Personal Perspective
Retraction: Etiopathology of preeclampsia — Recent progress from the perspective of a poor/ischemic placenta
2014 年 2 巻 2 号 p. 98-107

詳細
2014 年 2 巻 2 号 p. 98-107
Research on the etiopathology of preeclampsia has rapidly progressed since the mid-20th century. However, the pathogenesis of this disease remains poorly understood. The etiology of preeclampsia may begin from poor placentation in early gestation, which can lead to placental ischemia/hypoxia. Placental hypoxia induces antiangiogenesis, angiotensin II type 1 receptor agonistic antibodies, and cytokines, resulting in vasculoendothelial dysfunction. This reduces the production of nitric oxide and increases the secretion of endothelin-1. In addition, maternal constitutional factors, such as environmental, genetic, and immunological factors, as well as oxidative stress and inflammation, are likely to contribute to the pathogenesis of abnormal placentation and maternal sensitivity to antiangiogenic factors. Therefore, early-onset preeclampsia is considered more of a placental disease. On the other hand, late-onset preeclampsia results from an imbalance of placental blood supply and fetal demand when the uterine capacity approaches its limit. Accordingly, late-onset preeclampsia is considered more of a maternal disease.
It is generally recognized that preeclampsia is cured with the delivery of the placenta. Therefore, the presence of placental tissue is required for the development of the disorder, but not the fetus.1,2) However, clinical observations of patients with preeclampsia suggest that the fetus may play a role in the maternal manifestations of this complication.3,4) A striking example of the role of the fetus is the remission of preeclampsia following the death of a growth-restricted fetus in discordant twins or after correction of fetal hydrops in mirror syndrome associated with parvovirus infection. In the latter case, improvement in fetal status and presumably subsequent improvement in fetal perfusion of the placenta leads to the resolution of preeclampsia without the need for placental delivery.5)
Placental ischemia/hypoxia is generally regarded as the initiating event in preeclamptic syndrome. It results in the secretion of a variety of factors from the placenta, such as soluble fms-like tyrosine kinase 1 (sFlt-1), angiotensin II type 1 receptor agonistic antibodies (AT1-AA), and cytokines, which lead to dysfunction in the maternal vascular endothelium.6)
Although the placenta plays a crucial role in the development of preeclampsia syndrome, its onset, severity, and progression are significantly affected by the maternal response to placenta-derived factors.
The major objective of this review is to discuss recent insights into the etiopathology of preeclampsia from the perspective of poor/ischemic placentation (see Figure 1).
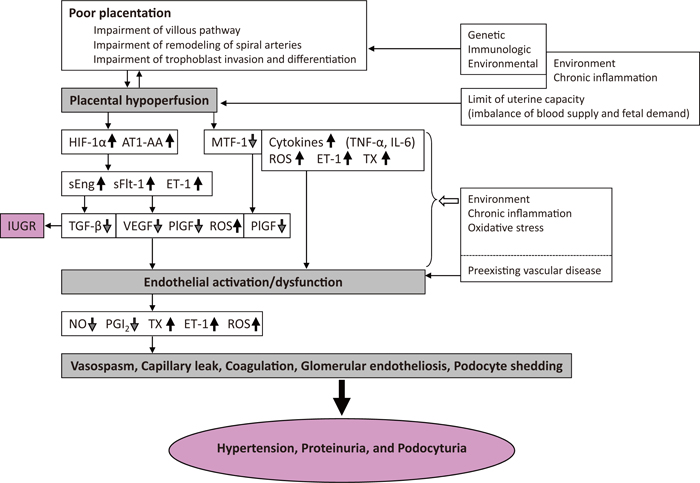
Possible pathogenic mechanisms underlying preeclampsia.
HIF-1α, hypoxia inducible factor-1α; AT1-AA, angiotensin II type 1 receptor agonistic antibody; sEng, soluble endoglin; sFlt-1, soluble fms-like tyrosine kinase 1; ET-1, endothelin-1; TGF-β, transforming growth factor-β; VEGF, vascular endothelial growth factor; PlGF, placental growth factor; NO, nitric oxide; PGI2, prostacyclin; TNF-α, tumor necrosis factor alpha; MTF-1, metal transcription factor-1; ROS, reactive oxygen species; IL-6, interleukin-6; TX, thromboxane.
During human placental development, trophoblast cells differentiate through two major pathways. In the villous pathway, cytotrophoblast cells fuse to form multinucleated syncytiotrophoblasts (STB). In the extravillous pathway, cytotrophoblast cells acquire an invasive phenotype and differentiate into either 1) interstitial extravillous trophoblasts (EVTs), which invade the decidua and a portion of the myometrium, or 2) endovascular EVTs, which remodel the maternal vasculature. These differentiation events are coordinated by the interplay of oxygen tension, transcription factors, hormones, growth factors, and other signaling molecules. miRNAs expressed in the placenta regulate trophoblast cell differentiation, proliferation, apoptosis, invasion/migration, and angiogenesis.7)
During early pregnancy, placentation occurs in a relatively hypoxic environment which is essential for appropriate embryonic development. Intervillous blood flow increases at around 10–12 weeks of gestation and results in exposure of the trophoblast to increased oxygen tension. This increase is temporally associated with downregulation of transforming growth factor-β3 (TGF-β3) levels and increased trophoblast invasion. Prior to this, low oxygen appears to prevent trophoblast differentiation towards an invasive phenotype.8)
Oxygen tension effects are mediated by hypoxia inducible factor-1 (HIF-1). The ontogeny of HIF-1α subunit expression during the first trimester parallels that of TGF-β3, an inhibitor of early trophoblast differentiation. Both molecules are highly expressed in early pregnancy and their levels fall at around 10 gestational weeks (GW) when Po2 levels are believed to increase. That is, physiological hypoxia promotes trophoblast proliferation rather than differentiation along the invasive pathway. Induction of HIF-1α by low Po2 (around 5–8 GW) may upregulate TGF-β3 expression, which in turn blocks trophoblast invasion.3)
As development continues, cytotrophoblasts invade the uterine wall and plug the maternal vessels, a process that helps maintain a state of physiological hypoxia. Cytotrophoblasts migrate further up arteries than veins. By 10 to 12 GW, blood flow to the intervillous space begins, and the increase in Po2 reduces HIF-1α expression, which in turn downregulates trophoblast TGF-β3 expression, thereby removing the inhibition of trophoblast differentiation along the invasive pathway. This allows trophoblast cells to complete their invasion through the uterus to the spiral arteries and results in successful pregnancy.8)
As the endovascular component of cytotrophoblast invasion progresses, the cells migrate along the lumina of spiral arterioles, replacing the maternal endothelial lining. Cytotrophoblasts are also found in the smooth muscle walls of these vessels. In normal pregnancy, the process whereby placental cells remodel uterine arterioles involves the decidua and inner third of the myometrial portions of these vessels. As a result, the diameter of the arterioles expands to accommodate the dramatic increase in blood flow needed to support rapid fetal growth later in pregnancy.9) In particular, greater dilation of the distal segment of spiral arteries than that of the proximal segment in normal pregnancy reduces the velocity of blood coming into the intervillous space, and the residual momentum carries the blood into the central cavity of the lobe, from which it disperses evenly through the villous tree.10)
Impairment of trophoblast invasion and remodeling of spiral arteriesFailure in placental response of the switching to oxygen tension during early pregnancy or a defect in the ability of the trophoblast to respond to this switch could result in an upregulation of both HIF-1α and TGF-β3 expression. Persistence of low oxygen tension promotes the release of HIF-1α. HIF-1α promotes the expression of TGF-β3, which can lead to decreased trophoblast differentiation and decreased invasion at the beginning of pregnancy.11) If the invasion is defective, the spiral arteries retain their musculo-elastic properties and responsiveness to vasoactive substances.12) The retention of smooth muscle will also increase the risk of spontaneous vasoconstriction and ischemia-reperfusion injury, generating oxidative stress.10) Blood via the incompletely remodeled spiral arteries will enter the intervillous space as a turbulent jet, damaging villous architecture and rupturing anchoring villi.10)
The impairments described above are thought to lead to placental ischemia and eventually to endothelial dysfunction,13) resulting in the clinical syndrome of preeclampsia. There is extensive evidence in the literature that the reduction of uteroplacental blood flow results in a combination of hypoxia, upregulation of placental HIF-1α, imbalance of angiogenic (vascular endothelial growth factor [VEGF], placental growth factor [PlGF]) and antiangiogenic (sFlt-1) factors, inflammation, deranged immunity, and increased endothelin-114) and AT1-AA15) expression. Placental hypoperfusion appears to be both a cause and a consequence of abnormal placental development.
Animal models that successfully reproduce at least hypertension have been generated by mechanically reducing uteroplacental blood flow.16,17,18) A pathogenic reduction in placental perfusion of pregnant baboons results in the development of preeclampsia and an increase in circulating sFlt-1, which is derived from both placental and extra-placental sources.17) Additionally, animal placental ischemia may be a stimulus for AT1-AA.19)
There are many risk factors for preeclampsia, such as chronic hypertension, obesity, diabetes mellitus, vascular and connective tissue disorders like systemic lupus erythematosus and antiphospholipid antibodies, and age >35 at first pregnancy.14) However, it is unclear why the normal sequence of events that lead to the development of uteroplacental circulation does not occur in some pregnancies. Taking these into consideration, environmental, immunological, vascular, and genetic factors all appear to play a role.20) Furthermore, obstetrical risk conditions that increase placental mass without correspondingly increasing placental blood flow (e.g. hydatidiform mole, hydrops fetalis, twin gestation, heavy for date baby) result in relative ischemia and are associated with preeclampsia.21)
It is important to note that placental hypoperfusion becomes more pronounced as pregnancy progresses since the uterine vasculature may be unable to accommodate the normal increase in blood flow to the fetus/placenta with advancing gestational age.22,23) Until late pregnancy, a certain balance is kept between the increasing intrauterine volume and uterine wall tension arising from the uterine muscle, and that balance maintains the uterine resting tonus at almost a constant level. However, when the intrauterine volume reaches a critical point after which hypertrophy of the muscle fibers can no longer keep up with the increase in intrauterine volume, i.e., when the uterine capacity approaches its limit, the uterine wall is further stretched. This naturally causes elevation of the intrauterine resting tonus followed by placental and renal ischemia via the utero-renal reflex and blood pressure elevation. This may reflect late-onset preeclampsia.18,24) According to a very recent study supporting a similar viewpoint, when placental growth reaches its limit (uterine capacity) at term, terminal villi become overcrowded with diminished intervillous pore-size impeding intervillous perfusion with increasing intervillous hypoxia and STB stress (e.g. oxidative stress, inflammatory stress, endoplasmic reticulum stress, etc).25)
As a physiological response to these vital changes in late pregnancy, the uterus becomes round, resulting in shortening of the uterine longitudinal length (according to Laplace’s theorem), a clinically “lightening phenomenon”.18,24) Thus, it may be speculated that even a growing pregnant uterus has the limit of the growth potential (uterine capacity). When the uterine capacity approaches its limit, the intrauterine resting tonus rises in relation to the cervical factor (the unripe cervix), which reduces placental perfusion, and blood pressure rises soon thereafter. If delivery does not occurred, late onset preeclampsia will be attacked.
Placental ischemia is more remarkable in the supine position compared with the lateral, due to aortocaval compression by the heavy round tense uterus in the supine position followed by supine hypertension rather than hypotension. Utero-placental venous return is also disturbed by the pregnant uterus, which subsequently causes the “sluice flow” mechanism to appear in the feto-placental circulation.26)
As a result of placental ischemia in rabbits, there is a loss of refractoriness in women destined to develop preeclampsia, and pregnant vascular refractoriness depends on endothelial-derived relaxant factors (EDRFs).27,28) Increased production of nitric oxide (NO) is more related to the vascular refractoriness to angiotensin II during normal pregnancy than to prostacyclin (PGI2).29,30)
Placental ischemia results in the release of a variety of placental factors that profoundly affect blood flow and blood pressure regulation. These factors include molecules such as sFlt-1 and AT1-AA and cytokines such as tumor necrosis factor alpha (TNF-α). These factors cause widespread dysfunction in the maternal vascular endothelium. This dysfunction manifests itself in the enhanced production of factors such as endothelin-1, reactive oxygen species (ROS), thromboxane, and 20-hydroxyeicosatetraenoic acid, as well as augmented vascular sensitivity to angiotensin II.31)
The clinical features of preeclampsia can be explained as clinical responses to generalized endothelial dysfunction.32,33) Serum from preeclamptic women causes endothelial activation in human umbilical vein endothelial cells in vitro.34)
Impaired endothelial function can be demonstrated by brachial artery flow-mediated dilation three years after a preeclamptic pregnancy.35) It is unknown whether this is a cause or effect of the preeclamptic pregnancy.
The association between preexisting vascular disease and susceptibility to developing preeclampsia may be due to preexisting endothelial cell damage.36) Preexisting endothelial damage may also explain why women who develop preeclampsia are also at increased risk of developing cardiovascular disease later in life.37,38) Women with a history of preeclampsia are also at increased risk for end-stage renal disease and hypothyroidism in the long term.39,40)
A variety of proangiogenic (VEGF, PlGF) and antiangiogenic (sFlt-1) factors are secreted by the developing placenta, and the balance among these factors is important for normal placental development. Increased production of antiangiogenic factors disturbs this balance and results in the systemic endothelial dysfunction characteristic of preeclampsia.
sFlt-1, VEGF, and PlGFsFlt-1, or soluble vascular endothelial growth factor receptor-1 (sVEGFR-1), is a naturally occurring, circulating antagonist of VEGF. VEGF is an endothelial specific mitogen that has a key role in promoting angiogenesis.41) Its activities are mediated primarily by interaction with two high-affinity receptor tyrosine kinases, VEGFR-1 (VEGF receptor-1 or fms-like tyrosine kinase-1 [Flt-1]) and VEGFR-2 (kinase-insert domain region [KDR]/Flk-1), which are selectively expressed on the vascular endothelial cell surface.
VEGFR-1 has two isoforms: a membrane-spanning isoform and a soluble isoform (sFlt-1 or sVEGFR-1). PlGF is another member of the VEGF family that is expressed predominantly in the placenta. It also binds to the VEGFR-1 receptor. sFlt-1 antagonizes the proangiogenic activity of circulating VEGF and PlGF by binding to them and preventing their interaction with their endogenous receptors. Increased placental expression and secretion of sFlt-1 appear to play a central role in the pathogenesis of preeclampsia, based on the following observations.42,43,44,45,46,47,48,49,50)
Elevation of plasma sFlt-1 by infecting rats with an adenoviral vector carrying sFlt-1 results in hypertension and proteinuria resembling human preeclampsia. Histologically, kidneys from these animals show glomerular endotheliosis.42) The most likely trigger for increased sFlt-1 production by the placenta is placental ischemia.6)
sFlt-1 levels increase during pregnancy in all women. However, compared to normotensive controls, the increase begins earlier in gestation and reaches higher levels in women who may soon develop preeclampsia. Free PlGF and VEGF levels fall concurrently with the rise in sFlt-1, which may be related, in part, to binding by sFlt-1.47) Early- and late-onset preeclampsia are associated with altered plasma levels of sFlt-1 and PlGF. The alterations are more pronounced in early-onset, relative to late-onset, preeclampsia.45,47,50,51)
As for the relationship between placental lesions and the level of angiogenesis in preeclampsia, the most common pathohistological findings in the placenta of patients with preeclampsia are lesions consistent with villous changes and vascular lesions (according to the criteria of the Society for Pediatric Pathology).52) The earlier the onset of preeclampsia, the higher the frequency of placental lesions consistent with placental hypoperfusion. This phenomenon appears to be a continuum.53) Patients with late-onset preeclampsia and histological findings consistent with placental hypoperfusion have a significantly lower median plasma concentration of PlGF and PlGF/sFlt-1 ratio than those with late-onset preeclampsia without placental hypoperfusion lesions.52) On the other hand, no difference is observed in the levels of VEGF, PlGF, sFlt-1, and HIF-1α in a comparison of early-(≤34 GW) and late-onset preeclampsia.54,55) Differences in these results might be attributable to differences in the distribution rate of disease severity, as well as different definitions of the onset time of early and late preeclampsia.
The observation of low and high PlGF levels56) and, similarly a high sFlt-1/PlGF ratio ≥85 (angiogenic preeclampsia) and a low sFlt-1/PlGF ratio <85 (non-angiogenic preeclampsia),57) across pregnancy among women who develop preeclampsia may be an important finding in relation to the pathophysiology and study of preeclampsia. Perhaps these different patterns of PlGF indicate that half of the preeclampsia cases result from insufficient angiogenic signaling (low PlGF), whereas the other half result from insensitivity to angiogenic signaling (high PlGF) or are determined by a different pathogenic factor.56) These findings suggest the possibility of at least two kinds of predisposing factors. One may lead to the overproduction of sFlt-1, such as multiple pregnancy, hydatidiform mole, trisomy 13, and first pregnancy.58,59) The second set of predisposing factors may include disorders that sensitize the maternal vascular endothelium to sFlt-1 and thus lower the threshold of developing preeclampsia. Such factors may include obesity, diabetes, renal disease, and preexisting vasculitis.59,60) In particular, overweight women with preeclampsia appear to have lower levels of adiponectin and sFlt-1, and higher levels of PlGF than women with preeclampsia who are of normal weight.61) The proposed underlying mechanism of preeclampsia is placental hypoxia-driven imbalances in angiogenic and antiangiogenic factors, including sFlt-1, PlGF, VEGF, TGF-β, soluble endoglin (sEng), and others, resulting in endothelial dysfunction. Although abnormalities in these factors have been reported, there is no discernible pattern that characterizes this disorder. This suggests that a defect in an upstream regulator may contribute to the pathogenesis of preeclampsia.
Pregnant mice deficient in catechol-O-methyltransferase (COMT), a key enzyme in the degradation of both catecholamines and estrogens, show a preeclampsia-like phenotype resulting from an absence of 2-methoxyoestradiol (2-ME), a natural metabolite of oestradiol that is elevated during the third trimester of normal human pregnancy. 2-ME ameliorates all preeclampsia-like features without toxicity in COMT(-/-) pregnant mice and suppresses placental hypoxia, HIF-1α expression, and sFlt-1 elevation. The levels of COMT and 2-ME may be significantly lower in women with severe preeclampsia and may correlate with elevated sFlt-1 levels.62)
Yet, severe early-onset preeclampsia is not associated with a decrease in placental COMT expression.63) Moreover, in late-onset preeclampsia, 2-ME levels are significantly increased compared to those in normal pregnant women at term. There is no significant difference in placental COMT expression between the two groups. Increased levels of 2-ME in patients with late-onset preeclampsia might be a compensatory mechanism in patients with late-onset preeclampsia.64) Whether the decreased COMT is the cause or consequence of abnormal placentation remains unclear. Moreover, the mechanisms by which glomerular endotheliosis, proteinuria, and podocyturia (glomerular epithelial cells) develop in the COMT(-/-) mouse model remain unknown.65) Further research is needed to better assess the role of this pathway in human disease.
Influence of placental hypoxia on antiangiogenesisPersistent placental hypoxia induces the expression of HIF-1α and AT1-AA, which promote the production of sFlt-1, sEng, and endothelin-1. These factors lead to endothelial dysfunction and the clinical manifestations of preeclampsia.15)
Hypoxia has been shown to be a major inducer of VEGF gene transcription.41) PlGF expression is decreased in trophoblasts under hypoxia in vitro66) and preeclampsia.67) HIF-1α is a transcription factor responsible for the regulation of VEGF, but not of PlGF. Metal transcription factor-1 (MTF-1) expression decreases under hypoxia in the trophoblast, trophoblast-derived cells, and preeclamptic placenta. A precise molecular mechanism of PlGF regulation via MTF-1 under hypoxic conditions in trophoblasts is also recognized. Collectively, these findings suggest that MTF-1 plays an important role in the regulation of PlGF in the trophoblast and that low serum levels of PlGF in patients with preeclampsia may come from the decrease in MTF-1 levels.68)
Soluble endoglin (sEng)Endoglin (Eng) is a coreceptor for transforming growth factor beta (TGF-β) and is highly expressed on cell membranes of the vascular endothelium and syncytiotrophoblasts.69) A novel placenta-derived soluble form of Eng, referred to as sEng, is an antiangiogenic protein that appears to be another important mediator of preeclampsia.70,71)
sEng inhibits the binding of TGF-β to its receptor, thereby inhibiting the downregulation of nitric oxide synthase.72) Thus, the combination of increased sFlt-1 and sEng levels appears to impair NO generation and activate endothelin-1 signaling with hypertension, resulting in maternal endothelial dysfunction.73)
The plasma concentration of sFlt-1 protein needed to produce the preeclampsia phenotype in rats was several fold higher than levels typically seen in patients with preeclampsia, and no coagulation or liver function abnormalities were reported in sFlt-1 treated animals.42)
Both sEng and sFlt-1 contribute to the pathogenesis of preeclampsia through separate mechanisms. sEng increases vascular permeability and induces hypertension in vivo. In pregnant rats, sEng appears to potentiate the vascular effects of sFlt-1 to induce a severe preeclampsia-like state, including the development of HELLP syndrome and restriction of fetal growth.69)
Fetuses of preeclamptic mothers do not have high circulating concentrations of either sEng74) or sFlt-1.75) This suggests that fetuses do not experience proteinuria or hypertension like their mothers, as they are not exposed to high concentrations of antiangiogenic factors.
Increased sensitivity to angiotensin II has been described in preeclampsia,76) and may be related to bradykinin (B2) receptor upregulation in preeclamptic patients. This upregulation leads to heterodimerization of B2 receptors with AT1 receptors, and this AT1/B2 heterodimer increases the responsiveness to angiotensin II in vitro.77)
Placental hypoxia leads to elevation of circulating AT1-AA,15) and patients with preeclampsia have increased levels of agonistic antibodies to the AT1 receptor. Angiotensin II is the endogenous ligand for the AT1 receptor, and thus increased activation of this receptor by autoantibodies could induce the hypertension and vascular injury observed in preeclampsia.78) Moreover, AT1-AA stimulates sFlt-1 secretion,79) and AT1-AA binds to endothelial and vascular cells, causing endothelial damage and vasoconstriction.80)
Preeclampsia typically manifests after 20 weeks gestation. However, the underlying mechanisms are likely to be triggered much earlier. This dichotomy of pathophysiological events has been proposed to represent a two-stage process: poor placental perfusion secondary to abnormal implantation and the development of the placental vasculature (Stage 1) leading to maternal responses evoked with varying degrees of clinical manifestations (Stage 2).81)
Recently, the Two Stage Model has been modified,82) based on the hypothesis that abnormal implantation/placentation occurs before abnormal vascular remodeling of spiral arteries.83) The hypothesis is as follows. 1) If the very first differentiation event of the trophoblast cell is affected during development from morula to blastocyst, this may result in a severe general defect of the trophoblast cell. This, in turn, may lead to a combination of preeclampsia and intrauterine growth restriction (IUGR) or even more severe outcomes such as spontaneous abortions. On the other hand, if the insult takes place slightly afterwards, when the blastocyst and trophoblast differentiates into the first cytotrophoblast and syncytiotrophoblast, the same dramatic outcome as described above may result. 2) If only the differentiation of the extravillous trophoblast pathway is affected, this may result in pure IUGR with typical characteristics such as failed invasion. 3) If only the villous pathway is affected, then preeclampsia may result with its typical characteristics such as release of syncytiotrophoblast fragments (STBM) and a maternal inflammatory response. In addition, maternal constitutional factors (genetic, behavioral, or environmental) may play a more extensive role than previously thought.
Clinically, IUGR is also accompanied by reduced placental perfusion. Despite the reduced perfusion, most women with IUGR infants do not necessarily develop preeclampsia. It is proposed that reduced placental perfusion, posited as secondary to failed remodeling of the maternal vessels supplying the intervillous space, is not sufficient to cause preeclampsia (Stage 2).84) Accordingly, Stage 2 of preeclampsia may also require maternal susceptibilities to factors secondary to reduced placental perfusion. The susceptibility is influenced by “maternal constitutional factors” such as obesity, hypertension, dyslipidemia, abnormal endothelial function, and diabetes, which result in increased sensitivity of the mother to the consequences of reduced placental perfusion.
Factors linking the two stages of preeclampsia, in addition to antiangiogenesis and AT1-AA, include the following factors released from the hypoxic placenta: activated blood components, excess STBM, activating immune cells and transmitting oxidized lipids, and excess inflammatory cytokines.82)
Another possible linking factor is the material released from the fetal-placental unit in order to overcome reduced placental perfusion and the subsequent reduced delivery of nutrients. Perhaps the placental-to-maternal linkage is a factor that modifies maternal metabolism to increase nutrient availability, thus acting on the placenta to facilitate nutrient transfer. Women who could not tolerate this modification would be much more likely to develop preeclampsia. Whether there are fetal/placental signals that increase nutrient availability is unknown.82)
Redman and Sargent85) also reviewed the two-stage model: poor placentation (Stage 1) and placental oxidative stress and inflammation (Stage 2). All inflammatory changes in normal pregnancy are exaggerated in preeclampsia and preeclampsia is not only an endothelial disease but the consequence of a wider systemic inflammatory response. The primary placental problem that leads to preeclampsia syndrome is likely to be oxidative stress rather than hypoxia.86) Oxidative stress is an inflammatory stimulus mediated by ROS. This provokes the release of sFlt-1 and possibly sEng via nuclear factor-kappa B as much as or more than hypoxia.86)
Two different types of preeclampsia exist. The early-onset type is a placental disease, whereas the late-onset type is a maternal (maternal reactions to the burden of pregnancy), rather than a placental, disease.87) There is also a mixture of conditions, ranging from mild preeclampsia with moderate placental involvement to gestational hypertension without placental dysfunction.88)
Late-onset preeclampsia is frequently associated with fetuses that are appropriate- or large-for gestational age,18,89) multiple gestation, gestational diabetes mellitus (GDM), and primigravida. In these cases, increased fetal demand for substrates that surpasses the placental ability to sustain fetal growth may induce fetal signaling for placental overproduction of antiangiogenic factors and subsequent ‘compensatory’ maternal hypertension. It is possible that relative uteroplacental ischemia due to a mismatch between limited uteroplacental blood flow and an increased fetal demand for nutrients may be central to the development of late-onset preeclampsia.5) On the other hand, late-onset preeclampsia may be regarded as a consequence of disrupted maternal homeostasis. That is, when the uterine capacity approaches its limit in late pregnancy,18,25) labor pains appear in order to maintain homeostasis. However, if labor pains do not appear, blood pressure elevates as a maternal warning sign that homeostasis is disrupted.18)
None.