Abstract
Thermococcus kodakarensis possesses two DNA polymerases, Pol B and Pol D. We generated a T. kodakarensis strain (DPB1) in which polB was completely deleted and a derivative of DPB1 in which polB was overexpressed; neither of the generated strains exhibited any growth delay, indicating that the lack or overexpression of Pol B in T. kodakarensis did not affect cell growth. We also found that DPB1 showed higher sensitivity to four DNA-damaging agents (ultraviolet C irradiation, γ-ray irradiation, methyl methanesulfonate, and mitomycin C) than the parental strain. The sensitivity of DPB1 was restored to the level of the parent strain by the introduction of a plasmid harboring polB, suggesting that the DNA damage-sensitive phenotype of DPB1 was due to the loss of polB. Collectively, these results indicate that Pol B is involved in DNA repair, but not DNA replication, which, in turn, implies that Pol D is the sole replicative DNA polymerase in Thermococcus species.
DNA polymerases are enzymes that synthesize complementary DNA strands of template DNA and, thus, are important for DNA replication and repair. Most living organisms possess several DNA polymerases, which are classified into the following 7 families based on their primary structures: A, B, C, D, E, X, and Y (2, 5, 24, 30).
Archaea, constituting the third domain of life, are useful for elucidating the mechanisms involved in the maintenance and transfer of genetic information, particularly from an evolutionary point of view. The study of archaeal DNA replication started in the 1980s, and once it was found that aphidicolin, a specific inhibitor of family B DNA polymerases, inhibits DNA synthesis and cell growth in halophilic and methanogenic archaea, it became apparent that DNA replication in archaea differs from that in bacteria (7, 46). This also suggested that archaeal DNA polymerases resemble the eukaryotic DNA polymerases Pols α, δ, and ɛ because these family B DNA polymerases were inhibited by aphidicolin. The amino acid sequences deduced from cloned genes from three different archaea (Thermococcus litoralis, Sulfolobus solfataricus, and Pyrococcus furiosus) showed sequence similarities with those of the family B DNA polymerases (31, 32, 41). Examinations of the first archaeal genome to be fully sequenced, that of Methanocaldococcus jannaschii, led to the conclusion that, in contrast to eukaryotes, archaea contain only a single Pol B homologue (3), even though two different family B DNA polymerases had been reported earlier in Pyrodictium occultum (42).
In a study to identify new DNA polymerases in P. furiosus, Uemori et al. constructed a genomic library that was expressed in Escherichia coli and then screened the expressed proteins for polymerase activity at a high temperature, which led to the discovery of the family D DNA polymerase Pol D (43). Pol D is an archaeal-specific DNA polymerase that does not have amino acid sequence similarity with any other DNA polymerase (5). Total genome sequences have shown that all archaeal species, except Crenarchaeota, possess both Pol B and Pol D. The biochemical properties of these enzymes, such as stimulation by proliferation cell nuclear antigen (PCNA) and 3′ → 5′ exonuclease proofreading activity, are consistent with those of replicative polymerases (18, 39). Knockout experiments on the genes encoding Pol B and Pol D in Halobacterium NRC-1 suggested that both DNA polymerases are involved in DNA replication and are essential for cell viability (1). In vitro biochemical characterization, such as primer usage and strand-displacement activity (16, 17), have suggested that Pol B replicates the leading strand, whereas Pol D replicates the lagging strand. This is similar to the activities of Pol ɛ, which replicates the leading strand, and Pol δ, which replicates the lagging strand, in eukaryotes, showing that the systems of DNA replication in archaea and eukaryotes are similar.
Thermococcales members are the most frequently isolated hyperthermophiles. They are widely distributed at deep-sea and shallow marine hydrothermal vents as well as terrestrial thermal springs. At elevated temperatures, spontaneous DNA reactions, such as the hydrolytic deamination of cytosines and adenines, hydrolytic depurination, the oxidation of guanines, methylation of bases and phosphates, and strand breakage, are accelerated (23). Since hyperthermophiles inhabit such extremely hostile conditions, they possess stress adaptation mechanisms to ensure recovery from heat damage. T. kodakarensis, belonging to Thermococcales, has been isolated from a solfatara on the shore of Kodakara Island, Japan (26). Genetic manipulation methods have been developed for T. kodakarensis. E. coli–T. kodakarensis shuttle vectors and strong constitutive promoters for gene expression may be used (21). In the present study, we generated a T. kodakarensis strain in which polB was completely deleted and investigated the effects of exposure to DNA-damaging agents on this mutant. Our results indicate that Pol B is involved in DNA repair, not DNA replication, in T. kodakarensis.
Materials and Methods
Strains, media, and plasmids
T. kodakarensis strain DAD (genotype, ΔpyrF ΔpdaD) was used as the parental strain (9). Strains were cultivated in a nutrient-rich medium (ASW-YT-S0), synthetic medium (ASW-AA-S0 medium), or medium for Southern and Western blotting (ASW-YT-Pyr) under strict anaerobic conditions at 85°C (37). ASW-YT-S0 medium contained 0.8×artificial seawater (ASW), 5 g L−1 yeast extract, 5 g L−1 tryptone, and 2 g L−1 elemental sulfur (S0). ASW-AA-S0 medium contained 0.8×ASW, a mixture of 20 amino acids, modified Wolfe’s trace minerals, a vitamin mixture, and 2 g L−1 elemental sulfur (34, 37). ASW-YT-Pyr medium contained 0.8×ASW, 5 g L−1 yeast extract, 5 g L−1 tryptone, and 5 g L−1 sodium pyruvate. Resazurin sodium salt, a redox indicator, was added to ASW-YT-S0, ASW-AA-S0, and ASW-YT-Pyr media at concentrations of 2, 1, and 2 mg L−1, respectively. Na2S solution (5%) was added to ASW-YT-S0, ASW-AA-S0, and ASW-YT-Pyr media at concentrations of 0.025, 0.0075, and 0.025%, respectively, to make the medium color transparent prior to the inoculation. To prepare solid ASW-YT media, 2 mL L−1 polysulfide solution (10 g of Na2S·9H2O and 3 g of elemental sulfur in 15 mL of H2O) instead of S0 and 1% gellan gum were added to liquid media (27).
E. coli strain DH5α and the plasmid pUC19 (Takara Bio, Kyoto, Japan) were used for DNA manipulation. E. coli cells harboring pUC19 or its derivatives were cultured at 37°C in 2×YT medium (16 g L−1 tryptone, 10 g L−1 yeast extract, and 5 g L−1 NaCl) supplemented with 50 mg L−1 ampicillin.
The plasmid pUD3 containing a pyrF marker gene cassette was used for targeted gene disruption (45). The E. coli–T. kodakarensis shuttle plasmid pTKG02, a derivative of pTK02 previously reported by Nagaoka et al. (27), was used for gene complementation.
Unless otherwise mentioned, all chemicals were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan), Sigma-Aldrich (St. Louis, MO, USA), or Nacalai Tesque (Kyoto, Japan).
DNA manipulation
DNA manipulations were performed according to standard methods, as previously described by Sambrook and Russell (35). Restriction endonucleases and modification enzymes were purchased from Takara Bio and Toyobo (Osaka, Japan). DNA amplifications were performed with KOD-Plus- ver. 2 DNA polymerase (Toyobo) and the oligonucleotide primers listed in Table 1. The extraction and purification of plasmids from E. coli and of DNA fragments from agarose gels were performed with a QIAprep Spin Miniprep Kit and QIAquick Gel Extraction Kit, respectively (QIAGEN, Hilden, Germany). The DNA sequences of all plasmids constructed in the present study were confirmed by DNA sequencing performed on our behalf by Eurofins Genomics (Tokyo, Japan). Disruption of polB in T. kodakarensis DAD

DNA fragments containing the 5′ and 3′ flanking regions (ca. 1.0 kbp each) of polB (NCBI locus tag: TK_RS00010) were amplified from the genomic DNA of T. kodakarensis using the primer sets tk0001-D1/tk0001-D2 and tk0001-D3/tk0001-D4, respectively. Amplified fragments were inserted into the HincII site of pUC19. The resulting plasmids were digested with SphI/PstI (5′ flanking) and PstI/EcoRI (3′ flanking) and inserted into pUC19 digested with SphI and EcoRI, and DNA fragments digested with SphI and EcoRI were then inserted into the gene disruption plasmid pUD3 containing the pyrF marker gene cassette (45). The resultant plasmid was designated pUDΔtk0001 (Fig. 1). The theoretical background for gene disruption by homologous recombination has been described previously (37).

The disruption of polB in T. kodakarensis DAD was performed as described previously (27) with slight modifications. Cells were cultivated for 17 h in ASW-YT-S0 medium supplemented with 0.5 mM agmatine sulfate (T. kodakarensis DAD is an agmatine auxotroph because it lacks pdaD) (9). Cells were harvested and resuspended in 200 μL of 0.8×ASW and kept on ice for 30 min. After the addition of 12 μg of pUDΔtk0001 to the cell suspension and a further incubation on ice for 1 h, treated cells were cultivated in ASW-AA-S0 medium with 0.5 mM agmatine sulfate at 85°C for 24 h to enrich transformants that displayed uracil prototrophy due to the integration of the plasmid into the chromosome by single-crossover recombination. After the incubation, cells were diluted with 0.8×ASW and spread onto solid ASW-YT medium supplemented with 0.5 mM agmatine sulfate, 0.75% (w/v) 5-fluoroorotic acid (5-FOA), and 45 mM NaOH, and then incubated at 85°C for 2 d. Since cells with an intact pyrimidine biosynthetic pathway convert 5-FOA to 5-fluoroorotidine-5′-phosphate, which is toxic, only pyrF-deficient cells via the second pop-out recombination grew under these conditions. Transformants were selected and their genotype was examined by a polymerase chain reaction (PCR) with the primer set tk0001-D1/tk0001DSEQ4-2 and DNA sequencing. The constructed ΔpolB strain was designated DPB1.
Southern blot analysis
Genomic DNA (20 μg) from strain DAD or DPB1 was digested with PvuII and subjected to 1% agarose gel electrophoresis. Nucleic acids were transferred to a nylon membrane (Hybond™-N+; GE Healthcare, Chicago, IL, USA) by semi-dry electroblotting. A DIG High Prime DNA Labeling and Detection Starter Kit I (for color detection with NBT/BCIP) (Roche Diagnostics, Rotkreuz, Switzerland) was used for labeling and detection.
To construct polB and polB-upstream probes, a DNA fragment encoding the entire polB gene was amplified from the genomic DNA of T. kodakarensis using the primer set tk0001-F2/tk0001-R and then cloned into pUC19 digested with HincII. The resulting plasmid was purified from E. coli DH5α and digested with SphI/BamHI; the DNA fragment obtained was used as the polB probe. The plasmid containing the fragment amplified with the primer sets tk0001-D1/tk0001-D2, as mentioned previously, was digested with SphI/PstI (5′ flanking); the DNA fragment obtained was used as the polB-upstream probe.
Western blot analysis
Cell extracts from strains in the stationary or exponential growth phase were subjected to SDS-PAGE. After electroblotting onto a polyvinylidene difluoride membrane, the proteins on the membrane were reacted with anti-TkoPol B, anti-PfuDP1, and anti-TkoPrimase antisera, which were prepared by immunizing rabbits with the recombinant proteins T. kodakarensis Pol B, P. furiosus Pol D (small subunit), and T. kodakarensis DNA primase, respectively. Regarding the detection of Pol B, Pol D, and DNA primase, anti-Rabbit IgG HRP (Rabbit TrueBlot; Rockland Immunochemicals, PA, USA) was used as the secondary antibody. Proteins were visualized with an enhanced chemiluminescence detection system (Merck Millipore, Burlington, MA, USA) and an LAS-3000 image analyzer (FUJIFILM, Tokyo, Japan). Regarding the detection of Pol B in polB-complemented cells, an anti-Rabbit IgG (Fc) AP-Conjugate (Promega, Madison, WI, USA) was used as the secondary antibody together with nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate.
DNA polymerase activity assay of cell-free extracts
Strains DAD and DPB1 were cultivated in ASW-YT-Pyr medium supplemented with 0.5 mM agmatine sulfate at 85°C for 9 h until they had reached an optical density of 0.35 at a wavelength of 660 nm. Harvested cells (2×1010 cells) were suspended in 1.5 mL of buffer A (50 mM Tris-HCl [pH 8.0], 1 mM ethylenediaminetetraacetic acid, 0.5 mM dithiothreitol, 10% glycerol) and disrupted by sonication. The supernatant was applied to an anion-exchange column (HiTrap Q HP, 1 mL; GE Healthcare), and the chromatograph was developed with a 0 to 1.0 M NaCl gradient in 10 mL of buffer A.
A nucleotide incorporation assay was used to detect the DNA polymerizing activity of chromatographic fractions. The reaction mixtures (20 μL) contained 20 mM Tris-HCl [pH 8.8], 5 mM MgCl2, 1 mM dithiothreitol, 0.2 mg mL−1 of activated calf thymus DNA, 10 μM deoxynucleoside triphosphate containing 200 nM thymidine 5′-[methyl-3H] triphosphate, and 2 μL of the cell fraction. The reaction mixture was incubated at 74°C for 5 min and acidinsoluble radioactivity bound to a DE81 filter was detected with a scintillation counter.
Construction of the plasmid for the complementation analysis of strain DPB1
We constructed the plasmid pUC-PTgdh to add the promoter (Pgdh) and terminator (Tgdh) regions of the gene encoding glutamate dehydrogenase (gdh) into polB. The intergenic region (ca. 0.6 kbp) containing Pgdh between the coding region of gdh (TK1431) and the adjacent gene TK1432 was amplified from the genomic DNA of T. kodakarensis by PCR with the primer set Pgdh-F/Pgdh-R, and the PCR product was used as the template for PCR with the primer set Pgdh-F/Pgdh-R2. A DNA fragment (ca. 0.1 kbp) containing Tgdh was amplified from the genomic DNA of T. kodakarensis by PCR with the primer set Tgdh-F2/Tgdh-R, and the PCR product was used as the template for PCR with the primer set PTgdh-S/Tgdh-R. Since the amplified Pgdh and Tgdh regions shared a segment with an identical sequence, the two fragments were joined by overlap extension PCR with the primer set Pgdh-F/Tgdh-R. The joined fragment was inserted into pUC19 digested with HincII, and amplified from the resulting plasmid with the primer set Pgdh-F/Tgdh-R. The unique NdeI recognition sequence, CATATG, in pUC19 was changed to CAAATG by inverse PCR using the primer set puc19NdeIF/puc19NdeIR, and the resulting plasmid was used as the template for inverse PCR with the primer set pucEcoRIX/pucHindIIIX to remove the multiple cloning site. The amplified fragment containing Pgdh and Tgdh was phosphorylated and ligated to the PCR product using the primer set puc19NdeIF/puc19NdeIR. The resulting plasmid was designated pUC-PTgdh and contained recognition sites for the restriction enzymes NdeI, NheI, BamHI, EcoRI, and XhoI between the Pgdh and Tgdh regions.
A DNA fragment encoding polB (Δintein-1, Δintein-2) was amplified from the plasmid pET-pol(mature) (38) by PCR with the primer set Tk-polB-F/Tk-polB-R and inserted into pUC19 digested with HincII. The polB (Δintein-1, Δintein-2) fragment digested with NdeI/BamHI was inserted into the plasmid pUC-PTgdh digested with NdeI/BamHI. A DNA fragment containing Pgdh, polB (Δintein-1, Δintein-2), and Tgdh was obtained by digestion with EcoRV and inserted into the E. coli–T. kodakarensis shuttle plasmid pTKG02 digested with EcoRV. The resulting plasmid was designated pTKG-polB.
Drop dilution assay
Strain DAD and its derivatives were cultured in ASW-YT-S0 medium supplemented with or without agmatine sulfate (0.5 mM) at 85°C for 17 h.
To elucidate the sensitivity of the strains to DNA damage by γ-rays, the incubated cells were harvested and resuspended in 0.8×ASW at a concentration of 1×106 cells μL−1, and then irradiated with γ-rays from a 60Co-source at dose rates of 1 or 1.5 kGy h−1 at the Takasaki Advanced Radiation Research Institute (Gunma, Japan). The cell suspension was kept on ice before and after irradiation.
To elucidate the sensitivity of the strains to agents that induce DNA alkylation and interstrand cross-linking, the incubated cells were resuspended in 0.8×ASW supplemented with methyl methanesulfonate (MMS) or mitomycin C (MMC), respectively, at a concentration of 1×106 cells μL−1 and incubated at room temperature for 4 h. Cell suspensions after the treatment with γ-rays, MMS, or MMC were then serially diluted 10-fold with 0.8×ASW and spotted on solid ASW-YT medium with or without agmatine sulfate (0.5 mM).
To elucidate the sensitivity to DNA damage caused by ultraviolet C (UV-C), incubated cells were harvested and resuspended in 0.8×ASW at a concentration of 1×106 cells μL−1. Serially diluted cells (2×106, 2×105, 2×104, 2×103, and 2×102 cells) were spotted on solid ASW-YT medium, and irradiated with a germicidal ultraviolet lamp (wavelength, 254 nm; irradiance, 0.4 W m−2, as calculated using an UVX Radiometer; UVP, Upland, CA, USA).
After an incubation at 85°C for 2 d, living cells were transferred to polyvinylidene difluoride membranes and visualized by staining with Coomassie Brilliant Blue R-250.
Results
Construction of a polB deletion mutant
To examine the physiological role of Pol B in T. kodakarensis, a polB deletion mutant was constructed. The plasmid pUDΔtk0001 harboring pyrF as a selectable marker and the 5′ and 3′ flanking regions of polB was constructed and introduced into the chromosomal DNA of strain DAD by single-crossover recombination (Fig. 1). Uracil-prototrophic cells, due to the integration of the plasmid pUDΔtk0001, were concentrated in ASW-AA-S0 medium without uracil. 5-FOA-resistant cells generated by the excision of the pyrF marker gene after popout recombination were then isolated on solid ASW-YT medium containing 0.75% (w/v) 5-FOA. The mutant genotype was confirmed by a PCR analysis with the primer set tk0001-D1/tk0001DSEQ4-2, which annealed outside the polB encoding region (Fig. 2A). A 1,390-bp DNA fragment corresponding to the ΔpolB locus formed by pop-out recombination was observed, indicating that polB was deleted (Fig. 2B). The resulting ΔpolB mutant was designated strain DPB1.

The deletion of polB in DPB1 was further confirmed by Southern blotting and a PCR analysis. Southern blotting with a polB-upstream probe revealed a decrease in the length of the PvuII fragment (from 7,551 to 2,550 bp) due to the deletion of polB (Fig. 2C), and Southern blotting with the polB probe revealed the absence of polB in the chromosome of DPB1 cells (Fig. 2D).
The absence of Pol B in strain DPB1 was confirmed by Western blotting (Fig. 3A). Pol D and DNA primase were detected with comparable signal intensities in strains DAD and DPB1 (Fig. 3B and C). In addition, after the detection of Pol B using anti-TkoPol B antiserum (Fig. 3A), the same membrane was exposed to anti-TkoPrimase antiserum as a blotting control (Fig. 3D) and Pol B was not detected in strain DPB1, whereas DNA primase was detected in DAD and DPB1 at similar signal intensities.

Gefter et al. discovered E. coli DNA polymerase III, a replicative DNA polymerase, in chromatographic fractions of a polA− cell extract (12). Therefore, to check for the existence of novel DNA polymerases in strains DAD and DPB1, cell extracts were fractionated by anion exchange chromatography and subjected to a nucleotide incorporation assay (Fig. 4). In strain DAD, major DNA polymerizing activity was detected in fractions containing Pol B, while minor DNA polymerizing activity was detected in fractions containing Pol D and DNA primase (Fig. 4A). In contrast, in strain DPB1, DNA polymerizing activity was only detected in fractions containing Pol D and DNA primase (Fig. 4B). In T. kodakarensis, Pol D and DNA primase may both synthesize DNA (11), although the activity of DNA primase is markedly lower than that of Pol D (17).
Construction of a polB-complemented strain
To express the Pol B protein in DPB1 cells, the promoter and terminator of gdh were used because the gdh promoter leads to strong constitutive gene expression in T. kodakarensis irrespective of culture conditions (e.g., media and growth temperature) (27, 33). Plasmid construction is summarized in Fig. 5.

The plasmid, pUC-PTgdh, containing a promoter-terminator cassette with recognition sites for the restriction enzymes NdeI, NheI, BamHI, EcoRI, and XhoI was constructed. polB in the chromosome DNA of T. kodakarensis contains two intervening sequences, intein-1 and intein-2, which are spliced out at the protein level in an autocatalytic manner. After excision of the intervening proteins, PI-PkoI and PI-PkoII, corresponding to intein-1 and intein-2, respectively, from the precursor protein, external protein segments were ligated to form a mature functional PolB protein. These spliced proteins, PI-PkoI and PI-PkoII, exhibit endonuclease activities at specific DNA regions (29, 38). In the gene complementation analysis, the PolB protein was produced from polB without any intervening sequences in DPB1 cells because the effects of the production of PI-PkoI and PI-PkoII on the viability of DPB1 cells need to be omitted. The fragment encoding polB without any intervening sequences was ligated with pUC-PTgdh for insertion between the promoter and terminator derived from the gdh gene.
The E. coli–T. kodakarensis shuttle plasmid pTKG02, a derivative of pTK02, was used for the expression of polB (27). pTKG02 contains pdaD, which encodes arginine decarboxylase, a catalyst of the synthesis of agmatine from arginine. Agmatine is essential for the growth of T. kodakarensis cells (9). T. kodakarensis strain DAD is an agmatine auxotroph that cannot grow in the absence of agmatine because it lacks pdaD; however, DAD cells harboring pTKG02 may grow in the absence of agmatine. The fragment containing the gdh promoter, polB (Δintein-1, Δintein-2), and gdh terminator was inserted into pTKG02 digested with EcoRV. The resulting plasmid was designated pTKG-polB.
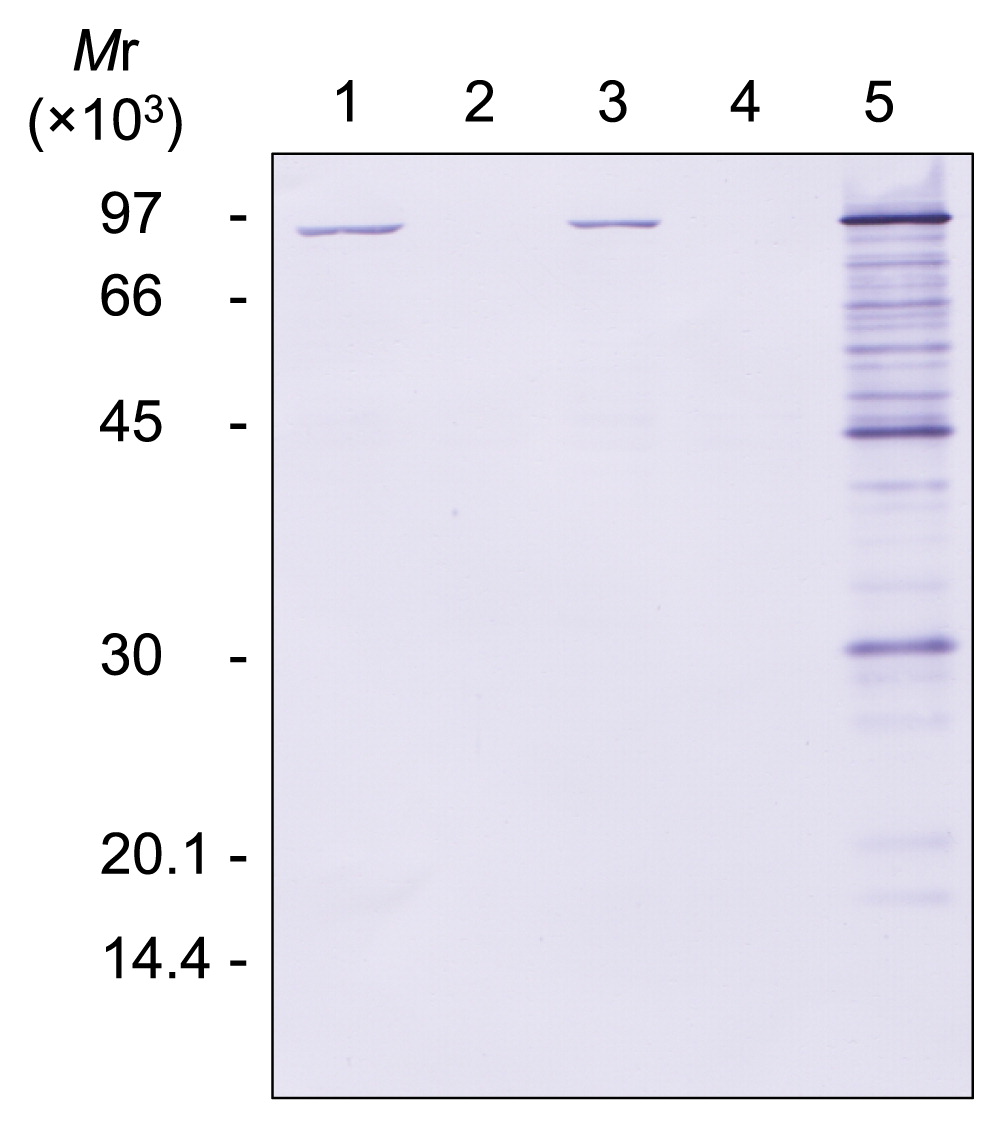
After the transformation of strain DPB1 with pTKG-polB, Western blotting was performed to confirm the expression of Pol B (Fig. 6). Pol B expression was detected in the polB-complemented strain (DPB1 harboring pTKG-polB) and in DAD with or without pTKG02, whereas it was not detected in DPB1 with or without pTKG02. The amount of Pol B expressed by the polB-complemented strain was higher than that expressed by DAD with or without pTKG02. Signals corresponding to the proteolytic degradation of Pol B were found in polB-complemented cells; however, DPB1 with or without pTKG-polB grew normally under the same conditions, as reported by Čuboňová et al. (6) (Fig. 7). These results indicated that the lack or overexpression of Pol B does not affect cell growth in T. kodakarensis.
Sensitivity of the polB disruptants and complementants to DNA damage
A drop dilution assay was performed to evaluate the sensitivity of the strains to DNA damage caused by exposure to UV-C, γ-rays, MMS, or MMC (Fig. 8).
When untreated, possession of the E. coli–T. kodakarensis shuttle plasmid pTKG02 did not affect cell viability. In addition, DPB1 with or without pTKG-polB showed the same cell viability as the parental strain, DAD. This result suggested that the lack or overexpression of Pol B did not affect cell viability in T. kodakarensis.
When exposed to UV-C irradiation, which mainly induces the formation of intrastrand cross-links that results in the production of cyclobutane pyrimidine dimers and pyrimidine–pyrimidine (6-4) photoproducts, the strains not harboring polB (i.e., DPB1 and DPB1/pTKG02) showed higher sensitivity than those harboring polB ( i.e., DAD, DAD/pTKG02, and DPB1/pTKG-polB) (Fig. 8A). DAD harboring pTKG02 showed higher sensitivity than DAD not harboring pTKG02, indicating that pTKG02 decreased the viability of cells exposed to UV-C irradiation. When exposed to MMC which induces the formation of interstrand cross-links in double-stranded DNA and inhibits replication fork progression, the strains harboring the plasmid pTKG02 showed higher sensitivity than when they were exposed to UV-C irradiation (Fig. 8D).
When exposed to γ-ray irradiation, which induces the formation of double-strand breaks, or to MMS, which introduces methylated bases (e.g., N7-methylguanine, N3-methyladenine, and O6-methylguanine) in DNA strands, DPB1 showed higher sensitivity than DAD, and the introduction of pTKG-polB to DPB1 restored resistance to the wild-type level (Fig. 8B and C).
Overall, DPB1 was more sensitive than DAD to all of the treatments and the presence of pTKG-polB in DPB1 restored resistance to DNA damage-related stress to an equivalent level to that of DAD harboring pTKG02. This result indicated that the observed sensitivity to DNA damage-related stress was due to the loss of polB.
Discussion
T. kodakarensis expresses two DNA polymerases, Pol B and Pol D. We herein successfully disrupted polB in the T. kodakarensis genome to generate a mutant, designated DPB1. DPB1 showed higher sensitivity to all of the tested DNA-damaging treatments (UV-C, MMS, MMC, and γ-rays) than the parental strain, DAD (Fig. 8). UV-C induces the formation of cyclobutane pyrimidine dimers and pyrimidine-pyrimidine (6-4) photoproducts in chromosomal DNA. These lesions block the progression of DNA synthesis by family A, B, C, D, or X DNA polymerases, although several enzymes belonging to family Y are able to bypass these lesions (30, 44); no gene encoding a family Y DNA polymerase has yet been reported in the T. kodakarensis genome (10). The present results also indicated that novel DNA polymerase activity was not found in strain DPB or DAD (Fig. 4). UV-induced lesions may be repaired either by light-dependent DNA repair enzymes called DNA photolyases or via the light-independent nucleotide excision repair (NER) pathway (20). In T. kodakarensis, UV-induced lesions appear to be repaired via the NER system because the T. kodakarensis genome does not contain any genes that encode DNA photolyases (10), and the disruption of genes associated with the NER pathway produces strains that are more sensitive to UV irradiation than their parental strains (8).
MMS introduces methylated bases into DNA strands, and MMC induces the formation of interstrand cross-links in double-stranded DNA and inhibits replication fork progression. This DNA damage appears to be repaired via the NER pathway in T. kodakarensis because the deletion of genes involved in the NER pathway produces strains that are more sensitive to these agents than their parental strains (8). In the present study, when strains were exposed to UV-C or MMC, DAD harboring pTKG02 showed higher sensitivity than DAD not harboring the plasmid. MMC is used for plasmid curing because it inhibits DNA replication and represses transcription due to the formation of interstrand cross-links in double-stranded DNA (40). Irradiation with UV-C also induces interstrand cross-links in double-stranded DNA (25). Therefore, a possible explanation for the higher sensitivities of the pTKG02-harboring strain to exposure to UV-C and MMC is the direct inhibition of plasmid replication by the formation of interstrand cross-links due to stalling replication forks and the indirect inhibition of plasmid replication through the repression of gene transcription related to plasmid replication on pTKG02, both of which cause the loss of plasmids in cells. Therefore, plasmid-cured cells no longer grow on a plate without agmatine.
γ-Ray irradiation induces double-strand breaks in DNA. Double-strand breaks in chromosomal DNA are considered to be repaired via the homologous recombination pathway. The sensitivity of DPB1 to DNA damage caused by UV-C, MMS, MMC, and γ-rays was suppressed by the introduction of a plasmid harboring polB into cells, suggesting that the exposure-sensitive phenotype of strain DPB1 was due to the loss of polB. In Thermococcus, Pol B exhibits strong strand-displacement activity and the ability to perform gap filling (13), which suggests that gap filling and strand-displacement synthesis by Pol B are important parts of the NER and homologous recombination pathways.
The growth of strain DPB1 was similar with that of the parental strain, DAD (Fig. 7). This is consistent with recently published findings (6, 36). The absence of Pol B did not affect cell growth, suggesting that Pol D is responsible for the synthesis of both the leading and lagging strands in DPB1 cells.
The RNA segment of Okazaki fragments must be removed for ligation to the preceding Okazaki fragment. Removal of the RNA segment at the 5′-terminus of Okazaki fragments was previously suggested to be accomplished by Fen1, RNase HII, or GAN (4). Fen1 is a DNA endonuclease specific for flap-structured DNA made by strand displacement in the RNA primer region of the former Okazaki fragment. However, the strand-displacement activity of Pol D is markedly weaker than that of Pol B. In vitro analyses revealed that the majority of lagging strand synthesis by Pol D with PCNA, RFC, Fen1, and DNA ligase was stopped before Okazaki fragment maturation (13, 15); therefore, it currently remains unclear whether the strand-displacement activity of Pol D is sufficient for Okazaki fragment processing in DPB1 cells. GAN forms a complex with Pol D in vivo and degrades single-stranded DNA in the 5′ → 3′ direction (22). However, in vitro experiments have shown that its activity is inhibited under metal ion concentrations that mimic those in T. kodakarensis (28). Furthermore, GAN cannot degrade RNA in vitro (22). In contrast to these findings, gene disruption experiments have suggested that GAN participates in the removal of the RNA segment of the Okazaki fragment (4). Even though RNase HII cleaves the RNA/DNA junction of Okazaki fragments, a single ribonucleotide remains at the 5′-terminus of the DNA segment (14). GAN may degrade the RNA segment of Okazaki fragments by associating with the replisome in T. kodakarensis.
The present study revealed that Pol B is important for DNA repair, not DNA replication. Further analyses are needed to fully elucidate the mechanisms underlying DNA replication in T. kodakarensis. However, the present study suggests that Pol D has the ability to synthesize both the leading and lagging DNA strands in T. kodakarensis; this result is consistent with previous findings (6). In the high temperature environments in which Thermococcales members live, cells are exposed to chemical stresses, such as the deamination of cytosines and adenines, depurination, the oxidation of guanines, and the methylation of bases and phosphates. DNA double-strand breaks are induced by heat as well as ionizing radiation. These DNA-damaging reactions are accelerated at elevated temperatures (23). Hence, hyperthermophiles have unique active and efficient repair systems to maintain their genomic integrity (19). The present results suggest that the function of Pol B is essential for these repair systems, which allow for survival in harsh environments.
Acknowledgements
This study was supported in part by the Inoue Enryo Memorial Foundation for Promoting Sciences.
References
- 1. Berquist, B.R., P.
DasSarma, and S.
DasSarma. 2007. Essential and non-essential DNA replication genes in the model halophilic Archaeon, Halobacterium sp. NRC-1. BMC Genet. 8:31.
- 2. Braithwaite, D.K., and J.
Ito. 1993. Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic Acids Res. 21:787-802.
- 3. Bult, C.J., O.
White, G.J.
Olsen, et al. 1996. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science. 273:1058-1073.
- 4. Burkhart, B.W., L.
Cubonova, M.R.
Heider, Z.
Kelman, J.N.
Reeve, and T.J.
Santangelo. 2017. The GAN exonuclease or the Flap endonuclease Fen1 and RNase HII are necessary for viability of Thermococcus kodakarensis. J Bacteriol. 199:e00141-17.
- 5. Cann, I.K.O., and Y.
Ishino. 1999. Archaeal DNA replication: identifying the pieces to solve a puzzle. Genetics. 152:1249-1267.
- 6. Čuboňová,, L., T.
Richardson, B.W.
Burkhart, Z.
Kelman, B.A.
Connolly, J.N.
Reeve, and T.J.
Santangelo. 2013. Archaeal DNA polymerase D but not DNA polymerase B is required for genome replication in Thermococcus kodakarensis. J Bacteriol. 195:2322-2328.
- 7. Forterre, P., C.
Elie, and M.
Kohiyama. 1984. Aphidicolin inhibits growth and DNA synthesis in halophilic archaebacteria. J Bacteriol. 159:800-802.
- 8. Fujikane, R., S.
Ishino, Y.
Ishino, and P.
Forterre. 2010. Genetic analysis of DNA repair in the hyperthermophilic archaeon, Thermococcus kodakaraensis. Genes Genet Syst. 85:243-257.
- 9. Fukuda, W., N.
Morimoto, T.
Imanaka, and S.
Fujiwara. 2008. Agmatine is essential for the cell growth of Thermococcus kodakaraensis. FEMS Microbiol Lett. 287:113-120.
- 10. Fukui, T., H.
Atomi, T.
Kanai, R.
Matsumi, S.
Fujiwara, and T.
Imanaka. 2005. Complete genome sequence of the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1 and comparison with Pyrococcus genomes. Genome Res. 15:352-363.
- 11. Galal, W.C., M.
Pan, Z.
Kelman, and J.
Hurwitz. 2012. Characterization of DNA primase complex isolated from the archaeon, Thermococcus kodakaraensis. J Biol Chem. 287:16209-16219.
- 12. Gefter, M.L., Y.
Hirota, T.
Kornberg, J.A.
Wechsler, and C.
Barnoux. 1971. Analysis of DNA Polymerases II and III in mutants of Escherichia coli thermosensitive for DNA synthesis. Proc Natl Acad Sci USA. 68:3150-3153.
- 13. Greenough, L., Z.
Kelman, and A.F.
Gardner. 2015. The roles of family B and D DNA polymerases in Thermococcus species 9°N Okazaki fragment maturation. J Biol Chem. 290:12514-12522.
- 14. Haruki, M., Y.
Tsunaka, M.
Morikawa, and S.
Kanaya. 2002. Cleavage of a DNA-RNA-DNA/DNA chimeric substrate containing a single ribonucleotide at the DNA-RNA junction with prokaryotic RNases HII. FEBS Lett. 531:204-208.
- 15. Heider, M.R., B.W.
Burkhart, T.J.
Santangelo, and A.F.
Gardner. 2017. Defining the RNase H2 enzyme-initiated ribonucleotide excision repair pathway in Archaea. J Biol Chem. 292:8835-8845.
- 16. Henneke, G., D.
Flament, U.
Hübscher, J.
Querellou, and J.-P.
Raffin. 2005. The hyperthermophilic euryarchaeota Pyrococcus abyssi likely requires the two DNA polymerases D and B for DNA replication. J Mol Biol. 350:53-64.
- 17. Ishino, S., and Y.
Ishino. 2006. Comprehensive search for DNA polymerase in the hyperthermophilic archaeon, Pyrococcus furiosus. Nucleosides, Nucleotides Nucleic Acids. 25:681-691.
- 18. Ishino, Y., and S.
Ishino. 2001. DNA polymerases from Euryarchaeota. Methods Enzymol. 334:249-260.
- 19. Ishino, Y., and I.
Narumi. 2015. DNA repair in hyperthermophilic and hyperradioresistant microorganisms. Curr Opin Microbiol. 25:103-112.
- 20. Jones, D.L., and B.K.
Baxter. 2017. DNA repair and Photoprotection: mechanisms of overcoming environmental ultraviolet radiation exposure in halophilic archaea. Front Microbiol. 8:1882.
- 21. Leigh, J.A., S.-V.
Albers, H.
Atomi, and T.
Allers. 2011. Model organisms for genetics in the domain archaea: methanogens, halophiles, Thermococcales and Sulfolobales. FEMS Microbiol Rev. 35:577-608.
- 22. Li, Z., M.
Pan, T.J.
Santangelo, W.
Chemnitz, W.
Yuan, J.L.
Edwards, J.
Hurwitz, J.N.
Reeve, and Z.
Kelman. 2011. A novel DNA nuclease is stimulated by association with the GINS complex. Nucleic Acids Res. 39:6114-6123.
- 23. Lindahl, T.
1993. Instability and decay of the primary structure of DNA. Nature. 362:709-715.
- 24. Lipps, G., S.
Röther, C.
Hart, and G.
Krauss. 2003. A novel type of replicative enzyme harboring ATPase, primase and DNA polymerase activity. EMBO J. 22:2516-2525.
- 25. Love, J.D., H.T.
Nguyen, A.
Or, A.K.
Attri, and K.W.
Minton. 1986. UV-induced interstrand cross-linking of d(GT)n·d(CA)n is facilitated by a structural transition. J Biol Chem. 261:10051-10057.
- 26. Morikawa, M., Y.
Izawa, N.
Rashid, T.
Hoaki, and T.
Imanaka. 1994. Purification and characterization of a thermostable thiol protease from a newly isolated hyperthermophilic Pyrococcus sp. Appl Environ Microbiol. 60:4559-4566.
- 27. Nagaoka, E., R.
Hidese, T.
Imanaka, and S.
Fujiwara. 2013. Importance and determinants of induction of cold-induced DEAD RNA helicase in the hyperthermophilic archaeon Thermococcus kodakarensis. J Bacteriol. 195:3442-3450.
- 28. Nagata, M., S.
Ishino, T.
Yamagami, H.
Ogino, J.-R.
Simons, T.
Kanai, H.
Atomi, and Y.
Ishino. 2017. The Cdc45/RecJ-like protein forms a complex with GINS and MCM, and is important for DNA replication in Thermococcus kodakarensis. Nucleic Acids Res. 45:10693-10705.
- 29. Nishioka, M., S.
Fujiwara, M.
Takagi, and T.
Imanaka. 1998. Characterization of two intein homing endonucleases encoded in the DNA polymerase gene of Pyrococcus kodakaraensis strain KOD1. Nucleic Acids Res. 26:4409-4412.
- 30. Ohmori, H., E.C.
Friedberg, R.P.P.
Fuchs, et al. 2001. The Y-family of DNA polymerases. Mol Cell. 8:7-8.
- 31. Perler, F.B., D.G.
Comb, W.E.
Jack, et al. 1992. Intervening sequences in an Archaea DNA polymerase gene. Proc Natl Acad Sci USA. 89:5577-5581.
- 32. Pisani, F.M., C.
De Martino, and M.
Rossi. 1992. A DNA polymerase from the archaeon Sulfolobus solfataricus shows sequence similarity to family B DNA polymerases. Nucleic Acids Res. 20:2711-2716.
- 33. Rahman, R.N.Z.A., S.
Fujiwara, M.
Takagi, and T.
Imanaka. 1998. Sequence analysis of glutamate dehydrogenase (GDH) from the hyperthermophilic archaeon Pyrococcus sp. KOD1 and comparison of the enzymatic characteristics of native and recombinant GDHs. Mol Gen Genet. 257:338-347.
- 34. Robb, F.T., and A.R.
Place. 1995. Media for thermophiles, p.167-168. In F.T.
Robb, and A.R.
Place (ed.), Archaea: A Laboratory Manual—Thermophiles. Cold Spring Harbor Laboratory Press, New York.
- 35. Sambrook, J., and D.W.
Russell. 2001. Molecular Cloning: A Laboratory Manual, 3rd ed. Cold Spring Harbor Laboratory Press, New York.
- 36. Sarmiento, F., J.
Mrázek, and W.B.
Whitman. 2013. Genome-scale analysis of gene function in the hydrogenotrophic methanogenic archaeon Methanococcus maripaludis. Proc Natl Acad Sci USA. 110:4726-4731.
- 37. Sato, T., T.
Fukui, H.
Atomi, and T.
Imanaka. 2003. Targeted gene disruption by homologous recombination in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J Bacteriol. 185:210-220.
- 38. Takagi, M., M.
Nishioka, H.
Kakihara, M.
Kitabayashi, H.
Inoue, B.
Kawakami, M.
Oka, and T.
Imanaka. 1997. Characterization of DNA polymerase from Pyrococcus sp. strain KOD1 and its application to PCR. Appl Environ Microbiol. 63:4504-4510.
- 39. Tori, K., M.
Kimizu, S.
Ishino, and Y.
Ishino. 2007. DNA Polymerase BI and D from the hyperthermophilic archaeon, Pyrococcus furiosus both bind to proliferating cell nuclear antigen with their C-terminal PIP-box motifs. J Bacteriol. 189:5652-5657.
- 40. Trevors, J.T.
1986. Plasmid curing in bacteria. (Curing; plasmids; protoplasts; intercalating dyes; incompatibility). FEMS Microbiol Rev. 32:149-157.
- 41. Uemori, T., Y.
Ishino, H.
Toh, K.
Asada, and I.
Kato. 1993. Organization and nucleotide sequence of the DNA polymerase gene from the archaeon Pyrococcus furiosus. Nucleic Acids Res. 21:259-265.
- 42. Uemori, T., Y.
Ishino, H.
Doi, and I.
Kato. 1995. The hyperthermophilic archaeon Pyrodictium occultum has two α-like DNA polymerases. J Bacteriol. 177:2164-2177.
- 43. Uemori, T., Y.
Sato, I.
Kato, H.
Doi, and Y.
Ishino. 1997. A novel DNA polymerase in the hyperthermophilic archaeon, Pyrococcus furiosus: gene cloning, expression, and characterization. Genes Cells. 2:499-512.
- 44. Yang, W.
2005. Portraits of a Y-family DNA polymerase. FEBS Lett. 579:868-872.
- 45. Yokooji, Y., H.
Tomita, H.
Atomi, and T.
Imanaka. 2009. Pantoate kinase and phosphopantothenate synthetase, two novel enzymes necessary for CoA biosynthesis in the Archaea. J Biol Chem. 284:28137-28145.
- 46. Zabel, H.-P., H.
Fischer, E.
Holler, and J.
Winter. 1985. In vivo and in vitro evidence for eukaryotic α-type DNA-polymerases in Methanogens. Purification of the DNA-polymerase of Methanococcus vannielii. Syst Appl Microbiol. 6:111-118.