Abstract
The rumen contains a complex microbial ecosystem that degrades plant materials, such as cellulose and hemicellulose. We herein reconstructed 146 nonredundant, rumen-specific metagenome-assembled genomes (MAGs), with ≥50% completeness and <10% contamination, from cattle in Japan. The majority of MAGs were potentially novel strains, encoding various enzymes related to plant biomass degradation and volatile fatty acid production. The MAGs identified in the present study may be valuable resources to enhance the resolution of future taxonomical and functional studies based on metagenomes and metatranscriptomes.
The rumen is a forestomach in the ruminant digestive tract that comprises a complex microbial ecosystem of bacteria, ciliate protozoa, fungi, methanogens, and viruses (Morgavi et al., 2013). These microbes degrade plant materials, such as cellulose and hemicellulose (Terry et al., 2019), to yield volatile fatty acids (VFAs), which are a major energy source for ruminants. Therefore, a more detailed understanding the structural and functional characteristics of the rumen microbiome will facilitate enhancements in the efficacy of ruminant production. Recent studies identified several rumen metagenome-assembled genomes (MAGs) (e.g., 4,941 MAGs from Stewart et al., 2019; 1,200 from Wilkinson et al., 2020; and 2,809 from Anderson and Fernando, 2021), which has markedly expanded the rumen microbial database.
Bacterial community compositions in animals appear to exhibit regional variations, which may be because of differences in diet, climate, and farming practices (Henderson et al., 2015). Despite this, we are the only group to have reconstructed rumen MAGs from cattle in Japan. We previously reconstructed 114 rumen MAGs from the shotgun metagenome data of Japanese Black (JB) and F1 crossbred (JB sire×Holstein dam) steers in Japan (Sato et al., 2021). We herein further expanded the catalogs of rumen microbial genomes in Japan with 146 MAGs from the rumen of JB, Japanese Shorthorn (JS), and F1 steers.
The experimental design and protocol were approved by the Kyoto University Animal Ethics Committee (Permit No. R2-119). This study was the first to use JB (n=2), JS (n=2), and F1 (n=6) steers from two farms (for subject data, see Table S1). Rumen contents were collected and processed according to previously reported methods (Sato et al., 2021). To extract DNA, 1.5-mL rumen samples were thawed and centrifuged at 12,000×g at 4°C for 15 min. Supernatants were discarded and pellets were treated as previously reported (Frias-Lopez et al., 2008), with modifications from Sato et al. (2021). Extracted DNA was stored at –20°C until it was required for further analyses. The metagenomic library was prepared with a Nextera XT DNA library preparation kit (Illumina) and sequenced on an Illumina Hiseq X Ten platform (2×150 bp).
Raw reads in a previous study (Sato et al., 2021; accession number DRA011676) and the present study were trimmed with Trimmomatic version 0.39 (Bolger et al., 2014). To remove host DNA contamination, trimmed reads were mapped with BWA-MEM (Li, H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. https://arxiv.org/abs/1303.3997) to the bovine reference genome ARS-UCD1.2/bosTau9. Filtered reads were assembled in SPAdes version 3.13.0 (Bankevich et al., 2012) using the “--meta” option (Nurk et al., 2017), pooled and co-assembled in MEGAHIT version 1.2.9 (Li et al., 2015), then mapped back to contigs with BWA-MEM (Li, H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. https://arxiv.org/abs/1303.3997). We binned MAGs with contigs using MetaBAT2 version 2.15 (Kang et al., 2019), MaxBin2 version 2.2.7 (Wu et al., 2016), and CONCOCT version 1.1.0 (Alneberg et al., 2014). Output bins were subsequently dereplicated and aggregated using DAS Tool version 1.1.4 (Sieber et al., 2018). The quality (completeness and contamination) of MAGs was evaluated in CheckM version 1.1.3. (Parks et al., 2015), retaining genomes with ≥50% completeness and <10% contamination. Retained MAGs were dereplicated in dRep version 3.2.2 (Olm et al., 2017) with the “dRep dereplicate” command at 99% and 95% average nucleotide identities (ANI) using a default clustering algorithm, which were hereafter called RUG1ANI99% and RUG1ANI95%, respectively, with MAGs in these groups being defined as nonredundant strains and species, respectively (Stewart et al., 2019).
Filtered reads were mapped against RUG1ANI95% MAGs using BamM (https://github.com/Ecogenomics/BamM) to prevent arbitrary mapping between similar genomes (Robbins et al., 2021). Genome relative abundance was calculated using the CoverM version 0.4.0 “genome” (https://github.com/wwood/CoverM) with the following parameters: -m relative_abundance --min-read-percent-identity 0.95 --min-read-aligned-percent 0.75. We taxonomically classified RUG1ANI99% MAGs in GTDB-tk version 2.1.1 (Chaumeil, P.A., et al. 2022. GTDB-Tk v2: memory friendly classification with the Genome Taxonomy Database. bioRxiv. https://doi.org/10.1101/2022.07.11.499641) with GTDB release 207. We then built a phylogenetic tree in PhyloPhlAn version 3.0.60 (Asnicar et al., 2020). The tree was visualized in iTOL version 6.1.2 (Letunic and Bork, 2021). To elucidate whether RUG1ANI99% MAGs represented potential novel species and strains, we compared them with the genomes of rumen prokaryotes (hereafter called RUG2), including Hungate1000 (Seshadri et al., 2018), and previously reported rumen MAGs (Stewart et al., 2019; Anderson and Fernando, 2021) using dRep (Olm et al., 2017). Genomes were defined as novel species (<95%) and strains (<99%) using the ANI outputs by dRep and GTDB-tk.
We predicted the proteins encoded by RUG1ANI99% MAGs with Prodigal version 2.6.3 (Hyatt et al., 2010), and then searched them against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database through GhostKOALA (Kanehisa et al., 2016). Carbohydrate-active enzyme (CAZyme) families, which are associated with cell wall degradation and essential for efficient lignocellulose processing in ruminants, were annotated using dbCAN2 (Zhang et al., 2018). This software integrates three tools (HMMER, DIAMOND, and Hotpep), and we only retained the CAZyme domains annotated by at least two of the three. The predicted protein sequences were then mapped to the Pfam database (Finn et al., 2016) using HMMER version 3.3.2 (Mistry et al., 2013) with an E value <10–5 to search cohesin (PF00963) and dockerin (PF00404) domains. We used PULpy (Stewart, R.D., et al. 2018 Open prediction of polysaccharide utilisation loci (PUL) in 5,414 public Bacteroidetes genomes using PULpy. bioRxiv. https://doi.org/10.1101/421024) to predict polysaccharide utilization loci (PUL), linked gene clusters that encode the cell envelope-associated enzymes required for sensing, binding, and degrading polysaccharide substrates (Bjursell et al., 2006). All novel sequence raw data were deposited in DDBJ (accession number DRA014084). The 146 RUG1ANI99% MAGs are available at https://doi.org/10.6084/m9.figshare.20705449.
Filtered samples yielded 706 M reads, and we retained 291 bins. Of these, 202 MAGs had ≥50% completeness and <10% contamination, with 54 being high quality (≥90% completeness and <5% contamination). Following dereplication at 99% ANI, we generated 146 nonredundant RUG1ANI99% MAGs with 36 high-quality MAGs. The range of their sum genome length was 0.41–5.84 Mb, with N50 ranging between 1.66 and 93.2 kb (Table S2). A comparison with MAGs in the RUG2 (8,153 genomes) and GTDB (317,542 genomes) databases revealed 16 and 130 RUG1ANI99% MAGs that were potential novel species and strains, respectively (Table S2). Therefore, the RUG2 and GTDB databases did not completely cover the diversity of the rumen microbiota from the tested cattle.
After aligning filtered reads against RUG1ANI95% MAGs, we identified 32 MAGs that were present (relative abundance >0) in >90% of the tested cattle (n=20), suggesting that they are core rumen bacteria in Japan (Table S3). The average mapping rate was 43.9% per sample, indicating that RUG1ANI95% MAGs did not cover the full rumen microbial diversity of these animals. This conclusion is also apparent because we did not recover MAGs assigned to core rumen microbial taxa, such as Fibrobacter. These MAGs may be unobtainable because we had less data than other studies (Stewart et al., 2019; Anderson and Fernando, 2021).
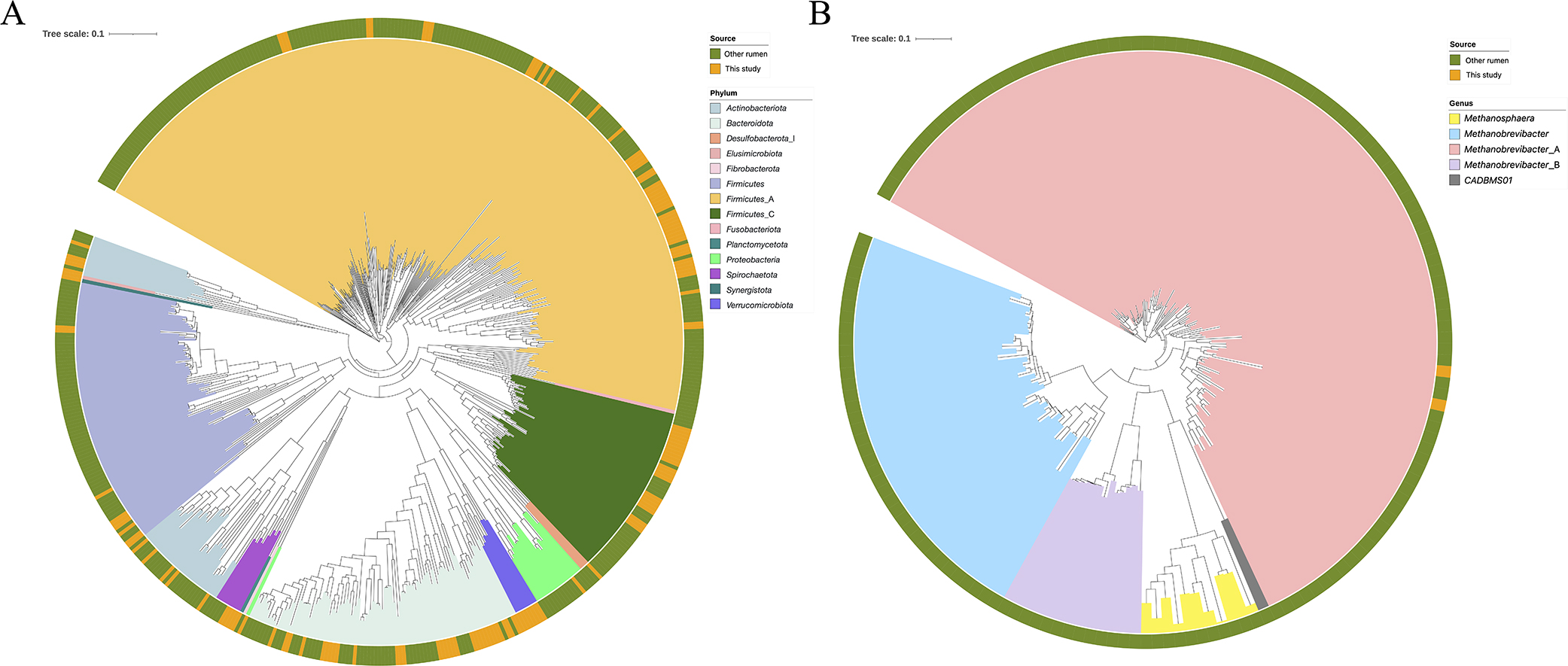
The 32 and 50 RUG1ANI99% MAGs were classified as members of Bacteroidota and Firmicutes_A, respectively (Fig. 1 and Table S2); this taxonomic assignment is consistent with that reported in a previous study in which many MAGs reconstructed from the cattle rumen were classified into these phyla (Gharechahi et al., 2021). The remaining RUG1ANI99% MAGs were assigned to the following 12 phyla: Firmicutes_C (23 MAGs), Actinobacteriota (10 MAGs), Firmicutes (9 MAGs), Verrucomicrobiota (7 MAGs), Spirochaetota (4 MAGs), Patescibacteria (3 MAGs), Proteobacteria (2 MAGs), Methanobacteriota (2 MAGs), Planctomycetota (1 MAG), Synergistota (1 MAG), Desulfobacterota_I (1 MAG), and Elusimicrobiota (1 MAG). In Bacteroidota MAGs, 10 RUG1ANI99% MAGs were classified as Prevotella, which is the most abundant bacterial genus in the rumen (Henderson et al., 2015), and its abundance increases with high-concentrate diets (Luo et al., 2017). Most of the animals tested were offered high-concentrate diets (Table S1), resulting in the reconstruction of Prevotella MAGs. In the present study, 14 Lachnospiraceae MAGs were reconstructed; however, we previously recovered only 3 Lachnospiraceae MAGs (Sato et al., 2021). Lachnospiraceae is associated with feed efficiency in ruminants (Li and Guan, 2017) and is crucial for shaping the characteristics of JB rumen microbiomes (Ogata et al., 2019; Sato et al., 2021). Therefore, the Lachnospiraceae MAGs obtained herein may be useful for metagenomic and metatranscriptomic research in ruminants, particularly JB cattle.
Prodigal (Hyatt et al., 2010) predicted 330,575 CDS from RUG1ANI99% MAGs, with a range of 537–8,276 (Table S2). We found 156,599 CDS (47.4%) in the KEGG Orthology (KO) database. Consistent with our previous findings (Sato et al., 2021), a KO analysis indicated that most RUG1ANI99% MAGs were roughly clustered according to their phylum (Fig. S1). We focused on genes related to VFA production because VFAs are a major energy source for ruminants (Table 1 and Fig. S2). We identified 113 RUG1ANI99% MAGs (77.4%) with at least one gene related to acetate production (K00925: acetate kinase, K00625: phosphate acetyltransferase). These genes were absent from Firmicutes_C RUG1ANI99% MAGs. Rumen propionate is produced via the succinate and acrylate pathways (Jeyanathan et al., 2014). As markers of the succinate pathway, we selected genes encoding methylmalonyl-CoA mutase (K01847, K01848, and K01849), methylmalonyl-CoA epimerase (K05606), and methylmalonyl-CoA decarboxylase (K11264); acrylate pathway markers were genes encoding lactoyl-CoA dehydratase (K20626 and K20627). Succinate pathway marker genes were present in 52 RUG1ANI99% MAGs; most were MAGs belonging to Bacteroidota and Firmicutes_C, including Prevotella and Succiniclasticum MAGs (10 and 15 MAGs, respectively). Prevotella (Wirth et al., 2018) and Succiniclasticum (Van Gylswyk, 1995) both produce propionate from succinate in the rumen. In contrast, none of the RUG1ANI99% MAGs possessed the acrylate pathway marker gene.
Table 1.
The number of RUG1
ANI99% MAGs associated with volatile fatty acid production
| Phylum1 |
Acetate2 |
Propionate3 |
Butyrate4 |
| Succinate pathway |
Acrylate pathway |
| Actinobacteriota (10) |
9 |
2 |
0 |
4 |
| Bacteroidota (32) |
29 |
22 |
0 |
15 |
| Desulfobacterota_I (1) |
1 |
0 |
0 |
0 |
| Elusimicrobiota (1) |
1 |
0 |
0 |
1 |
| Firmicutes (9) |
8 |
0 |
0 |
4 |
| Firmicutes_A (50) |
48 |
4 |
0 |
24 |
| Firmicutes_C (23) |
0 |
23 |
0 |
22 |
| Patescibacteria (3) |
3 |
0 |
0 |
0 |
| Planctomycetota (1) |
1 |
0 |
0 |
0 |
| Proteobacteria (2) |
2 |
0 |
0 |
1 |
| Spirochaetota (4) |
4 |
0 |
0 |
1 |
| Synergistota (1) |
0 |
1 |
0 |
1 |
| Verrucomicrobiota (7) |
7 |
0 |
0 |
2 |
| Methanobacteriota (2) |
0 |
0 |
0 |
2 |
1 The number after the phylum name shows MAG numbers of the phylum in RUG1ANI99% MAGs.
2 Numbers of MAGs that have genes assigned as K00925 and/or K00625.
3 Numbers of MAGs that possessed at least one succinate pathway marker gene (K01847, K01848, K01849, K11264, and K05606) or acrylate pathway marker gene (K20626 and K20627).
4 Numbers of MAGs with at least one gene related to butyrate production (K00634, K00929, K01034, K00626, and K00074).
Among 146 RUG1ANI99% MAGs, 136 and 112 possessed the genes encoding CAZymes GH13 (α-amylase) and GH77 (4-α-glucanotransferase), respectively, both of which are involved in starch degradation (Fig. S3 and Table S4). Furthermore, oligosaccharide-degrading GH2 (β-galactosidase) and GH3 (β-glucosidase) were present in 92 and 98 RUG1ANI99% MAGs, respectively. GH2, GH3, and GH13 were among the most dominant CAZymes in the cattle rumen (Wang et al., 2019). Additionally, Prevotella RUG1ANI99% MAGs had multiple genes related to CAZymes (Fig. S3), which is in accordance with the ability of the genus to degrade fiber sources, such as hemicellulose and pectin (Stewart et al., 1997). All Prevotella RUG1ANI99% MAGs possessed genes encoding GH2 (β-galactosidase), GH3 (β-glucosidase), GH5 (endo-β-1,4-glucanase, endo-β-1,4-xylanase, and β-mannosidase), GH13 (α-amylase), GH26 (endo-β-1,4-mannanases), GH32 (invertase), GH36 (α-galactosidase), GH43 (β-xylosidase), GH73 (lysozyme), GH94 (cellobiose phosphorylase), and GH97 (α-glucosidase).
Cellulosomes (Bayer et al., 2004; Artzi et al., 2017) and amylosomes (Ze et al., 2015) consist of scaffolding subunits that contain cohesion modules and dockerin modules, which append to an enzyme for the degradation of lignocellulose and starch, respectively. Cohesin modules specifically interact with dockerin modules, and the cohesin–dockerin interaction is responsible for the integration of enzymes into the complex (Bayer et al., 2004; Artzi et al., 2017). Therefore, we searched cohesin and dockerin domains in RUG1ANI99% MAGs. The proteins containing cohesin and dockerin domains are summarized in Table S5. In total, 200 dockerin and 18 cohesin domains were present in 25 RUG1ANI99% MAGs, including 2 Ruminococcus and 11 Ruminococcus_E MAGs. Sixteen dockerin-containing proteins carried CAZyme domains, mostly including GH families containing α-amylase (GH13_14, GH13_19, and GH13_28). Among the 25 RUG1ANI99% MAGs, 13 had both cohesin- and dockerin-containing proteins, while 9 had only dockerin-containing proteins. Dassa et al. (2014) reported that Ruminococcus albus 8, which degrades cellulosic substrates, harbored no cohesion-containing protein. Therefore, we cannot rule out the possibility that RUG1ANI99% MAGs, which had no cohesion- and some dockerin-containing proteins, used an alternative mechanism for the immobilization of dockerin-containing enzymes onto carbohydrates, similar to Ruminococcus albus 8 (Dassa et al., 2014).
In Bacteroidetes, CAZymes are often organized in PULs. We herein identified 215 PULs in Bacteroidota RUG1ANI99% MAGs, and 93 lacked any known CAZyme domains (Table S6). The most common PUL was a single susC/susD-like pair, which was consistent with previous findings (Stewart et al., 2019). Among 32 Bacteroidota MAGs, 29 had at least one PUL. The number of PULs per Bacteroidota MAG ranged between 1 and 18, with the highest number being observed in Prevotella MAG and Cryptobacteroides MAG (JB_Ai_91_bin_120 and F1_Ai_424_bin_68, respectively). Ninety-eight and 80 PULs were identified in Cryptobacteroides MAG and Prevotella MAGs, respectively, and contained various CAZymes. Overall, the present results suggest that Cryptobacteroides and Prevotella are important for rumen function (specifically carbon degradation) in cattle in Japan.
In summary, we reconstructed 146 rumen-specific MAGs from cattle in Japan, with many being potentially novel. These MAGs are valuable resources for enhancing the resolution of future metagenome- and metatranscriptomic-based taxonomical and functional studies.
Citation
Sato, Y., Takebe, H., Oishi, K., Yasuda, J., Kumagai, H., Hirooka, H., and Yoshida, T. (2022) Identification of 146 Metagenome-assembled Genomes from the Rumen Microbiome of Cattle in Japan. Microbes Environ 37: ME22039.
https://doi.org/10.1264/jsme2.ME22039
Acknowledgements
The present study was partly supported by a Grant-in-Aid from the Japan Society for the Promotion of Science (JSPS) Research Fellow Number 20J15021 and JSPS KAKENHI Grant Number JP21H05057. Super-computing resources were provided by the Human Genome Center, Institute of Medical Science, University of Tokyo. We thank members of the Iwate Agricultural Research Center Animal Industry Research Institute for helping us collect rumen samples.
References
- Alneberg, J., Bjarnason, B.S., De Bruijn, I., Schirmer, M., Quick, J., Ijaz, U.Z., et al. (2014) Binning metagenomic contigs by coverage and composition. Nat Methods
11: 1144–1146.
- Anderson, C.L., and Fernando, S.C. (2021) Insights into rumen microbial biosynthetic gene cluster diversity through genome-resolved metagenomics. Commun Biol
4: 818.
- Artzi, L., Bayer, E.A., and Moraïs, S. (2017) Cellulosomes: bacterial nanomachines for dismantling plant polysaccharides. Nat Rev Microbiol
15: 83–95.
- Asnicar, F., Thomas, A.M., Beghini, F., Mengoni, C., Manara, S., Manghi, P., et al. (2020) Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat Commun
11: 2500.
- Bankevich, A., Nurk, S., Antipov, D., Gurevich, A.A., Dvorkin, M., Kulikov, A.S., et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol
19: 455–477.
- Bayer, E.A., Belaich, J.P., Shoham, Y., and Lamed, R. (2004) The cellulosomes: multienzyme machines for degradation of plant cell wall polysaccharides. Annu Rev Microbiol
58: 521–554.
- Bjursell, M.K., Martens, E.C., and Gordon, J.I. (2006) Functional genomic and metabolic studies of the adaptations of a prominent adult human gut symbiont, Bacteroides thetaiotaomicron, to the suckling period. J Biol Chem
281: 36269–36279.
- Bolger, A.M., Lohse, M., and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics
30: 2114–2120.
- Dassa, B., Borovok, I., Ruimy-Israeli, V., Lamed, R., Flint, H.J., Duncan, S.H., et al. (2014) Rumen cellulosomics: divergent fiber-degrading strategies revealed by comparative genome-wide analysis of six ruminococcal strains. PLoS One 9: e99221.
- Finn, R.D., Coggill, P., Eberhardt, R.Y., Eddy, S.R., Mistry, J., Mitchell, A.L., et al. (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res
44: D279–D285.
- Frias-Lopez, J., Shi, Y., Tyson, G.W., Coleman, M.L., Schuster, S.C., Chisholm, S.W., et al. (2008) Microbial community gene expression in ocean surface waters. Proc Natl Acad Sci U S A
105: 3805–3810.
- Gharechahi, J., Vahidi, M.F., Bahram, M., Han, J.L., Ding, X.Z., and Salekdeh, G.H. (2021) Metagenomic analysis reveals a dynamic microbiome with diversified adaptive functions to utilize high lignocellulosic forages in the cattle rumen. ISME J
15: 1108–1120.
- Henderson, G., Cox, F., Ganesh, S., Jonker, A., Young, W., Abecia, L., et al. (2015) Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci Rep
5: 14567.
- Hyatt, D., Chen, G.-L., LoCascio, P.F., Land, M.L., Larimer, F.W., and Hauser, L.J. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf
11: 119.
- Jeyanathan, J., Martin, C., and Morgavi, D.P. (2014) The use of direct-fed microbials for mitigation of ruminant methane emissions: a review. Animal
8: 250–261.
- Kanehisa, M., Sato, Y., and Morishima, K. (2016) BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol
428: 726–731.
- Kang, D.D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., et al. (2019) MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ
7: e7359.
- Letunic, I., and Bork, P. (2021) Interactive Tree of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res
49: W293–W296.
- Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T.W. (2015) MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics
31: 1674–1676.
- Li, F., and Guan, L.L. (2017) Metatranscriptomic profiling reveals linkages between the active rumen microbiome and feed efficiency in beef cattle. Appl Environ Microbiol
83: e00061-17.
- Luo, D., Gao, Y., Lu, Y., Qu, M., Xiong, X., Xu, L., et al. (2017) Niacin alters the ruminal microbial composition of cattle under high-concentrate condition. Anim Nutr
3: 180–185.
- Mistry, J., Finn, R.D., Eddy, S.R., Bateman, A., and Punta, M. (2013) Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res
41: e121.
- Morgavi, D.P., Kelly, W.J., Janssen, P.H., and Attwood, G.T. (2013) Rumen microbial (meta)genomics and its application to ruminant production. Animal
7: 184–201.
- Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P.A. (2017) metaSPAdes: a new versatile metagenomic assembler. Genome Res
27: 824–834.
- Ogata, T., Makino, H., Ishizuka, N., Iwamoto, E., Masaki, T., Ikuta, K., et al. (2019) Long-term high-grain diet altered the ruminal pH, fermentation, and composition and functions of the rumen bacterial community, leading to enhanced lactic acid production in Japanese Black beef cattle during fattening. PLoS One
14: e0225448.
- Olm, M.R., Brown, C.T., Brooks, B., and Banfield, J.F. (2017) dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J
11: 2864–2868.
- Parks, D.H., Imelfort, M., Skennerton, C.T., Hugenholtz, P., and Tyson, G.W. (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res
25: 1043–1055.
- Robbins, S.J., Song, W., Engelberts, J.P., Glasl, B., Slaby, B.M., Boyd, J., et al. (2021) A genomic view of the microbiome of coral reef demosponges. ISME J
15: 1641–1654.
- Sato, Y., Takebe, H., Tominaga, K., Oishi, K., Kumagai, H., Yoshida, T., et al. (2021) Taxonomic and functional characterization of the rumen microbiome of Japanese Black cattle revealed by 16S rRNA gene amplicon and metagenome shotgun sequencing. FEMS Microbiol Ecol
97: fiab152.
- Seshadri, R., Leahy, S.C., Attwood, G.T., Teh, K.H., Lambie, S.C., Cookson, A.L., et al. (2018) Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection. Nat Biotechnol
36: 359–367.
- Sieber, C.M.K., Probst, A.J., Sharrar, A., Thomas, B.C., Hess, M., Tringe, S.G., et al. (2018) Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol
3: 836–843.
- Stewart, C.S., Flint, H.J., and Bryant, M.P. (1997) The rumen bacteria. In The Rumen Microbial Ecosystem. New York, NY: Springer, pp. 10–72.
- Stewart, R.D., Auffret, M.D., Warr, A., Walker, A.W., Roehe, R., and Watson, M. (2019) Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat Biotechnol
37: 953–961.
- Terry, S.A., Badhan, A., Wang, Y., Chaves, A.V., and McAllister, T.A. (2019) Fibre digestion by rumen microbiota—a review of recent metagenomic and metatranscriptomic studies. Can J Anim Sci
99: 678–692.
- Van Gylswyk, N.O. (1995) Succiniclasticum ruminis gen. nov., sp. nov., a ruminal bacterium converting succinate to propionate as the sole energy-yielding mechanism. Int J Syst Evol Microbiol
45: 297–300.
- Wang, L., Zhang, G., Xu, H., Xin, H., and Zhang, Y. (2019) Metagenomic analyses of microbial and carbohydrate-active enzymes in the rumen of holstein cows fed different forage-to-concentrate ratios. Front Microbiol
10: 649.
- Wilkinson, T., Korir, D., Ogugo, M., Stewart, R.D., Watson, M., Paxton, E., et al. (2020) 1200 high-quality metagenome-assembled genomes from the rumen of African cattle and their relevance in the context of sub-optimal feeding. Genome Biol
21: 229.
- Wirth, R., Kádár, G., Kakuk, B., Maróti, G., Bagi, Z., Szilágyi, Á., et al. (2018) The planktonic core microbiome and core functions in the cattle rumen by next generation sequencing. Front Microbiol
9: 2285.
- Wu, Y.W., Simmons, B.A., and Singer, S.W. (2016) MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics
32: 605–607.
- Ze, X., Ben David, Y., Laverde-Gomez, J.A., Dassa, B., Sheridan, P.O., Duncan, S.H., et al. (2015) Unique organization of extracellular amylases into amylosomes in the resistant starch-utilizing human colonic Firmicutes bacterium Ruminococcus bromii. mBio
6: e01058-15.
- Zhang, H., Yohe, T., Huang, L., Entwistle, S., Wu, P., Yang, Z., et al. (2018) dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res
46: W95–W101.