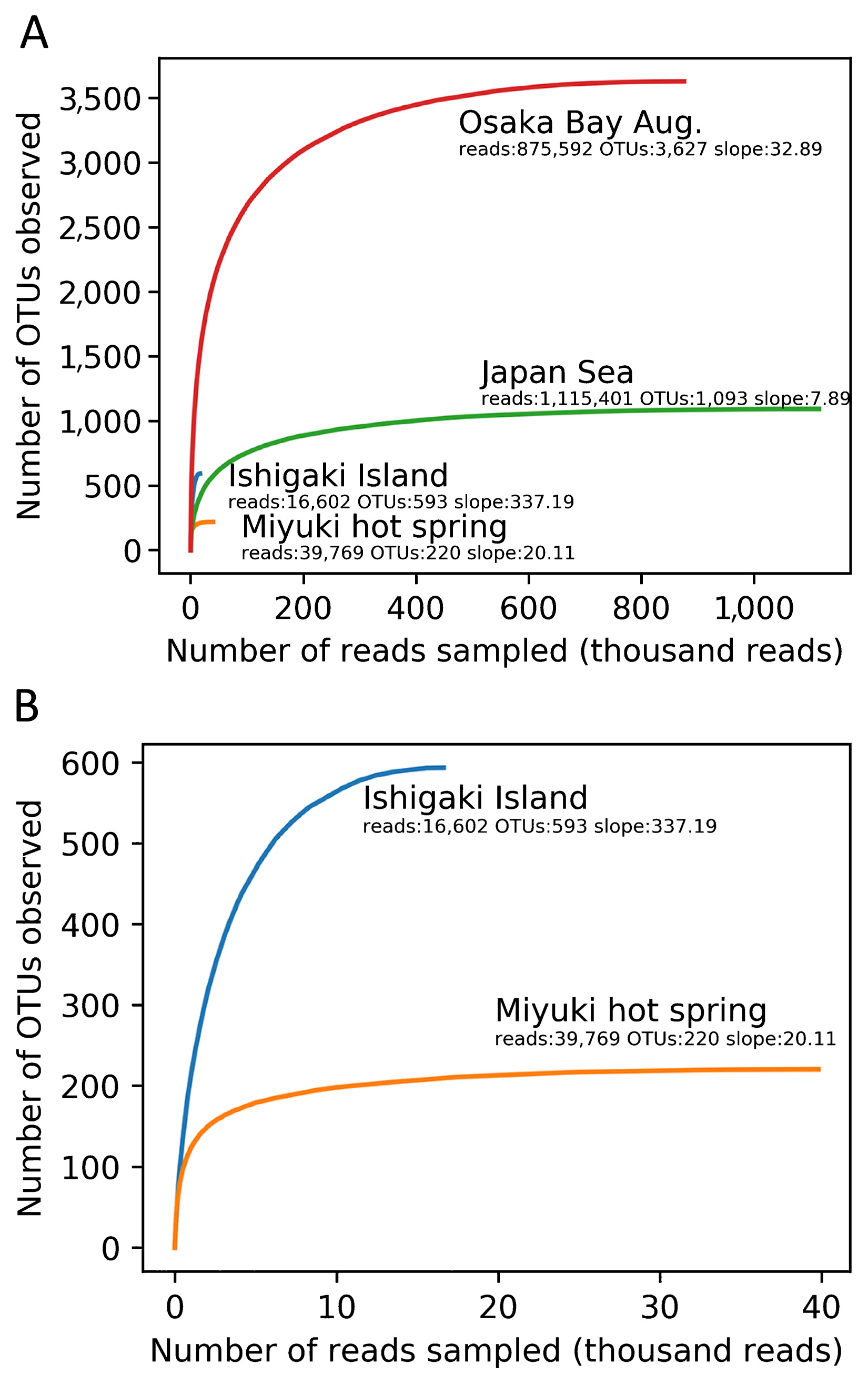
The resulting high quality ‘Megaviridae’ polB fragments were grouped into 3,627, 1,093, 593, and 220 non-singleton OTUs for Osaka Bay Aug., Japan Sea, Ishigaki Island, and Miyuki hot spring, respectively (Table 2). Rarefaction curves indicated that the number of OTUs was close to the plateau under the sequencing depth examined in the present study (Fig. 2). Unweighted Unifrac distances were calculated between ‘Megaviridae’ community structures in five samples by including previously generated data from Osaka Bay (22) and by selecting the 200 most abundant OTUs from each sample. A multidimensional scaling (MDS) analysis showed that the two samples from Osaka Bay were similar to each other. Other samples (Japan Sea, Ishigaki Island, and Miyuki hot spring) were distantly placed from the Osaka Bay samples in the MDS plot (Fig. 3).
Table 2
Number of operational taxonomic units (OTUs) for each sample.
| Sample |
Total number of OTUs |
Number of singleton OTUs |
Number of non-singleton OTUs |
Number of sequences included in non-singleton OTUs |
| Osaka Bay Aug. |
5,653 |
2,026 |
3,627 |
875,592 |
| Japan Sea |
2,206 |
1,113 |
1,093 |
1,115,401 |
| Ishigaki Island |
881 |
288 |
593 |
16,602 |
| Miyuki Hot spring |
304 |
84 |
220 |
39,769 |

We then performed phylogenetic analyses of representative sequences. The tree revealed dozens of diverse clades (clades ii to xii in Fig. 4) for ‘Mesomimivirinae’ (a proposed subfamily of ‘Megaviridae’), which includes known viruses of unicellular algae. The tree also revealed the detection of sequences belonging to ‘Megamimivirinae’ (another proposed subfamily including mimiviruses, Cafeteria roenbergensis virus and klosneuviruses; clade i in Fig. 4). However, we did not detect any sample-specific clades, which was unexpected because the hot freshwater spring (Miyuki) appeared to be ecologically distinct and isolated from other sites. In other words, each clade was found to contain OTUs from all or nearly all samples, although there were also OTUs specific to individual samples (as indicated by triangles in the outer ring of Fig. 4). Furthermore, a large proportion (78%; 330 OTUs) of the selected OTUs were found in more than one sample and 17 OTUs (4%) were discovered in all samples (Fig. 5). Regarding 7 out of 17 OTUs, their presence across five samples was supported at an identical read level (i.e. identical genotypes; star marks in Fig. 4).


The large number of OTUs shared among the samples tested prompted us to search metagenomic genes from the previous Tara Oceans expedition (34), which covered a large part of global oceans, for the non-singleton OTUs identified in the present study. Many of the OTUs were discovered in different oceanic regions, including the opposite side of the earth from Japan, such as the South Atlantic Ocean (Table 3).
Table 3
Number of OTUs identified in
Tara Oceans data.
| Sample |
Oceanic region* |
|
| MS |
RS |
IO |
SAO |
SO |
SPO |
NPO |
NAO |
| Osaka Bay Oct. |
35 |
29 |
146 |
119 |
1 |
224 |
73 |
53 |
| Osaka Bay Aug. |
37 |
9 |
96 |
79 |
0 |
159 |
58 |
28 |
| Japan Sea |
17 |
2 |
46 |
58 |
0 |
63 |
27 |
15 |
| Ishigaki Island |
13 |
1 |
28 |
21 |
0 |
26 |
12 |
5 |
| Miyuki hot spring |
6 |
2 |
18 |
12 |
0 |
19 |
9 |
11 |
* Abbreviations: MS, Mediterranean Sea; RS, Red Sea; IO, Indian Ocean; SAO, South Atlantic Ocean; SO, Southern Ocean; SPO, South Pacific Ocean; NPO, North Pacific Ocean; NAO, North Atlantic Ocean.
Previous studies isolated highly similar giant viruses in different countries. The genome of Mimivirus shirakomae (GenBank: AP017645) isolated in Japan (35) was nearly identical (nucleotide sequence identity of ~99.9%) to the genome of the first mimivirus (GenBank: NC_014649) isolated in England (29). The genome of a marseillevirus isolated in Shanghai (GenBank: MG827395) was nearly identical (~98.5%) to the genome of a marseillevirus isolated from Cannes, France (GenBank: KF261120). These findings suggest the long distance dispersal of giant viruses across continents and oceans through unidentified mechanisms, possibly via microscale droplets (30, 36), wind (3, 19), or oceanic current systems (4), as suggested for other smaller viruses. In the present study, we revealed the existence of polB OTUs that may be observed in largely distinct aquatic ecosystems, which span seawater, a mangrove site (brackish water), and freshwater hot spring. Furthermore, some OTUs were found in different oceans. Therefore, the dispersal of ‘Megaviridae’ occurs across distant geographical locations on a global scale. The present results also imply a relatively wide habitat and niche for at least some of the viruses belonging to ‘Megaviridae’.
It is notable that we detected 220 ‘Megaviridae’ OTUs from Miyuki hot spring, at which the water temperature was 69.4°C. This result suggests the existence of diverse ‘Megaviridae’ in a high temperature environment. Giant viruses have not yet been isolated from an environment as hot as Miyuki spring, except for medusavirus recently isolated from a cooler, but still warm environment (43.4°C freshwater) (38). A previous study reported the genome sequences of ‘Megaviridae’ assembled from metagenomic samples from a hot spring site, Yellowstone Lake (39). However, the genomes were co-assembled from different metagenomic data derived from samples collected at different locations with varying temperatures between 10 and 96°C. Therefore, it currently remains unclear whether ‘Megaviridae’ exist in a hot or warm environment. Another related study on the same metagenomes revealed the existence of virophages, which are parasites of ‘Megaviridae’ viruses, in a high temperature ecosystem (40). Zhou et al. assembled seven virophage genomes from 42 samples from Yellowstone Lake, and all of the virophage genomes were detected in vent water metagenomic samples (between 40 and 68°C). In the literature, the upper temperature limit for a eukaryotic cell to reproduce has been described as 65°C (6). Therefore, there may be no actively replicating eukaryotic cells in hot water at 69.4°C. In the Miyuki hot spring sampling site, water runs into a drain open to the surrounding environment, including the atmosphere. Therefore, it currently remains unclear whether ‘Megaviridae’ actively infect eukaryotic hosts in a hot environment. The quantitativity of the MEGAPRIMER approach needs to be investigated as previously pointed out (22); however, the two Osaka Bay samples (collected in different years, but within similar periods) being placed closely to each other in the MDS plot (Fig. 3) corroborate the effectiveness of the MEGAPRIMER approach for comparisons of ‘Megaviridae’ communities across environments.
Acknowledgements
This work was supported by The Canon Foundation (No. 203143100025), JSPS/KAKENHI (Nos. 26430184, 17H03850, 18H02279, 16H06279), Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan (Nos. 16H06429, 16K21723, 16H06437), The Kyoto University Foundation, and the Collaborative Research Program of the Institute for Chemical Research, Kyoto University (Nos. 2015-125, 2016-28, 2017-25, 2018-31). Computational work was completed at the SuperComputer System, Institute for Chemical Research, Kyoto University. We thank the Tara Oceans consortium, the projects Oceanomics and France Genomique (grants ANR-11-BTBR-0008 and ANR-10-INBS-09), and people and sponsors who supported the Tara Oceans Expedition (http://www.embl.de/taraoceans/) for making data accessible. This is contribution number 88 of the Tara Oceans Expedition 2009–2013.
References
- 1. Abrahao, J., L.
Silva, L.S.
Silva, et al. 2018. Tailed giant Tupanvirus possesses the most complete translational apparatus of the known virosphere. Nat Commun. 9:749.
- 2. Altschul, S.F., T.L.
Madden, A.A.
Schaffer, J.
Zhang, Z.
Zhang, W.
Miller, and D.J.
Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-402.
- 3. Baylor, E.R., M.B.
Baylor, D.C.
Blanchard, L.D.
Syzdek, and C.
Appel. 1977. Virus transfer from surf to wind. Science. 198:575-580.
- 4. Brum, J.R., J.C.
Ignacio-Espinoza, S.
Roux, et al. 2015. Ocean plankton. Patterns and ecological drivers of ocean viral communities. Science. 348:1261498.
- 5. Carradec, Q., E.
Pelletier, C.
Da Silva, et al. 2018. A global ocean atlas of eukaryotic genes. Nat Commun. 9:373.
- 6. Clarke, A.
2014. The thermal limits to life on Earth. Int J Astrobiol. 13:141-154.
- 7. Claverie, J.M., and C.
Abergel. 2018. Mimiviridae: An expanding family of highly diverse large dsDNA viruses infecting a wide phylogenetic range of aquatic eukaryotes. Viruses. 10:506.
- 8. Clouthier, S., E.
Anderson, G.
Kurath, and R.
Breyta. 2018. Molecular systematics of sturgeon nucleocytoplasmic large DNA viruses. Mol Phylogenet Evol. 128:26-37.
- 9. Deeg, C.M., C.T.
Chow, and C.A.
Suttle. 2018. The kinetoplastid-infecting Bodo saltans virus (BsV), a window into the most abundant giant viruses in the sea. Elife. 7:e33014.
- 10. Fischer, M.G., M.J.
Allen, W.H.
Wilson, and C.A.
Suttle. 2010. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc Natl Acad Sci USA. 107:19508-19513.
- 11. Fu, L., B.
Niu, Z.
Zhu, S.
Wu, and W.
Li. 2012. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 28:3150-3152.
- 12. Gallot-Lavallee, L., A.
Pagarete, M.
Legendre, S.
Santini, R.A.
Sandaa, H.
Himmelbauer, H.
Ogata, G.
Bratbak, and J.M.
Claverie. 2015. The 474-Kilobase-pair complete genome sequence of CeV-01B, a virus infecting Haptolina (Chrysochromulina) ericina (Prymnesiophyceae). Genome Announc. 3:e01413-15.
- 13. Ghedin, E., and J.M.
Claverie. 2005. Mimivirus relatives in the Sargasso Sea. Virol J. 2:62.
- 14. Hingamp, P., N.
Grimsley, S.G.
Acinas, et al. 2013. Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes. ISME J. 7:1678-1695.
- 15. Huerta-Cepas, J., F.
Serra, and P.
Bork. 2016. ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol Biol Evol. 33:1635-1638.
- 16. Hunter, J.D.
2007. Matplotlib: A 2D graphics environment. Comput Sci Eng. 9:90-95.
- 17. Johannessen, T.V., G.
Bratbak, A.
Larsen, H.
Ogata, E.S.
Egge, B.
Edvardsen, W.
Eikrem, and R.A.
Sandaa. 2015. Characterisation of three novel giant viruses reveals huge diversity among viruses infecting Prymnesiales (Haptophyta). Virology. 476:180-188.
- 18. Johannessen, T.V., A.
Larsen, G.
Bratbak, A.
Pagarete, B.
Edvardsen, E.D.
Egge, and R.A.
Sandaa. 2017. Seasonal dynamics of haptophytes and dsDNA algal viruses suggest complex virus-host relationship. Viruses. 9:84.
- 19. Klausner, Z., E.
Fattal, and E.
Klement. 2017. Using synoptic systems’ typical wind trajectories for the analysis of potential atmospheric long-distance dispersal of lumpy skin disease virus. Transbound Emerg Dis. 64:398-410.
- 20. La Scola, B., S.
Audic, C.
Robert, L.
Jungang, X.
de Lamballerie, M.
Drancourt, R.
Birtles, J.M.
Claverie, and D.
Raoult. 2003. A giant virus in amoebae. Science. 299:2033.
- 21. Larsen, J.B., A.
Larsen, G.
Bratbak, and R.A.
Sandaa. 2008. Phylogenetic analysis of members of the Phycodnaviridae virus family, using amplified fragments of the major capsid protein gene. Appl Environ Microbiol. 74:3048-3057.
- 22. Li, Y., P.
Hingamp, H.
Watai, H.
Endo, T.
Yoshida, and H.
Ogata. 2018. Degenerate PCR primers to reveal the diversity of giant viruses in coastal waters. Viruses. 10:496.
- 23. Lozupone, C., and R.
Knight. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 71:8228-8235.
- 24. Mihara, T., H.
Koyano, P.
Hingamp, N.
Grimsley, S.
Goto, and H.
Ogata. 2018. Taxon richness of “Megaviridae” exceeds those of bacteria and archaea in the ocean. Microbes Environ. 33:162-171.
- 25. Monier, A., J.M.
Claverie, and H.
Ogata. 2008. Taxonomic distribution of large DNA viruses in the sea. Genome Biol. 9:R106.
- 26. Monier, A., J.B.
Larsen, R.A.
Sandaa, G.
Bratbak, J.M.
Claverie, and H.
Ogata. 2008. Marine mimivirus relatives are probably large algal viruses. Virol J. 5:12.
- 27. Moniruzzaman, M., G.R.
LeCleir, C.M.
Brown, C.J.
Gobler, K.D.
Bidle, W.H.
Wilson, and S.W.
Wilhelm. 2014. Genome of brown tide virus (AaV), the little giant of the Megaviridae, elucidates NCLDV genome expansion and host-virus coevolution. Virology. 466–467:60-70.
- 28. Price, M.N., P.S.
Dehal, and A.P.
Arkin. 2009. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 26:1641-1650.
- 29. Raoult, D., S.
Audic, C.
Robert, C.
Abergel, P.
Renesto, H.
Ogata, B.
La Scola, M.
Suzan, and J.M.
Claverie. 2004. The 1.2-megabase genome sequence of Mimivirus. Science. 306:1344-1350.
- 30. Rastelli, E., C.
Corinaldesi, A.
Dell’Anno, et al. 2017. Transfer of labile organic matter and microbes from the ocean surface to the marine aerosol: an experimental approach. Sci Rep. 7:11475.
- 31. Sandaa, R.A., M.
Heldal, T.
Castberg, R.
Thyrhaug, and G.
Bratbak. 2001. Isolation and characterization of two viruses with large genome size infecting Chrysochromulina ericina (Prymnesiophyceae) and Pyramimonas orientalis (Prasinophyceae). Virology. 290:272-280.
- 32. Santini, S., S.
Jeudy, J.
Bartoli, et al. 2013. Genome of Phaeocystis globosa virus PgV-16T highlights the common ancestry of the largest known DNA viruses infecting eukaryotes. Proc Natl Acad Sci USA. 110:10800-10805.
- 33. Schvarcz, C.R., and G.F.
Steward. 2018. A giant virus infecting green algae encodes key fermentation genes. Virology. 518:423-433.
- 34. Sunagawa, S., L.P.
Coelho, S.
Chaffron, et al. 2015. Ocean plankton. Structure and function of the global ocean microbiome. Science. 348:1261359.
- 35. Takemura, M., T.
Mikami, and S.
Murono. 2016. Nearly complete genome sequences of two Mimivirus strains isolated from a Japanese freshwater pond and river mouth. Genome Announc. 4:e01378-16.
- 36. Wang, B., A.
Zhang, J.L.
Sun, H.
Liu, J.
Hu, and L.X.
Xu. 2005. Study of SARS transmission via liquid droplets in air. J Biomech Eng. 127:32-38.
- 37. Wilson, W.H., I.C.
Gilg, A.
Duarte, and H.
Ogata. 2014. Development of DNA mismatch repair gene, MutS, as a diagnostic marker for detection and phylogenetic analysis of algal Megaviruses. Virology. 466–467:123-128.
- 38. Yoshikawa, G., R.
Blanc-Mathieu, C.
Song, Y.
Kayama, T.
Mochizuki, K.
Murata, H.
Ogata, and M.
Takemura. 2019. Medusavirus, a novel large DNA virus discovered from hot spring water. J Virol. 93:e02130-18.
- 39. Zhang, W., J.
Zhou, T.
Liu, Y.
Yu, Y.
Pan, S.
Yan, and Y.
Wang. 2015. Four novel algal virus genomes discovered from Yellowstone Lake metagenomes. Sci Rep. 5:15131.
- 40. Zhou, J., D.
Sun, A.
Childers, T.R.
McDermott, Y.
Wang, and M.R.
Liles. 2015. Three novel virophage genomes discovered from Yellowstone Lake metagenomes. J Virol. 89:1278-1285.
