Abstract
The accessory nidamental gland (ANG) is part of the reproduction organ in the majority of female cephalopods, including the bigfin reef squid Sepioteuthis lessoniana, an economically important fishery product. Microbes in Alphaproteobacteria, Gammaproteobacteria, and Verrucomicrobia have been suggested to play a role in the maturation of the S. lessoniana ANG and are responsible for its color. However, the bacterial composition and dynamics of the different maturation stages of the ANG remain unclear. In the present study, we surveyed ANG-associated bacterial dynamics in wild-caught S. lessoniana at various developmental stages in different populations over 3 years. The results obtained showed that the ANG bacterial community shifted gradually and decreased in diversity throughout maturation. Verrucomicrobia occupied the ANG during the early stages in large numbers, and was replaced by Bacteroidia, Alphaproteobacteria, and Gammaproteobacteria in the later stages. Flavobacteriales and Alphaproteobacteria both appeared to contribute to pigmentation, while Bacteroidia, Alphaproteobacteria, and Gammaproteobacteria may be involved in enriching the heme biosynthesis pathway in the ANG with the maturation of S. lessoniana. The present results provide an open question of whether S. lessoniana actively selects the bacterial community in the ANG to adjust to its surrounding environment.
The bigfin reef squid Sepioteuthis lessoniana is a widespread loliginid squid species in the Indo-Pacific Ocean region, and represents one of the most economically important fishery products in Taiwan. This species is closely associated with coral reef habitats, and lays jelly-like egg strings in seagrasses, sea fans, coral rubble, and reefs (Norman, 2003). Between April and October in Taiwan, S. lessoniana swims in groups to the shore for spawning. The population structure of S. lessoniana in Taiwan has been investigated using life history parameters, statolith geochemical signatures, and genetic analyses (Ching et al., 2019; Chiang et al., 2020), and one predominant taxon was found in the water of northeastern Taiwan.
S. lessoniana trading still mainly relies on capture fishing; however, culturing techniques have also been established due to the high demand for this species. Aquaculture breeding techniques for this species need to be improved in order to reach market volumes. Current bottlenecks around the aquaculture production of cephalopods may be due to a lack of detailed information on the processes of maturation, reproduction, and hatching in natural environments and under cultivation conditions (reviewed in O’Brien et al., 2018), with limited information being available on microbial communities.
The accessory nidamental gland (ANG) is part of the reproduction organ that is present in the majority of female cephalopods. Alphaproteobacteria, Gammaproteobacteria, and Verrucomicrobia are common microbial taxa that associate with different species of cephalopod ANG (Pichon et al., 2005; Collins et al., 2012; Kerwin and Nyholm, 2017). Since juvenile cephalopods do not have a developed ANG, the microbial community in the ANG is considered to undergo horizontal transfer from the environment (Kaufman et al., 1998). The microbial community composition may be related to ANG maturity. During maturation, the ANG turns from white to an orange color that originates from the carotenoids produced by the microbiome (Lum-Kong and Hastings, 1992). One of the functions of the ANG is to provide bacteria in the egg jelly coat (JC) as a defense mechanism against microorganisms, such as those in the ANG of the Hawaiian bobtail squid, Euprymna scolopes (Collins et al., 2012; Kerwin and Nyholm, 2017).
Female S. lessoniana also have an ANG; however, information on the microbial community associated with this ANG is limited. Li et al. (2019) hypothesized that as bacteria move from marine environments into the ANG, the transferrin (TF)-like protein of the host may play an essential role in microbial selection. During the maturation process, the ANG gradually transfers bacteria from the outer epithelial cell layer, invaginating bacteria into the ANG and forming tubule structures for bacterial colonies (Li et al., 2019). The TF-like protein, which is rich in the epithelial cell layer and the early stages of the ANG (Li et al., 2019), may be the key for initial bacterial selection. However, the microbial composition in different ANG maturation stages and the dynamic composition throughout the maturation process remain unknown. Therefore, the present study surveyed ANG-associated bacterial dynamics in S. lessoniana at various developmental stages in different populations over 3 years. This is the first survey of the ANG of wild-caught populations. Our three-year field sampling of different stages of ANG maturation tracked resident bacterial communities using NGS amplicon sequencing to provide more detailed insights into the core organ-specific microbiome in certain ANG development stages.
Materials and Methods
Squid collection
Bigfin reef squids were purchased from a fisherman on Hoping Island, Keelung City, Taiwan. Squids were collected at night by hand jigging on a boat in the northeast coast of Taiwan (Fig. S1), and were maintained using a fresh seawater system on the boat. They were transferred to a seawater tank (2.5 tons FRP tank) with a circulating seawater system the next morning until tissue sampling (less than 2 hrs). The ANG of squids were collected after 6–12 h of fishing on the boat, which ensured that additional bacteria were not acquired in captivity. Squids were anesthetized in seawater containing 5% ethanol at room temperature and then euthanized by cutting off the head. All procedures and investigations were approved by the National Taiwan Ocean University Institutional Animal Care and Use Committee and were performed in accordance with standard guidelines.
To elucidate the relationship between the microbiota of the ANG and the female reproductive cycle, different ANG at various stages were collected for histological and amplicon analyses. Based on the techniques used for squid collection (hand jigging on a boat), it is difficult to collect seawater samples in natural habitats. Three batches of female squids were used in the present study between 2015 and 2018: autumn 2015 to spring 2016 (batch 1), autumn 2016 to spring 2017 (batch 2), and autumn 2017 to spring 2018 (batch 3) (Table 1). The ANG stage was assessed based on morphological characteristics in accordance with our previous study (Li et al., 2019). Four developmental stages of the ANG were recognized in the present study (Fig. 1): a colorless ANG without any bacterial aggregates in juvenile squids (stage 1), a colorless ANG with bacterial colonies in immature squids (stage 2), a white/light-orange ANG with bacterial colonies in maturing squids (stage 3), and a pigmented ANG with a large number of bacterial colonies in mature squids (stage 4).
Table 1.
Characteristics of sampled squids
| Batch |
Stage |
Sampling date |
ML (cm) |
BW (g) |
GW (g) |
GSI (%) |
Gonadal stage |
Reproductive phase |
| 1 |
2 |
Nov, 2015 |
15.5 |
216.5 |
0.4 |
0.2 |
PO |
Immature female |
| 1 |
2 |
Nov, 2015 |
17.0 |
195.7 |
0.5 |
0.2 |
PO |
Immature female |
| 1 |
2 |
Dec, 2015 |
23.5 |
774.5 |
0.7 |
0.1 |
PO |
Immature female |
| 1 |
3 |
Nov, 2015 |
26.0 |
938.3 |
1.3 |
0.1 |
PVO |
Maturing female |
| 1 |
3 |
Feb, 2016 |
22.0 |
611.8 |
1.0 |
0.2 |
PVO |
Maturing female |
| 1 |
3 |
Dec, 2015 |
25.0 |
818.3 |
2.7 |
0.3 |
PVO |
Maturing female |
| 1 |
4 |
Dec, 2015 |
26.0 |
986.8 |
>10 |
— |
MO |
Mature female |
| 1 |
4 |
Feb, 2016 |
26.0 |
883.6 |
>10 |
— |
MO |
Mature female |
| 1 |
4 |
Oct, 2015 |
21.5 |
522.2 |
>10 |
— |
MO |
Mature female |
| 2 |
1 |
Sep, 2016 |
15.5 |
196.0 |
<0.1 |
— |
PO |
Juvenile female |
| 2 |
1 |
Sep, 2016 |
14.5 |
211.4 |
<0.1 |
— |
PO |
Juvenile female |
| 2 |
2 |
Oct, 2016 |
18.0 |
308.3 |
0.3 |
— |
PO |
Immature female |
| 2 |
2 |
Oct, 2016 |
17.5 |
310.7 |
<0.1 |
— |
PO |
Immature female |
| 2 |
2 |
Nov, 2016 |
26.0 |
912.0 |
0.6 |
0.1 |
PO |
Immature female |
| 2 |
3 |
Nov, 2016 |
23.0 |
717.0 |
0.8 |
0.1 |
PVO |
Maturing female |
| 2 |
3 |
Dec, 2016 |
n.d. |
n.d. |
n.d. |
— |
PVO |
Maturing female |
| 2 |
3 |
Dec, 2016 |
21.0 |
542.0 |
n.d. |
— |
PVO |
Maturing female |
| 2 |
3 |
Dec, 2016 |
n.d. |
n.d. |
n.d. |
— |
PVO |
Maturing female |
| 2 |
3 |
Feb, 2017 |
24.0 |
735.5 |
0.5 |
0.1 |
PVO |
Maturing female |
| 2 |
3 |
Feb, 2017 |
27.0 |
1120.0 |
0.7 |
0.1 |
PVO |
Maturing female |
| 2 |
4 |
Feb, 2017 |
25.5 |
991.0 |
>10 |
— |
MO |
Mature female |
| 2 |
4 |
Mar, 2017 |
25.0 |
744.0 |
9.0 |
1.2 |
VO |
Mature female |
| 2 |
4 |
Mar, 2017 |
33.0 |
1787.0 |
>10 |
— |
MO |
Mature female |
| 2 |
4 |
Mar, 2017 |
32.0 |
1650.0 |
>10 |
— |
MO |
Mature female |
| 3 |
2 |
Nov, 2017 |
22.0 |
683.6 |
0.8 |
0.1 |
PO |
Immature female |
| 3 |
2 |
Dec, 2017 |
29.0 |
1321.8 |
1.9 |
0.1 |
PO |
Immature female |
| 3 |
2 |
Jan, 2018 |
23.5 |
680.8 |
1.4 |
0.2 |
PO |
Immature female |
| 3 |
2 |
Jan, 2018 |
27.5 |
977.7 |
1.7 |
0.2 |
PO |
Immature female |
| 3 |
3 |
Jan, 2018 |
28.7 |
1098.2 |
1.7 |
0.2 |
PVO |
Maturing female |
| 3 |
3 |
Jan, 2018 |
26.4 |
859.3 |
1.5 |
0.2 |
PVO |
Maturing female |
| 3 |
3 |
Jan, 2018 |
28.7 |
1252.9 |
1.7 |
0.1 |
PVO |
Maturing female |
| 3 |
3 |
Mar, 2018 |
24.7 |
695.1 |
1.3 |
0.2 |
PVO |
Maturing female |
| 3 |
4 |
Nov, 2017 |
29.0 |
1214.3 |
>10 |
— |
MO |
Mature female |
| 3 |
4 |
Dec, 2017 |
31.0 |
1265.7 |
4.9 |
0.4 |
VO |
Mature female |
| 3 |
4 |
Jan, 2018 |
23.5 |
891.5 |
>10 |
— |
MO |
Mature female |
| 3 |
4 |
Mar, 2018 |
26.5 |
1039.6 |
>10 |
— |
MO |
Mature female |
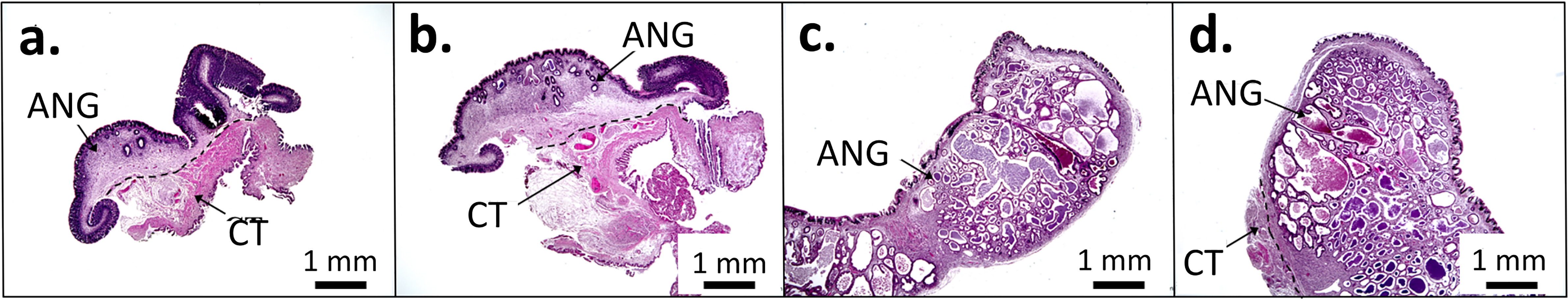
More than three potential bigfin reef squid taxa have been observed in its distribution area: Sepioteuthis sp. 1, sp. 2, sp. 2A, and sp. 3 (Cheng et al., 2014; Ching et al., 2021). The dominant taxon around northeastern Taiwan is Sepioteuthis sp. 1 (Cheng et al., 2014; Ching et al., 2021). To avoid differences in ANG bacterial communities due to lineage-specific selection, we used only one lineage—Sepioteuthis sp. 1—as the target for the present study. Total DNA was extracted using the QIAamp DNA Mini Kit (Qiagen) from the frozen muscle tissue of each bigfin reef squid, based on the manufacturer’s protocol. Multiplex cytochrome oxidase c subunit I (COI) haplotype-specific PCR (MHS-PCR) was performed to identify the potential lineages of squids using the following primers: AF-5′-TCTCATGCTGGACCTTCAGTA-3′; AR-5′-TGCTCCTGCTAAAACAGGAAG-3′; BF-5′-ATTGGGGGTTTTGGTAACTGG-3′; BR-5′-GATGCTAAAAGGAGTGTGAGG-3′; C1F-5′-TTAGTTGGTACCTCACTAAGG-3′; C1R-5′-CTCTTTCAACTGCTGAGGAC-3′; C2F-5′-TTAGTTGGTACCTCACTAAGG-3′; C2R-5′-GTTGATATAGAATAGGGTCTCCC-3′. These haplotype-specific primers were designed based on bigfin reef squid samples collected around Taiwan (Hsiao et al., 2016; Shen et al., 2016). PCR was performed using a PCR Thermal Cycler (Gene Atlas), each with 9.8 μL of reaction solution containing 0.2 ng template DNA, 0.3 μM of each primer, 0.2 mM dNTP mixture, 10× PCR buffer (20 mM Tris–HCl pH 8.0, 15 mM KCl, and 15 mM MgCl2), and 5 U μL–1 DNA polymerase (Thermo Scientific). Reaction conditions were as follows: initial denaturation at 94°C for 5 min; 35 cycles of denaturation at 94°C for 45 s, annealing at 60°C for 1 min, and extension at 72°C for 1 min; and a final extension at 72°C for 10 min. Amplified products were confirmed using 1.5% agarose gel electrophoresis, and we observed a band corresponding to one of the lineages. The bands for Sepioteuthis sp. 1, sp. 2, sp. 2A, and sp. 3 were 207, 122, 281, and 680 bp, respectively.
Total DNA extraction, PCR, barcoding PCR, and sequencing
The ANG stages of all squids were assessed based on morphological characteristics (listed in Table 1). The microbiota of 36 female squids at different developmental stages were analyzed. ANG were homogenized in Trizol reagent (Invitrogen). This nucleoprotein complex was mixed with chloroform to separate RNA, DNA, and proteins in the upper aqueous phase, lower phenol-chloroform phase, and interphase, respectively. Total DNA was precipitated from the phenol-chloroform phase using 100% ethanol. Isolated total DNA was used for an amplicon analysis. The equipment used was sterilized and free of RNase to prevent contamination during the experiment.
PCR was performed using two bacterial universal primers—361F (5′-CCTACGGGNGGCWGCA-3′) and 806R (5′-GACTACHVGGGTATCTAATCC-3′)—which were designed for the bacterial V3V4 high variable region of the 16S ribosomal RNA gene (Nossa et al., 2010). Thirty cycles of PCR were performed as follows: an initial step of 94°C for 3 min, 94°C for 15 s, 55°C for 15 s, and 68°C for 1 min, with 2 min at 72°C as the final extension after the last cycle. PCR amplicons were checked by DNA agarose gel electrophoresis with a 1.5% agarose gel and 0.5× TAE buffer. PCR-amplified 350–450-bp fragments were purified using the QIAquick Gel Extraction Kit (Qiagen). The quality of DNA was assessed using a NanoDropTM 1000 spectrophotometer (Thermo Scientific). DNA library construction and Illumina sequencing were performed by Genomics. TruSeq DNA Sample Preparation Kits (Illumina) were used for library construction, and prepared libraries were quantified using a GeneRead Library Quant Kit (Qiagen) before loading them into the NGS sequencer, MiSeq (Illumina). Five hundred nanograms of each purified PCR product were subjected to Illumina-based high-throughput sequencing.
Bioinformatic and statistical analyses
An amplicon analysis was performed as described by Tandon et al. (2018). Amplicons were quality checked (average phred score ≥27) and poor quality reads were trimmed. The trimming of merged sequences was conducted by mothur software with a minimum length=440 bp and maximum length=466 bp (Schloss et al., 2009). After trimming, chimeric reads were detected by UCHIME (http://drive5.com/uchime) (Edgar et al., 2011). In the operational taxonomic unit (OTU) analysis, quality-filtered reads were pooled together and analyzed with the UPARSE pipeline (Edgar, 2013) without the chimera removal step. In UPARSE, de-replication was performed with the options “–derepfulllength” and “–minsize 2” and OTUs were generated at 97% identity. Each OTU was searched (with global alignment) against the Silva132 database to find the corresponding taxonomy of the best hit using USEARCH (Edgar, 2013). Alignment was calculated using MOTHUR software to define OTUs with a 20% cut-off value for sequence dissimilarity. Defined OTUs were used to estimate the Shannon-Weaver diversity index, Chao1 estimator, and the Simpson index as well as to construct rarefaction curves and calculate the evenness and richness of the bacterial community. Unclassified OTUs were manually searched in NCBI using BLASTn. The relative abundance of each classified bacterial class in individual samples was incorporated into a matrix. A total of 2,310,443 qualified reads were obtained from the 36 samples and 594 OTUs were generated. After the primer was removed, sequencing reads were deposited into the NCBI Sequence Read Archive database under accession number PRJNA666041.
Biodiversity and statistical analyses
Results were presented in hierarchical clustering (CLUSTER) and non-Metric multi-Dimensional Scaling (nMDS) using R (PRIMER-E) to analyze the relationships between bacterial communities among samples.
Functional prediction
PICRUst2 (https://github.com/picrust/picrust2) was used to predict bacterial functions based on the bacterial OTUs of the ANG. The total OTU table, 191 core OTUs, Alphaproteobacteria OTUs, Gammaproteobacteria OTUs, and Bacteroidia OTUs were used to predict KEGG Orthology (KO) relative abundance. A metabolic module enrichment analysis was performed with a functional sets enrichment analysis (FSEA) as described by Liu et al. (2020). The ‘FSEA’ function in the MARco R package based on Liu et al. (2020) was applied (https://github.com/poyuliu/MARco). The metabolic module enrichment analysis was conducted with the R package gage (testing using the ‘gage’ function to assess metabolic module enrichment by comparing equal gene abundance distribution within groups and by comparing mean gene abundance among groups; significant modules were identified from one-tailed tests for up-regulation). The P values from multiple tests were adjusted with a false discovery rate (FDR), with a P value-adjusting function embedded in the gage package; the significance of enrichment analyses was defined by FDR q-value <0.05. Enrichment scores were calculated according to the gene-set enrichment analysis (GSEA) algorithm of DAVID bioinformatics resources.
Results
The rarefaction curve showed that all samples plateaued (Fig. S2). The range of species richness differed each year; however, richness always slightly declined from the earlier stage (stages 1 and 2) to the later stage of the ANG. Alpha diversity indicated no significant differences among the three years; therefore, bacterial richness/evenness was similar across the years (Fig. S3). On the other hand, bacterial compositions at the OTU level significantly differed among the years (ANOSIM, R2=0.286, P=0.01) (Fig. S4), although those in 2016 and 2017 were similar. This result implied that environmental conditions or bacterial diversity in seawater influences the basic bacterial constitution of the ANG of S. lessoniana.
In comparisons of diversity indices across sampling stages (Fig. S5), Chao 1 and Simpson showed that stages 1 and 2 had higher richness and evenness than the late stage. PCoA (Fig. 2a) revealed that the bacterial composition of each stage showed a change with a linear pattern along the first axis, whereas the majority overlapped. Stage 1 samples were separated from stage 4 samples, indicating bacterial succession in the ANG during maturation. In addition, the nMDS of the bacterial genus composition showed a similar pattern among stage 1, 2, and 3 individuals, but a markedly different pattern among stage 4 individuals (Fig. 2b).

Regarding bacterial compositions, the S. lessoniana ANG mainly comprised Proteobacteria (59%), Bacteroidetes (25%), Actinobacteria (6.7%), and Firmicutes (5.3%) (Fig. 3a). The phyla in stage 1 markedly differed from those in other stages. In stage 1, Firmicutes was the dominant phylum, while the abundance of Proteobacteria and Bacteroidetes increased in later stages (Fig. 3b). In all stages, the dominant class were Gammaproteobacteria, Alphaproteobacteria, and Bacteroidia, and the abundance of Bacteroidia slightly increased in stages 3 and 4. Some classes decreased throughout the maturation stages: Verrucomicrobiae, Mollicute, Clostridia, Bacilli, Oxyphotobacteria, KD4-96 (Chloroflexi), and Actinobacteria (Fig. 3c).
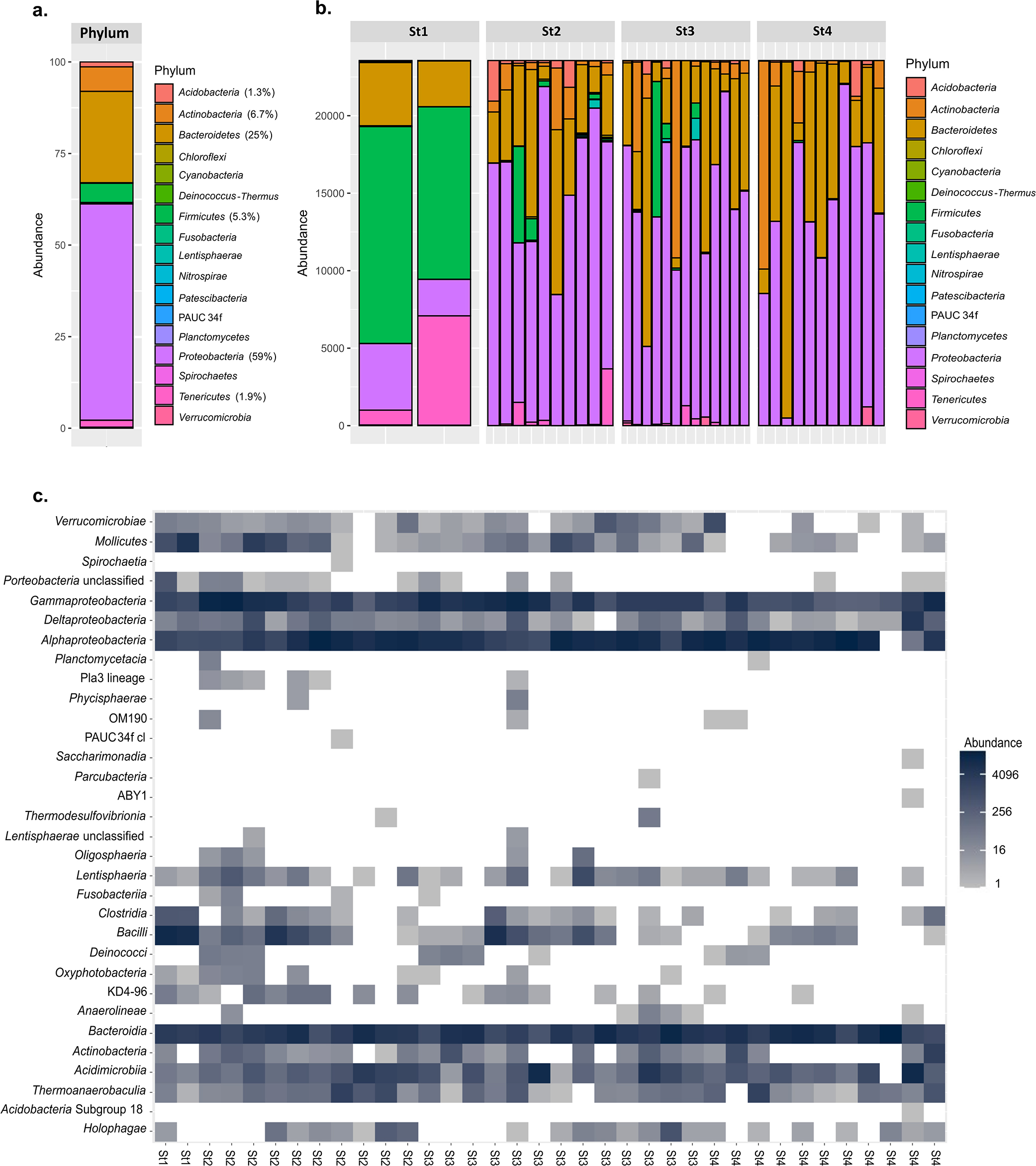
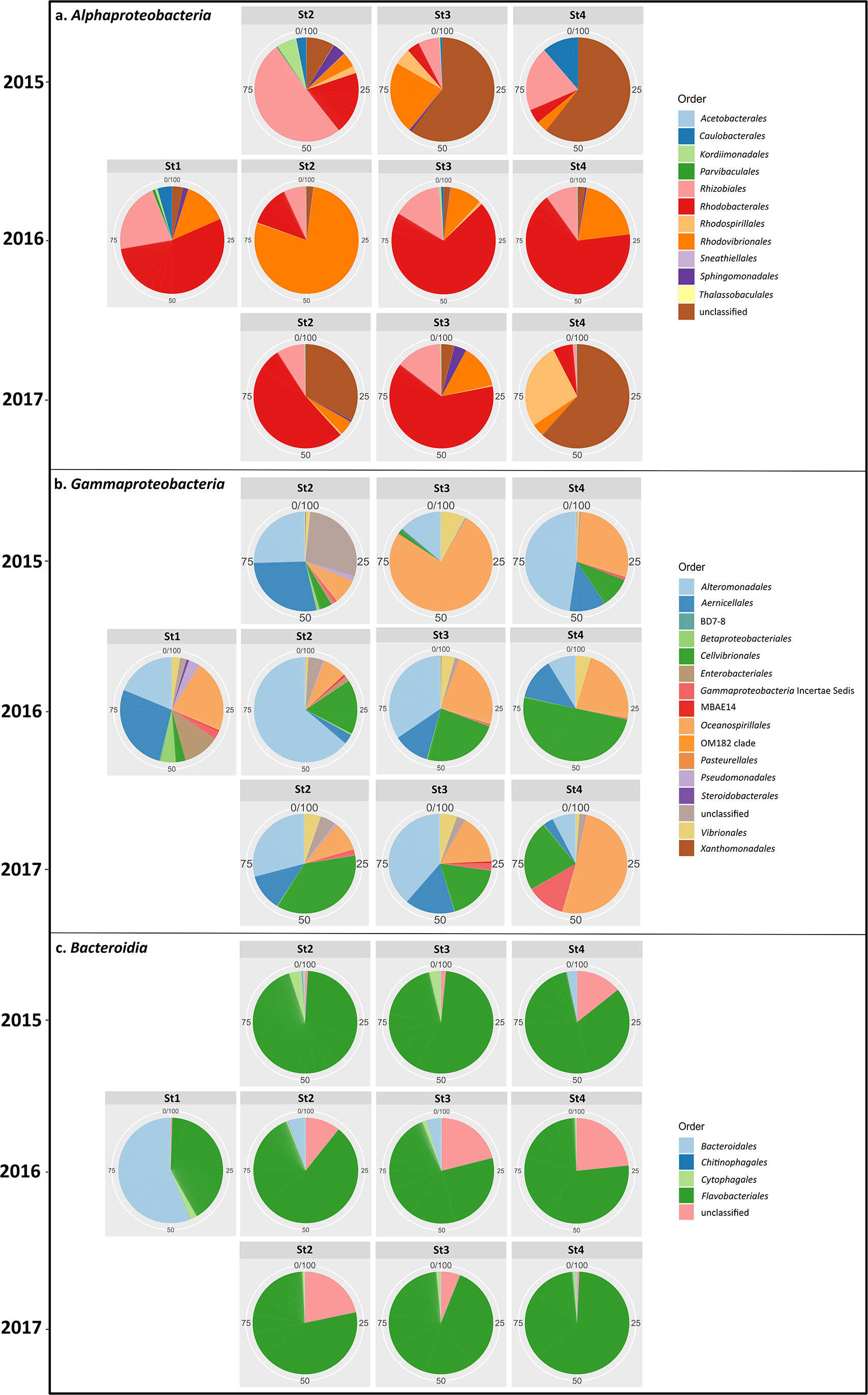
We also investigated the dynamics of the three dominant classes—Gammaproteobacteria, Alphaproteobacteria, and Bacteroidia—across all maturation stages (Fig. 4a, b, and c). In Alphaproteobacteria and Gammaproteobacteria, bacteria were more diverse in earlier stages (stages 1 and 2); however, the order Flavobacteriales in Bacteroidia remained dominant throughout all stages.
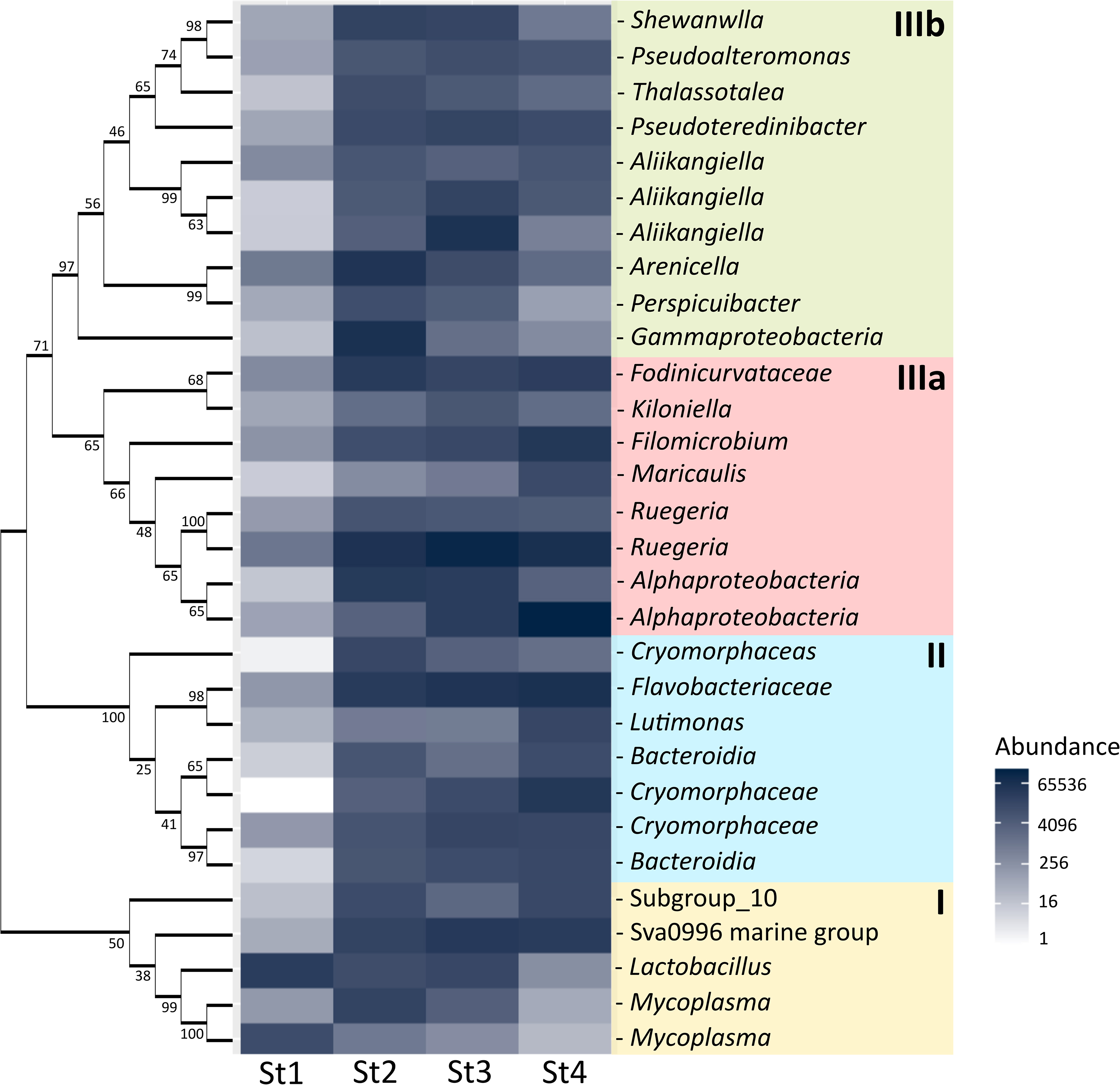
There were 191 OTUs present in all stages, and each stage had multiple unique OTUs (Fig. S6). The earlier stages shared fewer OTUs with the late stages and more OTUs were shared among close stages. Stages 1 and 4 did not share any OTUs (Fig. S6). The core 191 OTUs may be resident bacteria and specific to the ANG. The differences observed in bacterial compositions across the years may be due to the environment (Fig. S4), which may also influence the core 191 OTUs. The abundance of the 191 bacterial OTUs also suggests a significant difference among the years (R2=0.126, P=0.01, Fig. S7). The heatmap plotted the top 30 of the core 191 OTUs at different stages, and clear shifts were observed within the three phylogeny groups (Fig. 5). Stage 1 was dominated by group I (Gram-positive), but not group II or III (Gram-negative). In group I, the abundance of Mycoplasma, Lactobacillus, and two other OTUs gradually decreased throughout the maturation process; however, this may have been natural succession. Group II contained classes of Bacteroidetes; Bacteroidia were abundant in stage 4, and the abundance of Flavobacteriaceae remained stable from stages 2 to 4. The dominant OTU (OTU12) in Flavobacteriales (Fig. S8) was the most closely related to the marine species Pseudofulvibacter geojedonensis (NR126234.1). Group III comprised Alphaproteobacteria (IIIa) and Gammaproteobacteria (IIIb), and their abundance shifted across the maturation stages—group IIIa gradually increased from stages 2 to 4, whereas IIIb showed the reverse.
After the PICRUSt2 functional prediction analysis, genes were categorized into KEGG metabolic modules. We found that NADH:quinone oxidoreductase, the citrate cycle, heme biosynthesis, KDO2-lipid A biosynthesis, and lysine biosynthesis had the highest scores (Fig. 6). The heme biosynthesis function had the highest score; it was low in stage 1 and increased throughout the maturation process. Functional predictions of the 191 core OTUs present in all stages showed that the heme biosynthesis function also had the highest score (Fig. S9). In addition, in the functional prediction of the dominant bacterial classes Alphaproteobacteria (Fig. S10), Gammaproteobacteria (Fig. S11), and Bacteroidia (Fig. S12), the heme biosynthesis function had a high score. Among these three classes, Gammaproteobacteria had the highest heme biosynthesis enrichment score (>3) in all stages, and Gammaproteobacteria and Alphaproteobacteria showed higher scores in stages 2 and 3. Although Bacteroidia had the lowest heme biosynthesis score, it increased with progressive ANG stages.

Discussion
The microbial community in the ANG of S. lessoniana is influenced by the environment, the stage of maturity, and the host. The majority of studies on the ANG of mature cephalopods have considered the complex microbial consortium to be predominantly influenced by the environment. Due to a lack of information on microbial dynamics throughout the maturation process, it remains unclear whether the diversity of the complex microbial consortium remains similar in different mature stages. The present study incorporated spatial and temporal factors that may influence the dynamics of the microbial consortium in the ANG, which made it possible to identify the predominant driving factors and synergistic effects of the environment and maturity levels affecting the microbial composition. The results obtained suggest that the environment affects the microbial compositions of individuals, while maturity levels exert progressive effects on microbial dynamics over time.
Alphaproteobacteria are generally dominant in mature cephalopod ANG (Grigioni et al., 2000; Barbieri et al., 2001; Pichon et al., 2005; Collins et al., 2012; Kerwin and Nyholm, 2017). In the present study, Alphaproteobacteria were also dominant in mature ANG, similar to the findings of Kerwin and Nyholm (2017) and Collins et al. (2012), followed by Gammaproteobacteria and Bacteroidia. However, Verrucomicrobia were abundant in the ANG of the Hawaiian bobtail squid, E. scolopes, and the class Opitutae has been regarded as a core symbiotic group (Collins et al., 2012; Kerwin and Nyholm, 2017). Although bacterial Verrucomicrobia were present in the early stages of some samples, they were not included in the top 30 suggested resident OTUs in S. lessoniana. The abundance of Verrucomicrobia in E. scolopes was consistent across locations and populations (Kerwin and Nyholm, 2017; 2018), indicating that they are species-specific symbionts. E. scolopes and S. lessoniana are from various orders and, thus, are expected to have different ANG core microbiomes. In S. lessoniana, we suggested that Verrucomicrobia are abundant in the earlier stage of ANG maturation, but are then replaced by other bacterial groups, such as Bacteroidia, Alphaproteobacteria, and Gammaproteobacteria in the mature stages.
In E. scolopes, the order Flavobacteriales in Bacteroidia was found at a consistent abundance of 10% (Collins et al., 2012; Kerwin and Nyholm, 2017; 2018), which is in agreement with the present results on S. lessoniana across all maturation stages. Many members of the marine Flavobacteriales produce carotenoids—e.g., Algibacter spp. (Park et al., 2013), Olley spp. (Lee et al., 2013), Lacinutrix venerupis (Lasa et al., 2015), and Mesoflavibacter zeaxanthinifaciens (Asker et al., 2007). Based on the phylogenetic results of Bacteroidia, the dominant OTU (OTU12) of Flavobacteriales was closely related to P. geojedonensis, which produces Flexirubin-type pigments (Yoon et al., 2013).
Flexirubin is also a common pigment in some Flavobacteriales members, which produce carotenoids and form yellow-orange-colored bacterial colonies (Achenbach et al., 1978)—e.g., Ichthyenterobacterium magnum (Shakeela et al., 2015), Mesoflavibacter sabulilitoris (Park et al., 2014), and Flavivirga amylovorans (Yi et al., 2012). During maturation, the cephalopod ANG turns red-orange and becomes darker; this has been attributed to ANG-associated Alphaproteobacteria, such as members of the family Rhodobacteraceae (Collins et al., 2012). Based on the present results, we suggest that Flavobacteriales and Alphaproteobacteria both contribute to the pigmentation of the mature ANG of S. lessoniana.
One of the major functions of the ANG was suggested to be its provision of a bacteria consortium on the egg jelly coat (JC) (Kerwin and Nyholm, 2018) as an antimicrobial defense (Barbieri et al., 1997; Kerwin et al., 2019; Suria et al., 2020). The high abundance of Alphaproteobacteria and Gammaproteobacteria found in the later stage of maturation in the present study may have had an antimicrobial function. Although the specific role of Alphaproteobacteria in the ANG of S. lessoniana remains unclear, the genus Roseobacter may produce antimicrobial compounds that inhibit other bacteria, such as Vibrio fischeri (Cude et al., 2012). The Gammaproteobacteria Pseudoalteromonas isolated from the ANG and JC of the Hawaiian bobtail squid was also found to exhibit antibacterial activity (Suria et al., 2020).
Bacteria diversity in the ANG may originate from the environment instead of parental transmission based on the variations observed in the bacterial community of S. lessoniana ANG in the present study over the three years of sampling. This may lead to not only different bacteria in different environments, but also different environments creating different selection pressures for cephalopods, which, in turn, may yield different ANG bacterial communities (Kerwin and Nyholm, 2018). Two distance populations of E. scolopes from Hawaii were found to have a subset of the bacterial community of the ANG characterized to one location; this may be due to differences in morphologies between the two host populations as well as the reef topology and biota diversity (Kerwin and Nyholm, 2018). Future studies are needed to identify the biotic and abiotic factors influencing the bacterial composition of the ANG.
The ANG microbial community shifts gradually throughout maturation, with diversity decreasing across the different stages of maturation. Microbial dynamics in other animal organs have different patterns. In the human gut, microbiome diversity increases with age (Radjabzadeh et al., 2020). In the Hawaiian E. scolopes, the microbiome of its light organ has been shown to undergo a “reduction of complexity” as it matures (McFall-Ngai, 2014). Other studies on the ANG of various cephalopods, such as squids and cuttlefish, also reported that it harbors a complex microbial consortium (Collins et al., 2012; Kerwin and Nyholm, 2017). In the case of S. lessoniana, this may be the result of host selection.
Li et al. (2019) indicated that the TF-like protein in the outer layer of the ANG played an important role in the selection of bacteria that are invaginated into the ANG of S. lessoniana. Even though the TF-like protein and lactotransferrin (LTF) may have antimicrobial functions in the host, both are considered to be essential for iron acquisition in Gram-negative bacteria. This may favor bacteria that acquire iron from their host using mechanisms such as heme and siderophores. Some strains of Leisingera (the Roseobacter clade) in the ANG of bobtail squids contain siderophore synthesis genes, which may help them acquire iron from their host’s environment (Collins et al., 2015). The TF-like protein is dominantly expressed in the outer layer (epithelium cells) of the ANG (Li et al., 2019), thereby facilitating the acquisition of iron by Bacteroidia, Alphaproteobacteria, and Gammaproteobacteria, which appear to be involved in the heme biosynthesis pathway. Furthermore, this protein may enhance divergence in bacterial compositions among more mature individuals (Fig. 2). In contrast, the TF-like protein stimulated a low iron environment in the outer layer of the ANG of S. lessoniana, which may, in turn, facilitate the use of manganese (Mn) by Lactobacillus for metabolism (Pandey et al., 1994; Imbert and Blondeau, 1998). In the outer layer of the ANG, this iron-independent Lactobacillus may be involved in the selection of bacteria during the early stage of ANG development. Nevertheless, further studies are needed to establish whether the host limits iron outside the ANG or attempts to enrich it in order to promote the involvement of bacteria in heme production for the host.
In conclusion, the bigfin reef squid is one of the highest-valued fishery products in Asia; however, S. lessoniana trade still relies on catching the species wild in open water. Since coral reef destruction is increasing, spawning habitats have been decreasing in recent years, and, thus, aquaculture breeding techniques are now being used. These breeding methods have not yet created sufficiently high yields to reach the market volume. In contrast to other cephalopod model organisms, S. lessoniana has not yet been examined in detail, particularly the process of female sexual maturation and the effects of resident microbial communities. The present results revealed that the microbial community in the ANG changes throughout maturation, which may influence the host’s color and iron regulation mechanisms. These results will facilitate the development of future aquaculture systems in multiple ways; these systems may be improved by observing changes in the ANG-associated bacterial community in the laboratory or aquaculture systems. The identification of the selection pressures involved in the bacterial communities that S. lessoniana take into their ANG and elucidating the underlying mechanisms are important goals for future investigations.
Citation
Yang, S.-H., Chen, C., Hsieh, Y. E., Yang, S.-Y., Li, H.-W., Ching, T.-Y., et al. (2021) Bacterial Dynamics in the Accessory Nidamental Gland of Sepioteuthis lessoniana throughout Maturation. Microbes Environ 36: ME21030.
https://doi.org/10.1264/jsme2.ME21030
Acknowledgements
This work was supported by grants from the Ministry of Science and Technology (MOST-105-2313-B-019-012-MY3 and MOST-109-2628-B-019-002-) and by the Center of Excellence for the Oceans, National Taiwan Ocean University, from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) of Taiwan. We thank Mr. Cyuan-Hong Syu for preserving the bigfin reef squid in Hoping Island, Keelung City, Taiwan. We greatly acknowledge Dr. Po-Yu Liu for his bioinformatic support. We also thank Noah Last of Third Draft Editing for his English language editing.
References
- Achenbach, H., Kohl, W., Wachter, W.
et al. (1978) Investigations of the pigments fromCytophaga johnsonae Cy
j1. Arch Microbiol
117: 253–257.
- Asker,
D., Beppu, T., and
Ueda,
K. (2007)
Mesoflavibacter zeaxanthinifaciens gen. nov., sp.
nov., a novel zeaxanthin-producing marine bacterium of the family
Flavobacteriaceae. Syst Appl Microbiol
30: 291–296.
- Barbieri, E., Barry, K., Child, A., and Wainwright, N. (1997) Antimicrobial activity in the microbial community of the accessory nidamental gland and egg cases of Loligo pealei (Cephalopoda: Loliginidae). Biol Bull (Chicago, IL, U S)
193: 275–276.
- Barbieri, E., Paster, B.J., Hughes, D., Zurek, L., Moser, D.P., Teske, A., and Sogin, M.L. (2001) Phylogenetic characterization of epibiotic bacteria in the accessory nidamental gland and egg of the squid Loligo pealei (Cephalopoda: Loliginidae). Environ Microbiol
3: 151–167.
- Cheng, S., Anderson, F.E., Bergman, A., Mahardika, G., Muchlisin, Z., Dang, B., et al. (2014) Molecular evidence for co-occurring cryptic lineages within the Sepioteuthis cf. lessoniana species complex in the Indian and Indo-West Pacific Oceans. Hydrobiologia
725: 165–188.
- Chiang, C.I., Chung, M.T., Shiao, J.C., Wang, P.L., Chan, T.Y., Yamaguchi, A., and Wang, C.H. (2020) Seasonal movement pattern of the bigfin reef squid Sepioteuthis lessoniana predicted using statolith delta O-18 values. Front Mar Sci
7: 249.
- Ching, T.Y., Chen, C.S., and Wang, C.H. (2019) Spatiotemporal variations in life history traits and statolith trace elements of Sepioteuthis lessoniana populations around northern Taiwan. J Mar Biol Assoc U K
99: 203–213.
- Ching, T.Y., Chen, C.S., Yagishita, N., Yamaguchi, A., Wang, C.H., and Shen, K.N. (2021) Variations in life-history traits and statolith shape for Sepioteuthis spp. in the waters off southwestern Japan. Fish Sci
87: 173–185.
- Collins, A.J., LaBarre, B.A., Won, B.S.W., Shah, M.V., Heng, S., Choudhury, M.H., et al. (2012) Diversity and partitioning of bacterial populations within the accessory nidamental gland of the squid Euprymna scolopes. Appl Environ Microbiol
78: 4200–4208.
- Collins, A.J., Fullmer, M.S., Gogarten, J.P., and Nyholm, S.V. (2015) Comparative genomics of Roseobacter clade bacteria isolated from the accessory nidamental gland of Euprymna scolopes. Front Microbiol
6: 123.
- Edgar, R.C., Haas, B.J., Clemente, J.C., Quince, C., and Knight, R. (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics
27: 2194–2200.
- Edgar, R.C. (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods
10: 996–998.
- Grigioni, S., Boucher-Rodoni, R., Demarta, A., Tonolla, M., and Peduzzi, R. (2000) Phylogenetic characterisation of bacterial symbionts in the accessory nidamental glands of the sepioid Sepia officinalis (Cephalopoda: Decapoda). Mar Biol (Heidelberg, Ger)
136: 217–222.
- Hsiao, C.D., Shen, K.N., Ching, T.Y., Wang, Y.H., Ye, J.J., Tsai, S.Y., et al. (2016) The complete mitochondrial genome of the cryptic “lineage A” big-fin reef squid, Sepioteuthis lessoniana (Cephalopoda: Loliginidae) in Indo-West Pacific. Mitochondrial DNA, Part A
27: 2433–2434.
- Imbert, M., and Blondeau, R. (1998) On the iron requirement of lactobacilli grown in chemically defined medium. Curr Microbiol
37: 64–66.
- Kaufman, M.R., Ikeda, Y., Patton, C., Van Dykhuizen, G., and Epel, D. (1998) Bacterial symbionts colonize the accessory nidamental gland of the squid Loligo opalescens via horizontal transmission. Biol Bull (Chicago, IL, U S)
194: 36–43.
- Kerwin, A.H., and Nyholm, S.V. (2017) Symbiotic bacteria associated with a bobtail squid reproductive system are detectable in the environment, and stable in the host and developing eggs. Environ Microbiol
19: 1463–1475.
- Kerwin, A.H., and Nyholm, S.V. (2018) Reproductive system symbiotic bacteria are conserved between two distinct populations of Euprymna scolopes from Oahu, Hawaii. mSphere
3: e00531-17.
- Kerwin, A.H., Gromek, S.M., Suria, A.M., Samples, R.M., Deoss, D.J., O’Donnell, K., et al. (2019) Shielding the next generation: symbiotic bacteria from a reproductive organ protect bobtail squid eggs from fungal fouling. mBio
10: e02376-19.
- Lasa, A., Diéguez, A.L., and Romalde
J.L. (2015) Description of Lacinutrix venerupis sp. nov.: a novel bacterium associated with reared clams. Syst Appl Microbiol
38: 115-119.
- Lee, M.H., Jung, Y.T., Park, S., and Yoon, J.H. (2013) Olleya namhaensis sp. nov., isolated from wood falls, and emended description of the genus Olleya Mancuso Nichols et al. 2005 emend. Lee et al. 2010. Int J Syst Evol Microbiol
63: 1610–1615.
- Li, H.W., Chen, C., Kuo, W.L., Lin, C.J., Chang, C.F., and Wu, G.C. (2019) The characteristics and expression profile of transferrin in the accessory nidamental gland of the bigfin reef squid during bacteria transmission. Sci Rep
9: 20163.
- Liu, P.Y., Cheng, A.C., Huang, S.W., Lu, H.P., Oshida, T., Liu, W., and Yu, H.T. (2020) Body-size scaling is related to gut microbial diversity, metabolism and dietary niche of arboreal folivorous flying squirrels. Sci Rep
10: 7809.
- Lum-Kong, A., and Hastings, T.S. (1992) The accessory nidamental glands of Loligo forbesi (Cephalopoda: Loliginidae): characterization of symbiotic bacteria and preliminary experiments to investigate factors controlling sexual maturation. J Zool
228: 395–403.
- McFall-Ngai, M.J. (2014) The importance of microbes in animal development: lessons from the squid-vibrio symbiosis. Annu Rev Microbiol
68: 177–194.
- Norman, M.D. (2003) Cephalopods: A World Guide. Harxheim, Germany: Conchbooks.
- Nossa, C.W., Oberdorf, W.E., Yang, L., Aas, J.A., Paster, B.J., DeSantis, T.Z., et al. (2010) Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. World J Gastroenterol
16: 4135–4144.
- O’Brien, C.E., Roumbedakis, K., and Winkelmann, I.E. (2018) The current state of cephalopod science and perspectives on the most critical challenges ahead from three early-career researchers. Front Physiol
9: 700.
- Pandey, A., Bringel, F., and Meyer, J.M. (1994) Iron requirement and search for siderophores in lactic acid bacteria. Appl Microbiol Biotechnol
40: 735–739.
- Park, S.C., Hwang, Y.M., Lee, J.H., Baik, K.S., and Seong, C.N. (2013) Algibacter agarivorans sp. nov. and Algibacter agarilyticus sp. nov., isolated from seawater, reclassification of Marinivirga aestuarii as Algibacter aestuarii comb. nov. and emended description of the genus Algibacter. Int J Syst Evol Microbiol
63: 3494–3500.
- Park, S., Park, J.M., Jung, Y.T., Seong, C.N., and Yoon, J.H. (2014) Mesoflavibacter sabulilitoris sp. nov., isolated from seashore sand. Int J Syst Evol Microbiol
64: 3743–3748.
- Pichon, D., Gaia, V., Norman, M.D., and Boucher-Rodoni, R. (2005) Phylogenetic diversity of epibiotic bacteria in the accessory nidamental glands of squids (Cephalopoda: Loliginidae and Idiosepiidae). Mar Biol (Heidelberg, Ger)
147: 1323–1332.
- Radjabzadeh, D., Boer, C.G., Beth, S.A., van der Wal, P., Kiefte-De Jong, J.C., Jansen, M.A.E., et al. (2020) Diversity, compositional and functional differences between gut microbiota of children and adults. Sci Rep
10: 1040.
- Schloss, P.D., Westcott, S.L., Ryabin, T., Hall, J.R., Hartmann, M., Hollister, E.B., et al. (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol
75: 7537–7541.
- Shakeela, Q., Shehzad, A., Tang, K., Zhang, Y., and Zhang, X.H. (2015) Ichthyenterobacterium magnum gen. nov., sp. nov., a member of the family Flavobacteriaceae isolated from olive flounder (Paralichthys olivaceus). Int J Syst Evol Microbiol
65: 1186–1192.
- Shen, K.N., Yen, T.C., Chen, C.H., Ye, J.J., and Hsiao, C.D. (2016) The complete mitochondrial genome of the cryptic “lineage B” big-fin reef squid, Sepioteuthis lessoniana (Cephalopoda: Loliginidae) in Indo-West Pacific. Mitochondrial DNA, Part A
27: 2100–2101.
- Suria, A.M., Tan, K.C., Kerwin, A.H., Gitzel, L., Abini-Agbomson, L., Bertenshaw, J.M., et al. (2020) Hawaiian bobtail squid symbionts inhibit marine bacteria via production of specialized metabolites, including new bromoalterochromides BAC-D/D’. mSphere
5: e00166-20.
- Tandon, K., Yang, S.H., Wan, M.T., Yang, C.C., Baatar, B., Chiu, C.Y., et al. (2018) Bacterial community in water and air of two sub-alpine lakes in Taiwan. Microbes Environ
33: 120–126.
- Yi, H., Cho, J.C., and Chun, J. (2012) Flavivirga jejuensis gen. nov., sp. nov., and Flavivirga amylovorans sp. nov., new members of the family Flavobacteriaceae isolated from seawater, and emended descriptions of the genera Psychroserpens and Lacinutrix. Int J Syst Evol Microbiol
62: 1061–1068.
- Yoon, J.H., Lee, M.H., and Jung, Y.T. (2013) Pseudofulvibacter geojedonensis gen. nov., sp. nov., a polysaccharide-degrading member of the family Flavobacteriaceae isolated from seawater, and emended description of the genus Fulvibacter. Int J Syst Evol Microbiol
63: 1696–1701.