Abstract
Background: Undifferentiated pleomorphic sarcoma (UPS) is a pleomorphic variant of undifferentiated, aggressive, and heterogeneous mesenchymal malignancy. Patient-derived cancer models comprise an invaluable preclinical tool for developing novel therapeutic strategies, although owing to the diversity in UPS, multiple cell lines are required for research of this disease. Therefore, we aimed to establish novel patient-derived xenografts (PDXs) from the tumor tissue of a patient with UPS, along with a cell line from an established PDX. Methods: The biological characteristics of the established cell line such as morphology, growth rate, colony formation capacity, and immunohistochemical characteristics were determined. Proteomic features were assessed by mass spectrometry. Results: The tumor tissues of PDXs showed a similar histological appearance to that of primary tumor tissue. The cells grew at a sufficient rate for conventional experiments. The cells exhibited colony-forming ability and expressed a typical marker of UPS. The mass spectrometric protein expression profiling revealed a unique spectrum of functions from the original tumor tissue, through patient-derived xenograft, to the cell line. The use of the established xenograft and cell line will facilitate our understanding of the molecular mechanisms underlying poor prognosis of UPS, and will contribute to the development of novel therapeutic strategies.
Introduction
Undifferentiated pleomorphic sarcoma (UPS) constitutes a heterogeneous and aggressive subgroup of sarcoma1). The clinical outcome remains dismal and the five-year survival rate is 65–70%2). The curative treatment for UPS is surgical resection with consideration of adjuvant treatment according to the surgical margins, location of tumor, and tumor-associated risk factors. UPS exhibits marked resistance to radiotherapy and chemotherapy. At present, a common standard treatment is administered for all patients with UPS, whereas individualized strategies should be developed owing to the heterogeneity of UPS3,4,5). As is the case for other sarcomas, large clinical trials to evaluate the efficacy of novel treatments have not yet been conducted for UPS1). To address to the limited number of patients with UPS and the clinical and pathological diversity of this disease, preclinical model systems are required to examine novel therapeutic approaches.
Patient-derived xenografts (PDXs) and cell lines are invaluable tools to understand the complex malignant features and the clinical and pathological diversity of tumors at the molecular level. The standard procedure of creating PDXs is to section fresh surgically resected tumor tissues into small pieces, which are then implanted into immune-deficient mice. Once the tumor tissue is sufficiently enlarged, it is transplanted to other mice. After several passages, the tumor tissues in PDXs exhibit rapid and permanent growth. The tumor tissue can then be frozen in liquid nitrogen until use. Subsequently, cell lines can be established from the PDX tumors. Such cell lines for UPS, previously termed malignant fibrous histiocytoma, were established in prior studies6,7,8,9,10,11), and patient-derived xenografts of UPS were also reported12,13,14). These models are useful resources to examine the effects of anti-cancer drugs. However, to address to the clinical and biological diversity of the disease, additional patient-derived cancer models are necessary in UPS.
Here, we report a novel PDX established from the surgical specimen of a patient with UPS, as well as a cell line that was established from the PDX. The histological appearance of the tumor tissue, the properties of the cell line, and global protein expression were examined to characterize this patient-derived model.
Materials and Methods
This study utilized tumor tissue from a patient with UPS. The use of clinical materials in this study was approved by the ethics committee of the National Cancer Center, Tokyo, Japan. Written informed consent was obtained from the patient. The protocol of animal experiments was in accordance with the guidelines for Animal Experiments of the National Cancer Center and approved by the Institutional Ethics Review Committee for Animal Experimentation. In a previous report, we established a cell line directly from the tumor tissue; detailed patient data was described therein15).
Patient information
The patient-derived xenografts were established from the tumor tissue of an 83-year-old male patient with UPS, who was admitted to the National Cancer Center Hospital, Tokyo, Japan. The patient exhibited soft tissue mass in his scapular back 2 months prior to being referred to the hospital. Pathological examination by needle biopsy was consistent with pleomorphic spindle cell sarcoma. Wide resection via pedicled latissimus dorsi flap surgery was performed four weeks after the initial diagnosis. Pathological examination of resected tumor tissues yielded a diagnosis of UPS. At four weeks after surgery, the patient developed multiple lung metastases with right pleural effusion and died of disease six weeks after surgery.
Histological observation
Histological observations were performed on the tumor tissues, which were sectioned from a representative paraffinized block of the tumor to four-micrometer-thick sections. Then, the tissue sections were deparaffinized and stained with hematoxylin and eosin (HE).
Establishment of PDXs and cell lines
The surgically excised tumor was cut into 2–3 mm in pieces (approximately 200 mm3), and implanted subcutaneously using a 13-gauge transplant needle to the hind bilateral flanks of 6- to 12-week-okd female severe immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Sug/Jic (NOD/Shi-scid, IL-2Rnull or NOG) mice (Central Institute for Experimental Animals, Kanagawa, Japan). When the tumors grew up to sufficient size (500–1,000 mm3), they were transplanted to additional mice. Tumor size was measured periodically using a digital caliper (SuperCaliper, Mitutiyo, Kanagawa, Japan). Tumor volume was calculated as 6/pie × length × width × thickness16). After two passages, the tumors were minced into 8 mm3 with seizer, and soaked in CELLBANKER 1 plus (TAKARA BIO Inc., Osaka, Japan) at –80ºC overnight. Then tumor tissues were cryopreserved in liquid nitrogen tanks. We considered that the PDX was established when the engraftment of the cryopreserved tissue after quickly thawing at 37ºC was confirmed by visual inspection. Histological observations were performed on the sectioned tissues using HE staining.
The PDX tumor tissues were then minced with scissors and transferred to a culture dish. The cells were maintained in RPMI 1640 medium supplemented with 5% (or 10%) fetal bovine serum (FBS), 100 U/mL penicillin G, and 100 mg/mL streptomycin (Invitrogen Co., Carlsbad, CA), in a humidified atmosphere containing 5% CO2 at 37ºC. When the cultured cells reached confluence, they were dispersed with a 0.25% trypsin solution and seeded in another dish. The passaging was serially performed more than 30 times until a cell line was established.
Authentication of the PDX and cell line
For authentication of the established PDX and cell line, short tandem repeats (STRs) were examined by PCR. Genomic DNA was extracted from the excised tissue or cell lines using DNeasy blood and tissue kits (Qiagen, Hilden, Germany) or AllPrep DNA/RNA mini kits (Qiagen). DNA concentrations were measured using a NanoDrop 8000 (Thermo Fisher Scientific Waltham, MA), and DNA samples were stored at –20ºC until use. PCR amplification was performed for 10 loci (Supplementary Table 1). The PCR products were separated using an ABI 3130 or 3500xL Genetic Analyzer (Applied Biosystems, Waltham, MA). STR multiplex assays were carried out using GenePrint 10 (Promega, Madison, WI), and the data analysis was accomplished using GeneMapper 5.0 (Applied Biosystems) or PeakScanner (Applied Biosystems) programs. The STR profiles were compared between the original tumor tissues, tumor tissues of PDXs, and established cell lines. Moreover, the STR profiles of cell lines were compared with those from the ATCC, DSMZ, or JRCB databases for reference matching.
Mycoplasma contamination screening
Mycoplasma DNA was examined to determine the presence of contamination. Tissue culture medium was examined according to the guideline from The International Cell Line Authentication Committee17). When the cells reached 70–90% confluence, DNA was recovered from the culture medium and amplified using an e-Myco Mycoplasma PCR Detection Kit (Intron Biotechnology, Gyeonggi-do, Korea). The PCR products were separated by 1.5% agarose gel electrophoresis and detected using Midori Green Advanced (Nippon Genetics, Tokyo, Japan). The image data were recorded and analyzed using an Amersham Imager 600 (GE Healthcare Biosciences, Chicago, IL).
Cell growth and characteristics
The established cells were seeded into 96-well culture plates at various cell numbers in triplicate. Cell numbers were counted at several time points over a 120 h period of incubation at 37ºC. Cell counting Kit 8 (CCK-8) reagent (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was added to each well, and after incubation for 2 h, absorbance values were measured at a wavelength of 450 nm using a microplate reader (Bio-Rad, Hercules, CA). Cell culture doubling population time was calculated within a logarithmic growth phase of an exponential fitting curve.
Colony formation assay
The bottoms of 35-mm dishes were coated with 0.5% low melting agarose (Noble agar, Sigma Aldrich, St. Louis, MO), with DMEM supplemented with 10% FBS and penicillin streptomycin (Thermo Fisher Scientific, San Jose, CA). Then the bottoms of wells were covered with 0.33% agarose containing 5 × 104 cells. After incubation at 37ºC for 4 weeks, colonies containing more than 50 cells were confirmed by microscopic observation (ZEISS Primo Vert Inverted Microscope, Carl Zeiss, Jena, Germany). All assays were performed in duplicate.
Immunohistochemical verification
The cells were suspended, solidified using iPGell (Genostaff, Tokyo, Japan), and cell-blocks were created. The cell-blocks were fixed with 10% formalin neutral buffer solution, embedded in paraffin, and four-micrometer-thick paraffin sections were prepared. The sectioned tissues were stained with HE or reacted with specific antibodies according to a previous study18). The primary antibodies used were mouse monoclonal antibodies; and antibodies against α-smooth muscle actin (SMA; 1: 100; 1A4; Dako, Glostrup, Denmark), cytokeratin (1: 100; AE1/AE3; Dako), desmin (1: 100; D33; Dako), and myogenin (1: 100; F5D; Dako). A rabbit polyclonal antibody against S-100 (1: 2000; Dako) was also used. The reactions were detected using the EnVision system (Dako).
Mass spectrometric protein expression profiling
Proteins were extracted from original tumor tissues, PDX tumor tissue, and the cell line using urea lysis buffer (6 M urea, 2 M thiourea, 3% CHAPS, 1% Triton X-100) according to our previous report19). The protein samples (25 μg) were processed into tryptic digests using the filter-aided sample preparation (FASP) method20). The tryptic digests were subjected to liquid chromatography coupled with nanoelectrospray tandem mass spectrometry (Finnigan LTQ Orbitrap XL mass spectrometer; Thermo Fisher, Waltham, MA). The Mascot software package (version 2.5.1; Matrix Science, London, UK) was used to search for the mass of each peptide ion peak against a SWISS-PROT database (Homo sapiens, Swiss prot_2015_09.fasta file containing 20,205 sequences) using the following parameters: 1 missed cleavage; variable modifications: oxidation (Met), phosphorylation (ST), phosphorylation (Y); peptide tolerance: 10 ppm; MS/MS tolerance: 0.8 Da; peptide charge: 2+ and 3+. Every sample was subjected to mass spectrometry five times, and the identified peptides and proteins were accumulated.
Data analysis
The R package “treemap” was used to illustrate the results of functional classification of identified proteins21). The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used to classify gene sets into their respective pathways22). Significance was indicated by the p-value for each category, and the process groups with p < 0.05 were considered significant. The KEGG pathway annotations for each protein group in our proteome data were inferred using Database for Annotation, Visualization and Integration Discovery (DAVID) software (http://david.abcc.ncifcrf.gov/).
Results
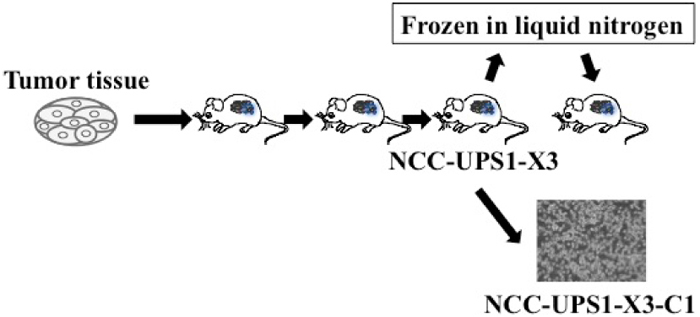
Workflow of establishment of a patient-derived cancer model
The workflow of experiments in this study is illustrated in Figure 1. The tumor tissue sections were inoculated onto the skin of mice, and when each reached a proper size, it was transplanted to another mouse. The tumor tissue of the third generation was frozen in liquid nitrogen, and inoculated to an additional mouse skin. We termed this third generation PDX model as NCC-UPS1-X3. The cell line was established from the third generation PDX tumor, and was termed as NCC-UPS1-X3-C1. The tumor cells were not isolated from the cell population by cloning. As the patient did not receive neo-adjuvant treatment, there was no possible effect of either chemotherapy or radiotherapy on the tumor from which the PDX was established.
Characteristics of the established PDX model
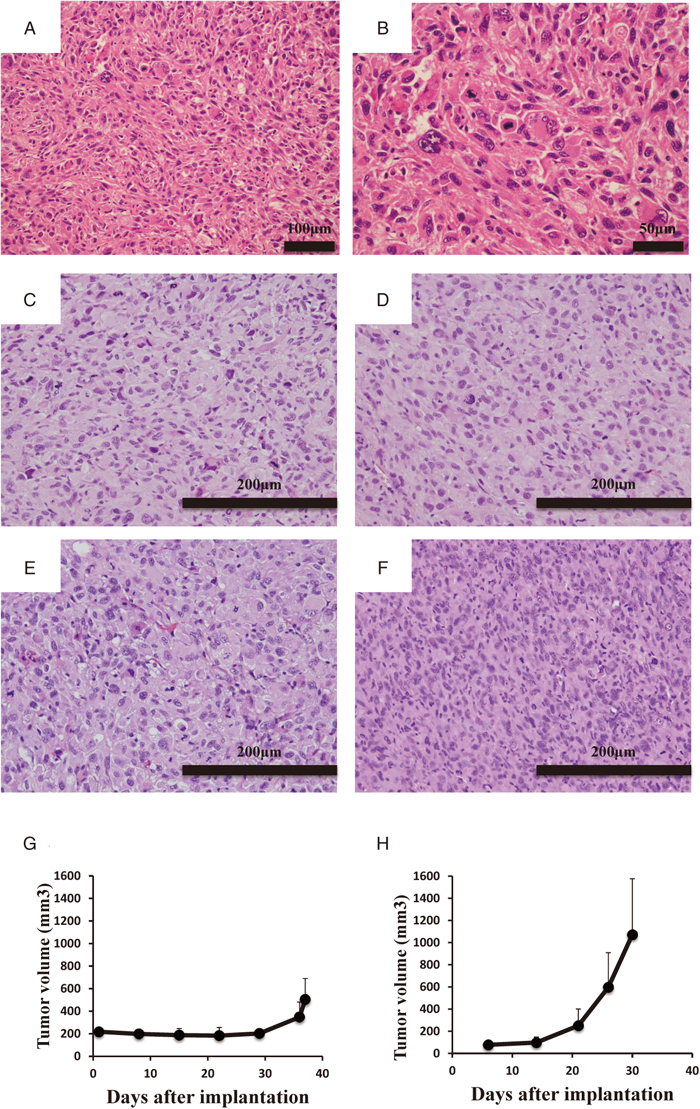
Figure 2A and B show the typical images of the resected tumor tissue from the patient with UPS in this study. The tumor tissue consisted of pleomorphic spindle cells and exhibited histological appearance that supported the diagnosis of UPS. The diffuse expression of SMA and negative expression of AE1/AE3, S100, desmin, caldesmon, myogenin, MDM2, and CDK2 was observed by immunohistochemical study (data not shown), being also consistent with the diagnosis of UPS.
We established a PDX model from the tumor tissue of a patient with UPS. The histological appearance of tumor tissues of different generation PDXs are shown in Figure 2C–F. The histological appearances of tumor tissues of PDXs from different generations were similar to those of the original tumor tissue (Figure 2A–F).
Growth status of the engrafted tumor tissues in mice was also examined. The growth curve of the PDX tumors in the first generation is shown in Figure 2G. At more than 30 days after implantation, the tumor sizes started to increase. The growth curve of PDX tumor tissues in the third generation is demonstrated in Figure 2H. The tumor size started to increase two weeks after transplantation, with the periods required for a tumor to reach 500 mm3 being apparently shorter than those of first generation tumors.
Authentication of the PDX and cell line and cytological assay
To confirm the identification of the origin of PDX tumors and the established cell line, we examined the status of STRs of ten microsatellites. We found that all of STRs except vWA (20) were identical to the other samples and to the surgically resected original tumor (Table 1). Through database search, we did not find any cell lines with the same STR patterns. We concluded that the PDX model NCC-UPS1-X3 and NCC-UPS1-X3-C1 cells comprised novel UPS models derived from the patient with UPS.
Table 1. Results of STR analysis
Microsatellite
(Chromosome) | Normal Tissue | Tumor Tissue | NCC-UPS1-X3 | NCC-UPS1-X3-C1
(P59) |
|---|
| Amelogenin (X Y) | X,Y | X,Y | X,Y | X,Y |
| TH01 (3) | 6,7 | 6, 7 | 6, 7 | 6,7 |
| D21S11 (21) | 28.2,29 | 28.2,29 | 28.2 | 28.2 |
| D5S818 (5) | 11 | 11 | 11 | 11 |
| D13S317 (13) | 9,12 | 9,12 | 12 | 12 |
| D7S820 (7) | 11 | 11 | 11 | 11 |
| D16S539 (16) | 9,12 | 9,12 | 9, 12 | 9,12 |
| CSF1PO (5) | 10,11 | 10,11 | 11 | 11 |
| vWA (12) | 16,20 | 16,20 | 20 | 20 |
| TPOX (2) | 8 | 8 | 8 | 8 |
Passage number of the cells were indicate.
We confirmed the absence of mycoplasma contamination by PCR. Mycoplasma-specific DNAs were not detected in conditioned media from the NCC-UPS1-X3-C1 cell culture (data not shown). We concluded that the cell line was free of mycoplasma contamination.
Phenotypic characterization of tissue cultured cells
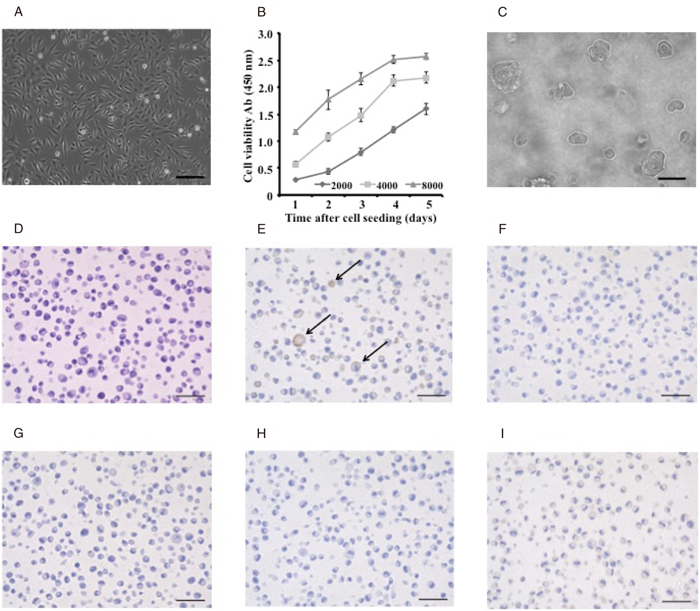
Morphological characterization was performed via microscopic observations. NCC-UPS1-X3-C1 cells were spindle, round, or polygonal in shape (Figure 3A). The growth status of NCC-UPS1-X3-C1 cells was examined (Figure 3B). Population doubling time was approximately 40 h. Colony formation capabilities of NCC-UPS1-X3-C1 cells were confirmed on soft agar plates (Figure 3C).
For immunocytochemical study, NCC-UPS1-X3-C1 cells were embedded in paraffin (Figure 3D) and the cells were reacted with antibodies against histological marker proteins. We found that the NCC-UPS1-X3-C1 cells were positive for smooth muscle actin (Figure 3E) but negative for cytokeratin (Figure 3F), S100 (Figure 3G), desmin (Figure 3H), and myogenin (Figure 3I).
Proteomic study
The protein expression profiles of primary tumor tissue, tumor tissue of the PDX model NCC-UPS1-X3, and NCC-UPS1-X3-C1 cells were obtained by mass spectrometry. The peptides obtained from protein identification are summarized in Supplementary Tables 1–3. To increase the proteome coverage, we subjected the same samples five times to mass spectrometry. We found that unique proteins were obtained each time; the relationship between the injection times and the increased number of proteins are illustrated in the format of a Venn diagram in Figure 4. As a consequence of repeated injection, we observed 1481, 1569, and 1693 proteins in primary tumor tissue (Figure 4A), tumor tissues of PDX model NCC-UPS1-X3 (Figure 4B), and the cell line NCC-UPS1-X3-C1 (Figure 4C) cells.

The protein expressions of tumor cells should be affected by the environment changes. To understand the alterations of molecular pathways during the process of model establishment, we performed the treemap analysis of KEGG pathways (Figure 5). The proteins are assigned to certain KEGG pathways and each treemap consists of unique KEGG pathways. The columns are colored according to the enrichment score of pathways, and the size of column is determined according to the number of proteins in that pathway. Figure 5 depicts a functional classification of identified proteins in a treemap format. Each treemap includes KEGG pathways with adjusted p-value < 0.05. Supplementary Table 4 summarizes the annotations of the proteins used in the treemaps. “Focal adhesion”, “Pathogenic Escherichia coli infection”, and “Ribosome” were mostly enriched in the original tumor tissues (Fig. 5A). “Ribosome”, “Proteasome” and “Pathogenic Escherichia coli infection” were mostly enriched in both PDX tumor tissue and cell line samples (Fig. 5B and C). “Ribosome”, “Pathogenic Escherichia coli infection” and “Proteasome” were commonly enriched in all samples (Fig. 5D). The enrichment scores for “Focal adhesion”, “ECM-receptor interaction”, “Pathogenic Escherichia coli infection”, “Complement and coagulation cascades”, and “Glycolysis/Gluconeogenesis” were decreased in the PDX tumor and cell line samples compared to those in the original tumor whereas the enrichment scores for “Ribosome” and “Aminoacyl-tRNA biosynthesis” were increased in the PDX tumor and cell line samples compared to those in the original tumor.
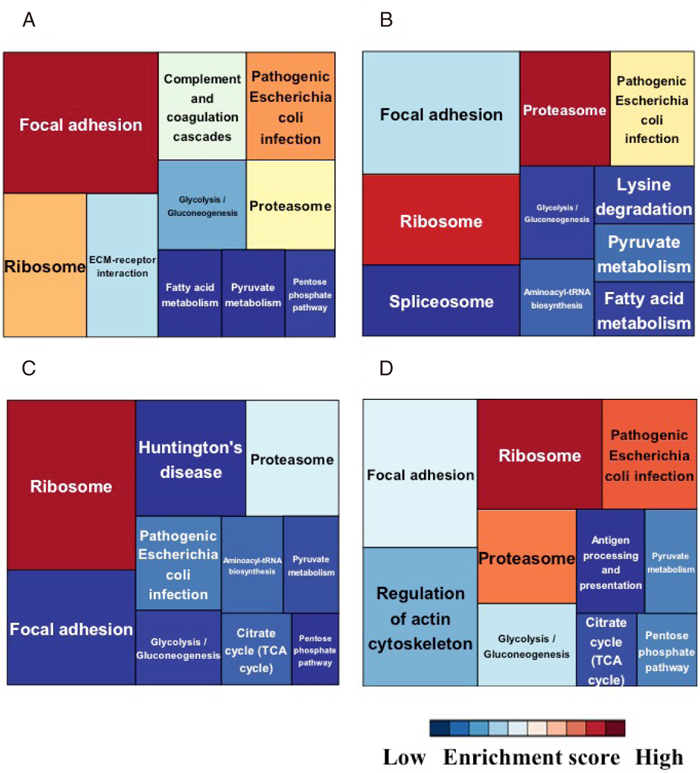
Based on observation of the treemap results, we found a suggestive spectrum of pathways, which changed during the process of model establishment. We categorized the enriched pathways according their possible significances (Table 2). In Table 2, the KEGG pathways are labeled with the ranking orders that were determined according to the enrichment score; the pathways with the higher enrichment score are highly ranked. We found a spectrum of pathways among the samples from tumor tissues, PDX tumor tissues, and the established cell lines. Pathways in Group A were consistently common in all samples. Pathways in Group B were common in all samples, but not consistent between PDX tumors and cell lines. Pathways in Group C were observed only in the original tumor tissue and PDX tumor tissues. The pathway in Group D was enriched in the original tumor tissue and the cell line. Pathways in Group E were observed only from the tumor tissue. Pathways in Group F were observed both in the PDX tumor tissues and the cell line consistently or irregularly. Pathways in Group G were observed consistently only in PDX tumor tissues. Pathways in Group H were observed only in the cell line. The different enrichment patterns of pathways may reflect the different phenotypes of each model. Furthermore, these observations may indicate important concerns regarding the patient-derived cancer model. For example, when we examined the biological features relating to pathways in group A, because those pathways are consistently observed in the primary tumor tissues, the PDX tumor tissues, and the cell line, all models are suitable for study. In contrast, we may need to avoid studying the pathways in Group E, because they were not observed in the models. The pathways in Groups G and H were enriched only in the PDX tissues and the cell line, respectively.
Table 2. Rank of KEGG pathways according to enrichment score, -log2 (p-value), in the proteome of different samples.
| No | Name of pathways | Name of samples | Groups |
|---|
| Tumor | NCC-UPS1-X3 | NCC-UPS1-X3-C1 | Common |
|---|
| 1 | Focal adhesion | 1 | 4 | 8 | 4 | A |
| 2 | Pathogenic Escherichia coli infection | 2 | 3 | 3 | 2 |
| 3 | Ribosome | 3 | 2 | 1 | 1 |
| 4 | Proteasome | 4 | 1 | 2 | 3 |
| 5 | Glycolysis/Gluconeogenesis | 7 | 8 | 7 | 5 |
| 6 | Pentose phosphate pathway | 8 | 12 | 9 | 7 |
| 7 | Fatty acid metabolism | 9 | 9 | 18 | 11 |
| 8 | Pyruvate metabolism | 10 | 5 | 6 | 8 |
| 9 | Regulation of actin cytoskeleton | 11 | 13 | 11 | 6 |
| 10 | Valine, leucine and isoleucine degradation | 13 | 11 | 14 | n.d. |
| 11 | Glutathione metabolism | 14 | 25 | 22 | 12 |
| 12 | Citrate cycle (TCA cycle) | 15 | 15 | 5 | 9 |
| 13 | Aminoacyl-tRNA biosynthesis | 25 | 6 | 4 | n.d. |
| 14 | Lysine degradation | 23 | 7 | 13 | n.d. |
| 15 | ECM-receptor interaction | 6 | 14 | n.d. | 16 | B |
| 16 | Fc gamma R-mediated phagocytosis | 20 | 21 | n.d. | 20 |
| 17 | Oocyte meiosis | 26 | 23 | n.d. | 17 |
| 18 | Antigen processing and presentation | 22 | 22 | n.d. | 10 |
| 19 | Systemic lupus erythematosus | 12 | n.d. | n.d. | 21 | C |
| 20 | Fatty acid elongation in mitochondria | 16 | n.d. | n.d. | n.d. |
| 21 | Complement and coagulation cascades | 5 | n.d. | n.d. | n.d. |
| 22 | Adherens junction | 17 | n.d. | n.d. | n.d. |
| 23 | Leukocyte transendothelial migration | 18 | n.d. | n.d. | 19 |
| 24 | Prion diseases | 19 | n.d. | n.d. | n.d. |
| 25 | PPAR signaling pathway | 21 | n.d. | n.d. | n.d. |
| 26 | Arginine and proline metabolism | 24 | n.d. | n.d. | n.d. |
| 27 | Starch and sucrose metabolism | 27 | n.d. | n.d. | 22 |
| 28 | Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 28 | n.d. | n.d. | n.d. |
| 29 | Spliceosome | n.d. | 10 | 12 | 13 | D |
| 30 | Huntington’s disease | n.d. | 20 | 10 | 18 |
| 31 | Parkinson’s disease | n.d. | 17 | 15 | n.d. |
| 32 | Propanoate metabolism | n.d. | 19 | 21 | n.d. |
| 33 | Glyoxylate and dicarboxylate metabolism | n.d. | 24 | n.d. | n.d. | E |
| 34 | Leukocyte transendothelial migration | n.d. | 16 | n.d. | n.d. |
| 35 | Endocytosis | n.d. | 26 | n.d. | n.d. |
| 36 | beta-Alanine metabolism | n.d. | 27 | n.d. | n.d. |
| 37 | Neurotrophin signaling pathway | n.d. | 28 | n.d. | 23 |
| 38 | Steroid biosynthesis | n.d. | 29 | n.d. | n.d. |
| 39 | Fructose and mannose metabolism | n.d. | 30 | n.d. | n.d. |
| 40 | Tryptophan metabolism | n.d. | 31 | n.d. | n.d. |
| 41 | Butanoate metabolism | n.d. | 18 | n.d. | n.d. |
| 42 | DNA replication | n.d. | n.d. | 20 | n.d. | F |
| 43 | Protein export | n.d. | n.d. | 17 | n.d. |
| 44 | Gap junction | n.d. | n.d. | 19 | n.d. |
| 45 | Cysteine and methionine metabolism | n.d. | n.d. | 23 | n.d. |
| 46 | Tight junction | n.d. | n.d. | 16 | n.d. |
| 47 | Terpenoid backbone biosynthesis | n.d. | n.d. | 24 | n.d. |
n.d.: not detected.
Group A: pathways in all samples
Group B: pathways in the original tumor and NCC-UPS1-X3
Group C: pathways unique to the original tumor
Group D: pathways in NCC-UPS1-X2 and NCC-UPS1-X3-C1
Group E: pathways unique to NCC-UPS1-X3
Group F: pathways unique to NCC-UPS1-X3-C1
Discussion
Our results indicate that the patient-derived cancer model appears to have promise as a research tool. The tumor tissues implanted in the mice may mimic the tumor tissues in the donor patients. Previous studies suggested that the response to treatment was common between the original tumor tissues and those of PDXs23,24). Patient-derived cells also have unique advantages as they are easy to handle and the effects of many possible anti-cancer drugs can be screened in a high throughput way25). To achieve the goal of personal medicine, the patient-derived cancer model may constitute an indispensable tool. However, although the patient-derived cancer model is expected to faithfully reproduce the features of original tumors, it is highly controversial whether such models retain the molecular features of original tumors. Wang et al. reported that somatic mutations, gene copy number, and expression profiles, as well as the phospho-tyrosine proteome landscape were similar between patient tumors and PDX tumors in lung cancer26). In contrast, Ben-David et al. stressed that copy number alterations rapidly accumulated during PDX passaging, owing to the clonal selection of preexisting minority tumor cells27). Dieter et al. pointed out that EBV-associated B-lymphoproliferation frequently develop following xenotransplantation28). In the present study, we found that the proteomic features were similar but not identical among the original tumor tissues, the tumor tissues of PDX, and the cell line. In addition, although the patient-derived cancer model established with PDX exhibited morphologically similar appearance to the original tumor tissue at the different passages, the growth rate of PDX tumors increased in the late passages. Our observations were consistent with those of a previous report29). During passage, the tumor cells may have adopted themselves to the circumstances of the mouse environment, or the tumor cells with advantageous traits were selected during the process of engraftment. Considering these reports and our observations, we doubt on the versatile utility of patient-derived cancer models. We thus need to assess the phenotype of PDXs and cell lines to verify the consistency of original properties of the tumors.
In conclusion, to generate a novel preclinical model of UPS, we successfully obtained a PDX model and a cell line. We consider that patient-derived cancer models, especially for rare cancers, should be shared in the research community. We hope that our models accelerate the development of novel clinical applications for patients with UPS.
Conflict of Interest
The authors declare that they have no conflicting interests.
Sources of Support
This research was supported by the National Cancer Center Research and Development Fund (26-A-3 and 26-A-9).
Acknowledgements
We would like to thank Editage (www.editage.jp) for English language editing and constructive comments on the manuscript.
References
- 1) Fletcher, C. D. M., Bridge, J. A., Hogendoorn, P., et al.: WHO Classification of Tumours of Soft Tissue and Bone (ed. fourth edition). Geneva, WHO Press, 2013.
- 2) Goldblum, J. R.: An approach to pleomorphic sarcomas: can we subclassify, and does it matter? Mod. Pathol., 27 Suppl 1, S39–46, 2014.
- 3) Radaelli, S., Stacchiotti, S., Casali, P. G., et al.: Emerging therapies for adult soft tissue sarcoma. Expert Rev. Anticancer Ther., 14, 689–704, 2014.
- 4) Reichardt, P.: Soft tissue sarcomas, a look into the future: different treatments for different subtypes. Future Oncol., 10, s19–27, 2014.
- 5) Versleijen-Jonkers, Y. M., Vlenterie, M., van de Luijtgaarden, A. C., et al.: Anti-angiogenic therapy, a new player in the field of sarcoma treatment. Crit. Rev. Oncol. Hematol., 91, 172–185, 2014.
- 6) Nakatani, T., Marui, T., Yamamoto, T., et al.: Establishment and characterization of cell line TNMY1 derived from human malignant fibrous histiocytoma. Pathol. Int., 51, 595–602, 2001.
- 7) Krause, A. K., Hinrichs, S. H., Orndal, C., et al.: Characterization of a human myxoid malignant fibrous histiocytoma cell line, OH931. Cancer Genet. Cytogenet., 94, 138–143, 1997.
- 8) Mairal, A., Chibon, F., Rousselet, A., et al.: Establishment of a human malignant fibrous histiocytoma cell line, COMA: Characterization by conventional cytogenetics, comparative genomic hybridization, and multiplex fluorescence in situ hybridization. Cancer Genet. Cytogenet., 121, 117–123, 2000.
- 9) Hakozaki, M., Hojo, H., Sato, M., et al.: Establishment and characterization of a new cell line, FPS-1, derived from human undifferentiated pleomorphic sarcoma, overexpressing epidermal growth factor receptor and cyclooxygenase-2. Anticancer Res., 26, 3393–3401, 2006.
- 10) Nishio, J., Iwasaki, H., Nabeshima, K., et al.: Establishment of a new human pleomorphic malignant fibrous histiocytoma cell line, FU-MFH-2: Molecular cytogenetic characterization by multicolor fluorescence in situ hybridization and comparative genomic hybridization. J. Exp. Clin. Cancer Res., 29:153, 2010.
- 11) Becker, M., Graf, C., Tonak, M., et al.: Xenograft models for undifferentiated pleomorphic sarcoma not otherwise specified are essential for preclinical testing of therapeutic agents. Oncol. Lett., 12, 1257–1264, 2016.
- 12) Steinstraesser, L., Jacobsen, F., Schubert, C., et al.: Establishment of a primary human sarcoma model in athymic nude mice. Hum. Cell, 23, 50–57, 2010.
- 13) Tilkorn, D. J., Daigeler, A., Hauser, J., et al.: A novel xenograft model with intrinsic vascularisation for growing undifferentiated pleomorphic sarcoma NOS in mice. J. Cancer Res. Clin. Oncol., 138, 877–884, 2012.
- 14) Tilkorn, D. J., Stricker, I., Hauser, J., et al.: Experimental murine model of primary high grade undifferentiated pleomorphic sarcoma not otherwise specified. In Vivo, 26, 559–563, 2012.
- 15) Sakumoto, M., Takahashi, M., Oyama, R., et al.: Establishment and proteomic characterization of NCC-LMS1-C1, a novel cell line of primary leiomyosarcoma in the bone. Jpn. J. Clin. Oncol., 47, 954–961, 2017.
- 16) Tomayko, M. M. and Reynolds, C. P.: Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol., 24, 148–154, 1989.
- 17) Capes-Davis, A., Dirks, W. G., MacLeod, R. A., et al.: Quality Matters: Cell lines and their use in research. GIT Lab. J. Eur., 17, 12–13, 2014.
- 18) Yoshida, A., Got,o K., Kodaira, M., et al.: CIC-rearranged sarcomas: A study of 20 cases and comparisons with ewing sarcomas. Am. J. Surg. Pathol., 40, 313–323, 2016.
- 19) Kondo, T. and Hirohashi, S.: Application of highly sensitive fluorescent dyes (CyDye DIGE Fluor saturation dyes) to laser microdissection and two-dimensional difference gel electrophoresis (2D-DIGE) for cancer proteomics. Nat. Protoc., 1, 2940–2956, 2007.
- 20) Wisniewski, J. R., Zougman, A., Nagaraj, N., et al.: Universal sample preparation method for proteome analysis. Nat. Methods., 6, 359–362, 2009.
- 21) Baehrecke, E. H., Dang, N., Babaria, K., et al.: Visualization and analysis of microarray and gene ontology data with treemaps. BMC Bioinf., 5:84, 2004.
- 22) Altermann, E. and Klaenhammer, T. R.: PathwayVoyager: pathway mapping using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. BMC Genomics, 6:60, 2005.
- 23) Tentler, J. J., Tan, A. C., Weekes, C. D., et al.: Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol., 9, 338–350, 2012.
- 24) Izumchenko, E., Paz, K., Ciznadija, D., et al.: Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol., 28, 2595–2605, 2017.
- 25) Barretina, J., Caponigro, G., Stransky, N., et al.: The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 483, 603–607, 2012.
- 26) Wang, D., Pham, N. A., Tong, J., et al.: Molecular heterogeneity of non-small cell lung carcinoma patient-derived xenografts closely reflect their primary tumors. Int. J. Cancer, 140, 662–673, 2017.
- 27) Ben-David, U., Ha, G., Tseng, Y. Y., et al.: Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet., 49, 1567–1575, 2017.
- 28) Dieter, S. M., Giessler, K. M., Kriegsmann, M., et al.: Patient-derived xenografts of gastrointestinal cancers are susceptible to rapid and delayed B-lymphoproliferation. Int. J. Cancer, 140, 1356–1363, 2017.
- 29) Pearson, A. T., Finkel, K. A., Warner, K. A., et al.: Patient-derived xenograft (PDX) tumors increase growth rate with time. Oncotarget, 7, 7993–8005, 2016.