Microbes inhabit numerous environments, including soil, water environments, plants, and animals. Humans host various complex commensal microbe communities in and on their bodies, such as the oral microbiota, skin microbiota, and gut microbiota (The Integrative HMP Research Network Consortium, 2019). Although there are up to 37 trillion cells in the human body, approximately 40 trillion gut microbes inhabit the gastrointestinal tract (Sender et al., 2016). The host employs a number of mechanisms to maintain intestinal homeostasis and prevent anomalous immune responses directed against the microbiota. One of these protective mechanisms involves a mucus layer that covers the epithelial surface of the colon, which functions as a barrier and separates epithelial cells from gut microbes (Tropini et al., 2017) (Fig. 1A). The mucus layer prevents pathogens from reaching and persisting on intestinal epithelial surfaces and, thus, it is an important component of host immunity. This layer is constantly renewed and acts as a trap for both commensal residents and pathogens, preventing their access to the epithelium (Johansson et al., 2014; Desai et al., 2016). The gut microbiota is present in different microhabitats and metabolic niches in the mucus layer secreted from the gut, the mucosa, and the surfaces of digestive residues in the gut lumen (Macfarlane and Dillon, 2007). There are several indications that gut microbes inhabit the colonic mucus as a polymicrobial biofilm, which is an extracellular matrix-enclosed aggregate form of microbes (Hooper and Gordon, 2001; Sonnenburg et al., 2004; Flemming et al., 2016). Bacteria in biofilms cooperate or compete with each other and form complex communities (Tytgat et al., 2019). Healthy mucosal biofilms benefit the host because they are involved in the exchange of nutrients on the epithelial surface, increase colonization resistance, and protect the host from invasion by intestinal pathogens (Hooper and Gordon, 2001; Sonnenburg et al., 2004). The disruption of healthy mucosal biofilms leads to the nearby or direct contact of pathogenic invasive biofilms with colonic epithelial cells, resulting in inflammation (Swidsinski et al., 2005; Dejea et al., 2014; Desai et al., 2016) (Fig. 1B).
MAMP- and metabolite-mediated gut microbiota-host interactions
Gut microbes interact with the host via gut microbe-derived products (Levy et al., 2017). Gut microbe-derived MAMPs are recognized by pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-like receptors (NLRs), on epithelial cells (Cani, 2018). TLRs are transmembrane proteins that localize to the host cell surface, whereas NLRs are cytosolic proteins (Cani, 2018). A stimulation from gut commensal microbes shapes and trains the immune system. MAMPs are recognized by PRRs and induce adaptive responses by the immune system, resulting in the memory effect of innate immune cells and enhancements in the immune defenses of the host (Quintin et al., 2012; Saeed et al., 2014; Netea et al., 2016). Although the immune stimulation by MAMPs has been extensively examined, this stimulation may also be caused by another type of microbe-derived substance called bacterial membrane vesicles (MVs). Bacterial MVs are “bubble”-like membrane structures released by bacteria that range between 20 and 400 nm in diameter (Toyofuku et al., 2019). Many gut microbes and pathogens have been reported to produce MVs (Shen et al., 2012; Obana et al., 2017; Canas et al., 2018; Chelakkot et al., 2018). Bacteroides fragilis is a commensal microbe in the human gastrointestinal tract that produces a type of capsular exopolysaccharide called polysaccharide A (PSA), which has an immunomodulatory function. PSA was previously shown to be effective in animal models of inflammatory bowel disease (IBD) and multiple sclerosis (Shen et al., 2012). PSA is carried by the MVs of B. fragilis and is delivered to the host immune system to promote regulatory T-cell (Treg) activity and anti-inflammatory cytokine production through TLR2 (Shen et al., 2012) (Fig. 2). In addition, Akkermansia muciniphila-derived MVs are present at higher amounts in healthy individuals than in patients with type 2 diabetes (T2D). The administration of A. muciniphila-derived MVs ameliorated T2D in a mouse model by enhancing the tight-junction function of epithelial cells, which act as a barrier and reduce gut permeability (Chelakkot et al., 2018) (Fig. 2).
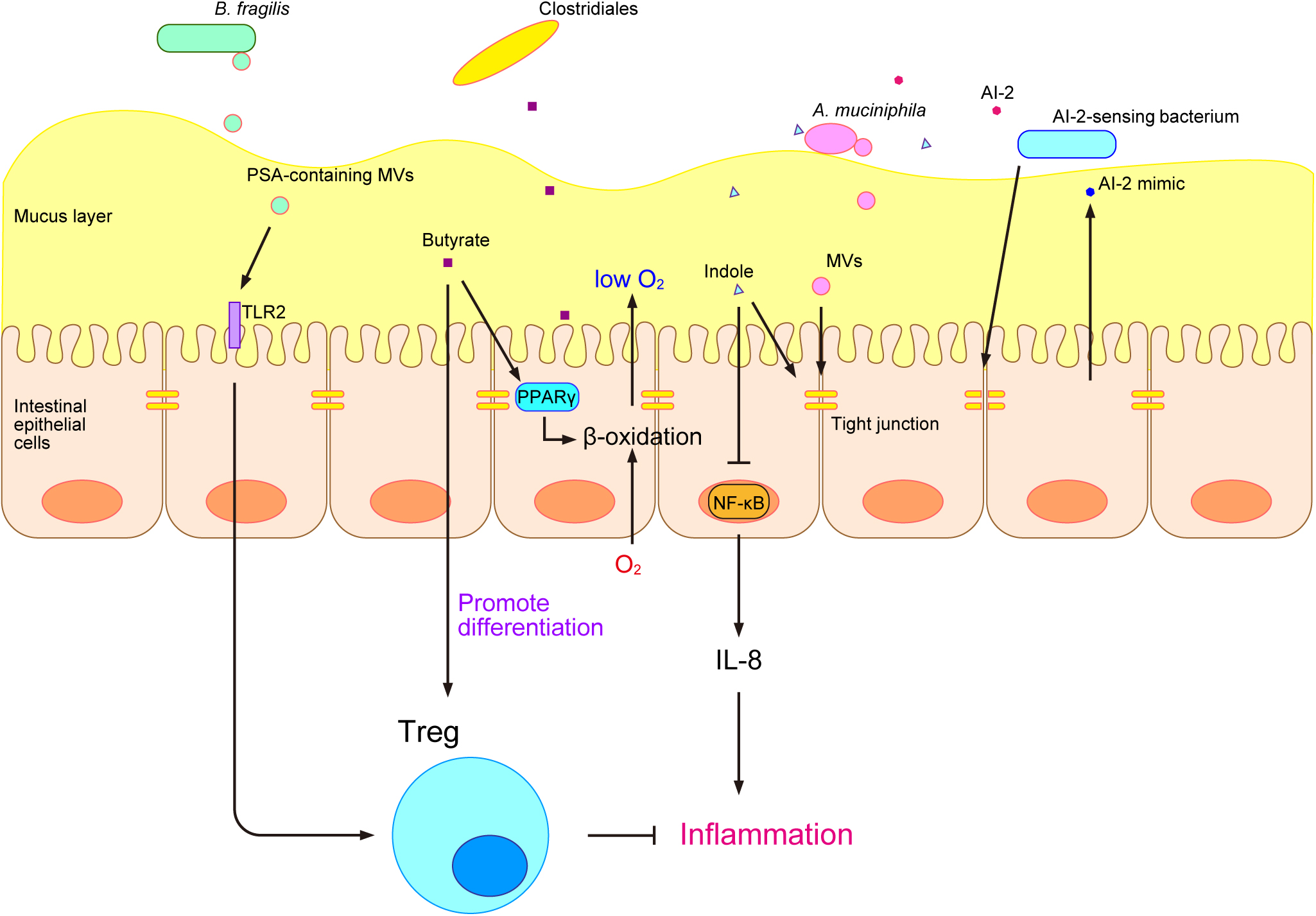
The development of metabolome analysis technologies has allowed us to understand that host-gut microbe interactions occur not only via microbe-derived substances, but also through various microbial metabolites, such as trimethylamine N-oxide (TMAO), secondary bile acids, and short-chain fatty acids (SCFAs) (Fukuda and Ohno, 2014). TMAO is produced by gut microbes from dietary phosphatidylcholine and promotes atherosclerosis (Wang et al., 2011). Bile acids are synthesized in the liver, secreted into the lumen of the small intestine, and facilitate the emulsification and absorption of lipids (Jia et al., 2018). Gut microbes convert bile acids into various secondary bile acids, which have been reported to exert inflammatory and carcinogenic effects (Jia et al., 2018). SCFAs, such as propionate, lactate, and butyrate, have been extensively examined and perform various functions in the host (Brown et al., 2003; Furusawa et al., 2013; Okada et al., 2013; Smith et al., 2013; Byndloss et al., 2017). Propionate and other SCFAs activate the orphan G protein-coupled receptors GPR41 and GPR43 and affect host energy metabolism through intestinal peptides, such as glucagon-like peptide-1 (GLP-1) (Brown et al., 2003). We previously showed that lactate induced the turnover of colonic epithelial cells in mice that were fed after starvation and promoted tumorigenesis in the colon (Okada et al., 2013). Furthermore, butyrate was shown to have an immunomodulatory function in a mouse model and induced the differentiation of colonic Treg (Furusawa et al., 2013) (Fig. 2). It also suppressed the growth of Enterobacteriaceae and protected the murine gut from Salmonella and Escherichia coli infections by activating β-oxidation and limiting luminal oxygen levels through the intracellular butyrate sensor peroxisome proliferator-activated receptor γ (PPARγ) (Byndloss et al., 2017) (Fig. 2). To obtain a more detailed understanding of the relationship between the gut microbiome and gut metabolites, we developed an integrated omics analysis called metabologenomics, which is a method that includes both microbiome and metabolome analyses (Ishii et al., 2018). This metabologenomic approach enables us to examine the complex network among the gut microbiota, gut microbiota-derived metabolites, and the host, thereby providing more comprehensive information about the gut environment (Ishii et al., 2018). An analysis of the localization of various gut microbes in polymicrobial biofilms may provide novel insights into the metabolic network in the gut environment because previous studies suggested that gut microbes engage in cross-feeding in the gut environment (Belenguer et al., 2006; Bunesova et al., 2018; Pareek et al., 2019), and biofilms are one of the locations at which cross-feeding occurs (Sztajer et al., 2014; Sakanaka et al., 2015).
In addition to microbial metabolites, bacterial cell-to-cell signaling molecules are also involved in interactions with the host. Bacteria produce several types of cell-to-cell signaling molecules for intraspecies and interspecies communication and regulate their gene expression and behavior (Yang et al., 2017a; Mukherjee and Bassler, 2019). The tryptophan metabolite indole is a major signaling molecule that is produced by various gut microbes and is involved in bacterial interspecies communication as well as interkingdom communication between the host and gut microbes (Jaglin et al., 2018). Indole increases the expression of genes involved in the function of the colonic mucosal barrier and in mucin secretion and increases tight-junction resistance while decreasing nuclear factor-κB (NF-κB) and the proinflammatory chemokine interleukin-8 (IL-8) in epithelial cells in vitro (Bansal et al., 2010) (Fig. 2). Short-term exposure to indole promotes GLP-1 secretion by mouse colonic L cells through the inhibition of K+ channels, whereas long-term exposure decreases its secretion by blocking NADH dehydrogenase (Chimerel et al., 2014). Autoinducer-2 (AI-2) is another cell-to-cell signaling molecule that modulates virulence production, biofilm formation, and gut colonization (Thompson et al., 2015). Its production has been reported in many bacteria, including various gut microbes (Thompson et al., 2015). AI-2 signaling has both interspecies and interkingdom communication functions. Notably, in the interspecies signaling of gut microbes, Blautia obeum has been suggested to protect the murine gut from Vibrio cholerae infection through AI-2 signaling (Hsiao et al., 2014). AI-2 may also alter the composition of the gut microbiota in antibiotic-treated mice and increase the abundance of Firmicutes, one of the major phyla of gut microbes (Thompson et al., 2015). In gut interkingdom signaling, epithelial cells sense the disruption of tight junctions by bacteria and produce a molecule that mimics AI-2, which may be detected by the bacterial AI-2 receptor and activates gene regulation controlled by AI-2 signaling (Ismail et al., 2016) (Fig. 2). Previous studies revealed that AI-2 signaling modulated biofilm formation by gut microbes in vitro (González Barrios et al., 2006; Ðapa et al., 2013; Laganenka and Sourjik, 2018); therefore, the host appears to use AI-2 signaling to maintain the composition of its mucosal biofilms.
Gut microbiota and host health
Gut microbiota and gastrointestinal tract infection and inflammation
The gut microbiota maintains homeostasis in the gastrointestinal tract, and the disruption of this balance results in infection and inflammation (Levy et al., 2017). We previously demonstrated that Bifidobacterium longum subsp. longum JCM 1217T produced acetate and protected host mice from E. coli O157:H7 infection by up-regulating gut epithelial barrier function to block the translocation of the Shiga toxin from the gut lumen into the blood (Fukuda et al., 2011). In addition, Clostridiales contributed to resistance to pathogen colonization and protected neonatal mice from infection (Kim et al., 2017b). In gnotobiotic mice, the restriction of dietary fiber decreased the nutrient supply to the colon, promoted the growth of mucin-degrading A. muciniphila, and reduced the thickness of the colonic mucus barrier, which resulted in easy access and infection by the mucosal pathogen Citrobacter rodentium (Desai et al., 2016).
IBD encompasses a group of autoimmune diseases in the gastrointestinal tract and mainly consists of Crohn’s disease and ulcerative colitis. The gut microbiota is related to IBD; the transition from symbiosis to dysbiosis in the gut results in gastric chronic inflammation (Nagao-Kitamoto and Kamada, 2017). Various species have been reported to cause inflammation. Although commensal B. fragilis is considered to be beneficial for IBD, enterotoxigenic strains of B. fragilis (ETBF) have been detected in the stools of 13.3% of IBD patients versus 2.9% of control groups (Nagao-Kitamoto and Kamada, 2017). ETBF produce toxins that bind to colonic epithelial cells, disrupt E-cadherin and the mucosal barrier, and increase IL-8 production, which results in inflammation (Nagao-Kitamoto and Kamada, 2017). Microscopic observations of gut samples from IBD patients revealed invasive biofilms abundant with B. fragilis covering the entire mucosa and in epithelial crypts (Swidsinski et al., 2005). Furthermore, colonization of the colon by the oral pathogen Klebsiella pneumoniae resulted in IBD (Atarashi et al., 2017). K. pneumoniae colonization results in the accumulation of T helper 1 (Th1) cells and a colitis phenotype in gnotobiotic mice. Furthermore, a metagenomic data analysis showed that Klebsiella was significantly more abundant in the feces of IBD patients, which provides further support for the involvement of this pathogen in the pathogenesis of IBD (Atarashi et al., 2017).
Gut microbes and their polymicrobial invasive biofilms strongly correlated with the incidence of colorectal cancer (CRC) (Dejea et al., 2014). Fusobacterium, a tumorigenic microbe, is enriched in CRC tumor tissue. The fluorescent in situ hybridization (FISH) staining of proximal CRC clinical samples showed that Fusobacterium-containing polymicrobial invasive biofilms existed directly on tumor tissue. Furthermore, clinical samples of tissues containing biofilms had lower levels of E-cadherin and higher expression levels of the inflammatory factor IL-6 and its activator Stat3 than those without biofilms (Dejea et al., 2014). The same group also discovered that biofilm communities on the colon epithelium prepared from CRC patients and even healthy individuals were carcinogenic in murine CRC models (Tomkovich et al., 2019). Moreover, biofilm-forming bacteria from CRC patients interacted with the host and altered host microRNA expression during the development of CRC in murine CRC models (Tomkovich et al., 2020). One species of Fusobacterium, Fusobacterium nucleatum, is frequently detected in liver metastases of Fusobacterium-associated CRC (Bullman et al., 2017). A treatment with the antibiotic metronidazole reduced the size of tumors in a mouse model of CRC, indicating that F. nucleatum is involved in CRC tumor growth (Bullman et al., 2017). F. nucleatum may also induce TLR4 and MYD88 signaling and activate autophagy in CRC, resulting in chemoresistance (Yu et al., 2017). We recently reported that F. nucleatum elevated the present form of carcinoma to a more advanced stage of CRC, while other bacteria in the early stage of CRC, namely, Atopobium parvulum and Actinomyces odontolyticus, co-occurred in intramucosal carcinomas (Yachida et al., 2019). ETBF and polyketide synthase (pks) genotoxic island-containing (pks+) E. coli have also been identified as tumorigenic bacteria (Dejea et al., 2018). The pks genotoxic island encodes the biosynthesis genes of a microbial carcinogen called colibactin, which alkylates DNA and forms colibactin-DNA adducts, resulting in DNA damage and genotoxicity (Wilson et al., 2019). pks+ E. coli and ETBF co-colonize the colons of patients with familial adenomatous polyposis, which is caused by a hereditary mutation (Dejea et al., 2018). A FISH analysis of clinical samples showed that pks+ E. coli and ETBF formed invasive biofilms on the mucosal tissues of FAP patients, and ETBF enhanced pks+ E. coli colonization in the colons of mice (Dejea et al., 2018). Co-colonization in mice increased DNA damage in colonic epithelial cells, colonic tumor formation, and mortality. This tumorigenic effect was attributed to IL-17-induced inflammation (Dejea et al., 2018). Taking these findings from CRC and IBD research into consideration, further studies on the comprehensive spatial information and microbiome compositions of polymicrobial biofilms are important for advancing the development of gut microbiota-targeted treatments.
Gut microbiota and atopic dermatitis/immunology
Atopic dermatitis (AD) is a skin disease caused by chronic inflammation and is characterized by severe itching, redness, and eczematous skin lesions (Novak et al., 2003). AD is initiated by the activation of T helper 2 (Th2) cells and suppression of Th1 (Grewe et al., 1998). Staphylococcus aureus interacts with various cells of the cutaneous immune system to penetrate the epidermis and dermis and perpetuate chronic inflammation (Leyden et al., 1974; Breuer et al., 2002). Alterations in the gut microbiota influence the immune system balance via metabolite production, which may cause an inflamed microenvironment in the specific microbiome of the gut (Zeng et al., 2017; Lee et al., 2018). AD was previously shown to be induced when enteric bacteria from AD mice were transplanted into healthy sterile mice (Zachariassen et al., 2017). Faecalibacterium, Oscillospira, Bacteroides, Parabacteroides, and Sutterella all have potential as gut microbe biomarkers for AD (Koga et al., 2016; Reddel et al., 2019). The abundance of Clostridium difficile, E. coli, and S. aureus was found to be higher in the gut microbiota of AD patients than in that of healthy individuals, whereas the amounts of Bifidobacterium and Bacteroides were lower (Kirjavainen et al., 2002; Penders et al., 2006; Abrahamsson et al., 2012; Nylund et al., 2015; Lee et al., 2016). In addition, the SCFA-producing gut microbiota was confirmed to be present in a larger proportion of AD patients than healthy individuals (Song et al., 2016).
In research on allergies in children who consume yogurt, yogurt consumption correlated with the prevention of AD (Shoda et al., 2017). Moreover, Faecalibacterium prausnitzii attenuated the symptoms of AD (Song et al., 2016). Similar findings were observed when Lactobacillus sakei WIKIM30 was orally administered to BALB/c mice with induced AD (Kwon et al., 2018). WIKIM30 regulated Th2 and ameliorated AD by increasing the relative abundance of the gut microbiota responsible for the generation of Treg. Lactococcus chungangensis CAU 28, which is in cream cheese, was found to attenuate AD symptoms because it contributed to the suppression of Treg and Th2 immune responses by adjusting SCFAs and the gut microbiota and reducing the number of eosinophils and mast cells as well as immunoglobulin E levels (Kim et al., 2019). In the feces of 11 healthy children and 28 infants who developed AD, the severity of AD inversely correlated with gut microbiota diversity and butyric acid-producing bacteria (Nylund et al., 2015).
A recent study suggested a relationship between AD and T helper 17 (Th17) cells (Sugaya, 2020). IL-17 and IL-22 are cytokines that are secreted by Th17 and serve as its marker (Ivanov et al., 2009). A previous study reported that the number of IL-22-producing Th17 cells was significantly increased in the skin of AD patients (Nograles et al., 2009). In another study, the number of IL-17-producing T cells was found to be increased in the peripheral blood and acute lesional skin of AD patients (Koga et al., 2008). In addition, Treg were shown to attenuate inflammation in AD mice (Kalekar and Rosenblum, 2019). Although there is currently no information on gut microbial biofilms in the field of AD, the adherence of bacteria and mucosal biofilms may be important for the immunology of AD. For example, segmented filamentous bacterium (SFB) and enterohemorrhagic E. coli (EHEC), which induce the differentiation of Th17, and Clostridia, which induce the differentiation of Treg, are intestinal epithelium-associated or -adhering bacteria (Ivanov et al., 2009; Atarashi et al., 2013; Furusawa et al., 2013; Atarashi et al., 2015). Therefore, researchers need to focus more on the connections between mucosal biofilms and immunology.
Gut microbiota and metabolic disorders
The prevalence of overweight and obese individuals is a growing epidemic health concern and affected an estimated 1.3 billion individuals worldwide in 2016 (NCD Risk Factor Collaboration, 2017). The progression of certain metabolic disorders, including T2D and atherosclerotic cardiovascular disease (ACVD), is linked to being overweight or obese. Obesity and T2D are both associated with a gut microbiota with an altered composition and function (Qin et al., 2012; Harsch and Konturek, 2018). Although many studies have attempted to elucidate the roles of the gut microbiota in obesity, the precise mechanisms remain unclear because of the complexity of the relationship between the host and microbiota. Diet is one of the environmental factors contributing to obesity, and has been extensively examined in connection with the gut microbiota (Carmody et al., 2015). For example, a Western diet, characterized by high fat and low dietary fiber, has been shown to influence the composition of the gut microbiota and reduce its diversity (He et al., 2018). The findings of a recent study on pre-obese children suggested that the composition of the gut microbiota in conjunction with long-term dietary habits may be helpful for predicting the development of obesity in children (Rampelli et al., 2018). These findings also indicated that an impaired gut microbiota causes metabolic dysfunction and ultimately obesity in the host (Rampelli et al., 2018). The composition of the gut microbiota of obese animals and humans differs from that of lean subjects (Hartstra et al., 2015; Castaner et al., 2018). The ratio of the phyla Firmicutes and Bacteroidetes was increased in obese subjects and considered to be associated with higher energy absorption from food and elevated low-grade inflammation (Turnbaugh et al., 2006). Additionally, metabolites of the gut microbiota, including SCFAs, LPS, and secondary bile acids, play a critical role in the modulation of metabolism and obesity (Baothman et al., 2016). Increased levels of Enterobacteriaceae and Streptococcus spp. have been identified in the stools of ACVD patients by a metagenome-based study (Jie et al., 2017). Furthermore, the gut bacterial metabolite TMAO produced from choline, l-carnitine, and phosphatidylcholine promoted the progression of ACVD (Wang et al., 2011; Koeth et al., 2013; Tang et al., 2013).
By using culture-based approaches developed in the past decade to examine the gut microbiota (Sommer, 2015), it is possible to investigate the relationship between specific bacterial species and various conditions, including obesity. Endotoxin-producing Enterobacter cloacae has been shown to promote obesity and insulin resistance in germ-free mice (Fei and Zhao, 2013). Similarly, Clostridium ramosum, promoted diet-induced obesity, possibly by enhancing nutrient absorption (Woting et al., 2014). In contrast, some bacterial species exert anti-obesogenic effects (Table 3). For example, supplementation with A. muciniphila ameliorated the symptoms of metabolic syndrome induced by a high-fat diet, including fat mass gain, chronic tissue inflammation, metabolic endotoxemia, and insulin resistance, and improved the inflammatory state and gut barrier (Everard et al., 2013). Due to its critical role in the maintenance of metabolic homeostasis, A. muciniphila has been proposed as a next-generation probiotic to combat obesity, diabetes, and cardiometabolic disorders (Cani and de Vos, 2017). Prevotella copri performs carbohydrate fermentation from complex polysaccharides in the diet and contributes to better glucose tolerance; therefore, it is regarded as a beneficial bacterium in high-fiber dietary interventions (Kovatcheva-Datchary et al., 2015; De Vadder et al., 2016). Two species of the genus Parabacteroides, Parabacteroides goldsteinii and Parabacteroides distasonis, were found to have the potential to correct obesity-associated abnormalities in mice fed a high-fat diet (Wang et al., 2019a; Wu et al., 2019). These findings on the roles of specific bacterial species in the gut provide important insights for understanding intricate gut microbiota-host interactions. The identification of these pro- and anti-obesogenic bacteria and a more detailed understanding of the mechanisms underlying this disease will enable us to explore effective approaches for the treatment of metabolic disorders, including obesity. The link between gut microbial biofilms and metabolic disorders currently remains unclear. However, gut microbial biofilms may play a key role because their composition and structure were previously shown to be changed by different diets (Earle et al., 2015).
Table 3.
Roles of specific bacterial species with anti-obesogenic effects
| Bacterium |
Subject |
Treatment period |
Outcomes |
Reference |
| Akkermansia muciniphila |
Mice |
4 weeks |
Alleviates high-fat diet-induced metabolic symptoms, including endotoxemia, fat mass gain, adipose tissue inflammation, and insulin resistance; improves inflammation, gut barrier function, and gut peptide secretion |
Everard et al., 2013 |
| Prevotella copri |
Mice |
2 weeks |
Performs carbohydrate fermentation from complex polysaccharides in the diet and contributes to better glucose tolerance |
Kovatcheva-Datchary et al., 2015 |
| Bifidobacterium breve B-3 |
Mice |
8 weeks |
Reduces the accumulation of body weight and epididymal fat and alleviates serum levels of fasting glucose, cholesterol, and insulin |
Kondo et al., 2010 |
| Bifidobacterium breve B-3 |
Pre-obesity adults |
12 weeks |
Reduces fat mass and alleviates parameters associated with liver functions and inflammation, including γ-glutamyltranspeptidase and high-sensitivity C-reactive protein |
Minami et al., 2015 |
| Bifidobacterium pseudocatenulatum |
Mice |
8 weeks |
Reduces body weight and fat mass, fasting glucose, and insulin resistance |
Zhao et al., 2018 |
| Lactobacillus gasseri |
Healthy adults with large visceral fat areas |
12 weeks |
Reduces abdominal visceral fat areas, the body mass index, hip and waist circumferences, and body fat |
Kadooka et al., 2013 |
| Bacteroides uniformis |
Mice |
7 weeks |
Suppresses body weight gain; improves liver function by reducing liver steatosis as well as liver cholesterol and triglycerides; reduces dietary fat absorption and reverses immune dysfunction |
Gauffin Cano et al., 2012 |
| Bacteroides acidifaciens |
Mice |
10 weeks |
Suppresses body weight and fat mass; ameliorates insulin resistance; increases serum GLP-1 and decreases gut dipeptidyl peptidase-4 |
Yang et al., 2017b |
| Bacteroides thetaiotaomicron |
Mice |
7 weeks |
In chow diet-fed mice, reduces fat mass and increases lean body mass; in high-fat diet-fed mice, suppresses body weight gain and adiposity, increases serum adiponectin and decreases leptin, up-regulates the expression of genes for fatty acid oxidation and lipolysis, and improves the inflammatory status |
Liu et al., 2017 |
| Eubacterium hallii |
db/db mice |
5 weeks |
Decreases insulin sensitivity and increases energy expenditure and fecal butyrate concentrations |
Udayappan et al., 2016 |
| Parabacteroides goldsteinii |
Mice |
8 weeks |
Ameliorates obesity and increases adipose tissue thermogenesis; enhances gut integrity; lowers the inflammatory status; increases insulin sensitivity |
Wu et al., 2019 |
| Parabacteroides distasonis |
Mice |
5 weeks |
Reduces weight gain, hyperglycemia, and hepatic steatosis; alters the bile acid profile; increases gut gluconeogenesis and insulin sensitivity |
Wang et al., 2019a |
The gut-brain axis is a system through which the gut microbiota affects brain development, neural activities, and brain disease (Mayer et al., 2014). In recent years, the effects of this relatively unknown system on physical and neurological conditions have been attracting increasing attention.
Many diseases and phenomena have been associated with the gut-brain axis (Schroeder and Bäckhed, 2016; Ye et al., 2018). One example is anxiety-like behavior. Recent studies reported that antibiotics alter the composition of the gut microbiota, which subsequently results in long-term changes in behavior (Burokas et al., 2017; De Palma et al., 2017). Another study showed the attenuation of anxiety-like behavior in the male offspring of pregnant mice exposed to a low dose of penicillin (Leclercq et al., 2017). During defeat sessions in which control group mice and penicillin-treated mice were physically subjected to stress by unfamiliar male aggressors, the penicillin-treated group fought back and had fewer scars than control mice, indicating that a deficiency in gut microbes was associated with a reduced fear response (Leclercq et al., 2017). Similarly, an imbalance in the hypothalamic-pituitary-adrenal axis caused by gut microbiome alterations may affect the neuroendocrine system, provoking anxiety-like behavior. After the application of a chronic restraint stress to germ-free and specific pathogen-free mice, specific pathogen-free mice exhibited more anxiety-like behavior than germ-free mice (Huo et al., 2017). In addition, microbiota-derived metabolites were found to contribute to the mental health of the host and relieved psychosocial stress (van de Wouw et al., 2018).
Autism spectrum disorder (ASD) is a severe neurodevelopmental disorder involving impaired social communication and interactions as well as repetitive behavior patterns. The gut microbiota has been suggested to play a critical role in the pathogenesis of ASD. The gut microbiota profiles of ASD patients markedly differ from those of typically developing controls (Kang et al., 2013; Tomova et al., 2015; Liu et al., 2019). Bifidobacterium, Blautia, Dialister, Prevotella, Veillonella, and Turicibacter levels were consistently lower in ASD patients than in healthy controls, whereas Lactobacillus, Bacteroides, Desulfovibrio, and Clostridium levels were higher (Yang et al., 2018; Liu et al., 2019). Similarly, an altered gut microbiota composition was observed in ASD mice (Hsiao et al., 2013; Buffington et al., 2016; Kim et al., 2017a). An altered gut microbiota composition resulted in the production of different metabolites, which may influence the progression of ASD (Wang et al., 2019b). Children with ASD had significantly higher levels of Actinobacteria, but significantly lower species richness than those exhibiting typical development (Wang et al., 2019b). Supplementation with probiotics and fecal microbiota transplantation alleviated the symptoms of ASD in children (Steenbergen et al., 2015; Kang et al., 2017). Interestingly, specific bacterial species, such as B. fragilis and Lactobacillus reuteri, have therapeutic potential in animal models of ASD (Hsiao et al., 2013; Buffington et al., 2016).
The effects of gut microbial communities on physical and neurological conditions have also been examined in Alzheimer’s disease (AZD). Accounting for ~70% of all dementia cases, AZD is the most common form of dementia (Ko et al., 2019). The diversity of the gut microbiota was previously reported to be lower in AZD patients than in age- and sex-matched control individuals (Vogt et al., 2017). Bile acids also play an important role in the development of AZD. A recent study compared the bile acid profiles of healthy adults, adults with early mild cognitive impairment, adults with late mild cognitive impairment, and AZD patients, and found that primary bile acid levels were lower and microbiota-produced secondary bile acid levels were higher in subjects with cognitive impairment (MahmoudianDehkordi et al., 2019). Therefore, specific changes in the gut microbial community were suggested to alter the levels of secondary bile acids, conceivably contributing to the pathogenesis of AZD (MahmoudianDehkordi et al., 2019).
Similar gut microbiota alterations have been observed in Parkinson’s disease (PD) patients (Scheperjans et al., 2015; Hill-Burns et al., 2017). The symptoms of PD, a progressive nervous system disorder, include motor deficits, such as tremors, rigid muscles, and bradykinesia (Nalls et al., 2014). In PD patients, gut bacteria from the family Prevotellaceae were 77.6% lower than in healthy individuals (Scheperjans et al., 2015). Another study transplanted the gut microbiota from PD patients to germ-free mice, and detected physical impairments due to neurodegeneration in recipient mice (Sampson et al., 2016). Collectively, these findings indicate that gut microbes contribute to the progression of PD.
Gut-innervating nociceptor neurons have been shown to inhibit Salmonella infection through the modulation of Peyer’s Patch microfold cells and gut adherence bacterium SFB levels (Lai et al., 2020). Although the relationships between bacterial adherence or biofilm formation and the brain currently remain unclear, some relationships appear to exist between the adherence of gut microbes and the neurological system.
Conclusions
Various omics technologies enable us to obtain extensive information in a high-throughput manner in gut microbe research. The development of the integrated omics analysis method, metabologenomics, has facilitated research on complex gut microbiota communities and gut microbiota-host interaction networks (Ishii et al., 2018). Large cohort studies and high-throughput omics techniques may be commonly used in this area of study. However, difficulties may be associated with investigating host-microbiota interactions by solely relying on an omics analysis. As shown in studies on IBD and CRC, spatial information on the intestinal mucosal surface is important for examining host-microbiota interactions. Therefore, gut microbiota research may need to shift its focus from the compositional traits and variations of gut microbiota and metabolites to integrating them into the spatial and temporal dynamics of gut microbial biofilms. Therefore, it is important to incorporate spatio-temporal characterizations using microscopic imaging and molecular biology analyses because they may contribute to clarifying the detailed mechanisms underlying gut microbiota-host interactions and host immune defenses on the intestinal mucosal barrier and also reveal new targets for drug/treatment development (Fig. 3) (Swidsinski et al., 2005; Dejea et al., 2014; Atarashi et al., 2017; Bullman et al., 2017; Yu et al., 2017; Dejea et al., 2018; Wilson et al., 2019). Novel synergy among these approaches will revolutionize the field of gut microbiology, thereby allowing us to make more contributions toward maintaining and promoting optimal host health for a higher quality of life.

