Review
Development challenges associated with rAAV-based gene therapies
2021 年 46 巻 2 号 p. 57-68
詳細
2021 年 46 巻 2 号 p. 57-68
The number of gene therapies in development continues to increase, as they represent a novel method to treat, and potentially cure, many diseases. Gene therapies can be conducted with an in vivo or ex vivo approach, to cause gene augmentation, gene suppression, or genomic editing. Adeno-associated viruses are commonly used to deliver gene therapies, but their use is associated with several manufacturing, nonclinical and clinical challenges. As these challenges emerge, regulatory agency expectations continue to evolve. Following administration of rAAV-based gene therapies, nonclinical toxicities may occur, which includes immunogenicity, hepatotoxicity, neurotoxicity, and the potential risks for insertional mutagenesis and subsequent tumorgenicity. The mechanism for these findings and translation into the clinical setting are unclear at this time but have influenced the nonclinical studies that regulatory agencies are increasingly requesting to support clinical trials and marketing authorizations. These evolving regulatory expectations and toxicities, as well as future nonclinical considerations, are discussed herein.
Gene therapy is the introduction of nucleic acids into a host cell to replace a lost gene, suppress an overactive gene or repair a mutated gene, or to introduce something novel all together. Gene therapies offer a substantial clinical benefit because they represent a potential cure for some diseases, many of which are categorized as rare conditions. The use of gene therapies as a modality to treat diseases continues to increase, as evidenced by the more than 900 investigational new drug applications for gene therapy clinical trials reported by the United States Food and Drug Administration (US FDA Press Release, 2020). There are 3 major types of gene therapies: gene augmentation, gene suppression, and genomic editing. Gene augmentation typically corrects a cell with a loss of function mutation, by introducing a functional gene next to the non-functioning gene. Gene suppression often corrects a cell that is producing too much of a toxic protein, by targeting the mRNA/DNA of the abnormal gene to knock down or prevent protein expression. Genomic editing typically introduces a double-strand break at the target site on the DNA, and following homology-directed repair or non-homologous end joining, the diseased cell function is corrected, the targeted protein is knocked down or knocked out, or a new functioning allele is introduced into the cell (Anguela and High, 2019). There are 2 primary approaches for delivering gene therapies. The in vivo approach administers the gene therapy by the intended clinical route to the patient, and following distribution to the target tissue, the gene therapy produces the desired effect. The ex vivo approach collects target cells from the patient, and following exposure to the gene therapy that results in gene transfer, the modified cells are introduced back into the patient, to produce the desired effect (Kaufmann et al., 2013).
Viral vectors have emerged as the most used approach for delivering gene therapies. These vectors differ in many ways, and there are advantages and disadvantages associated with each vector’s use (Lundstrom, 2018). Adeno-associated virus (AAV)-based vectors are one of the most frequently used to deliver gene therapies. AAV is a naturally occurring virus that contains a single-stranded DNA genome comprised of the 3 genes (Rep, Cap and aap), with coding sequences flanked by inverted terminal repeats (ITRs). The Rep gene encodes for proteins required for viral genome replication and packaging; the Cap gene encodes for viral proteins (VP1, VP2, and VP3) that form the outer capsid and assist with cell binding and internalization; and aap gene encodes the assembly-activating protein needed for capsid assembly and nuclear localization of the VP proteins for some, but not all, AAV serotypes (Naso et al., 2017). The only remaining portion of wildtype AAV present in recombinant AAV (rAAV) vectors used in gene therapies are the ITRs, which are required for packaging during vector production, and second strand vector DNA synthesis.
Following administration of rAAVs, the genetic material is inserted into host cells, via the AAV transduction pathway. As summarized by Wang et al. (2019), the rAAV interacts with glycosylated cell surface receptors of the host cell to trigger its internalization via clathrin-mediated endocytosis. Once inside the endosome, the VP1 and VP2 regions undergo a conformational change, and following phospholipase A2 domain-induced endosomal escape (Stahnke et al., 2011), the rAAV either undergoes proteolysis by proteasomes, or is transported into the nucleus, where it un-coats. rAAVs can be either single-stranded (ssAAV) or self-complementary (scAAV). ssAAVs are transcriptionally inert and must be converted to double-stranded DNA, via second strand synthesis (using host cell DNA polymerase) or strand annealing of sense and anti-sense genomes, prior to transcription. scAAV are already double-stranded and can be transcribed immediately after entering the host cell nucleus. The ITR-flanked coding sequence contained with the rAAVs form circular concatemers that primarily reside as episomes in the nucleus of transduced cells.
There are advantages and disadvantages to using rAAVs as the vector for a gene therapy. rAAVs are non-pathogenic, replication deficient gene therapy vectors that have been used in the clinical setting since 1995 (Flotte et al., 1996), and following administration, they have efficient transduction that results in stable and prolonged transgene expression in non-dividing cells. There are many different AAV serotypes (12 natural and > 100 synthetic variants), and the different serotypes transduce different tissues (Colella et al., 2018; Li and Samulski, 2020). As such, selection of an AAV serotype that has tropism for the target tissue is critical when designing an rAAV-based gene therapy. In addition, while an AAV serotype may have a preferred tropism for certain tissues, its distribution to tissues can be dose-related, and understanding this dose:distribution relationship is important for characterizing the safety and efficacy of a given serotype. The efficacy and selectivity of a rAAV-based gene therapy can be enhanced by using a different AAV serotype, modifying the capsid (i.e. site-directed mutagenesis with insertion of a ligand in the VP2 or VP3 regions), or by modifying elements of the expression cassette, such as the enhancer or promoter, or the size, activity or codon of the transgene. However, the use of rAAVs for gene therapies also has a few disadvantages; for example, AAVs have a packaging capacity of < 4.8 kilobases, which limits the size of the transgene that can be used in the gene therapy. Furthermore, with cell division, the episomal DNA of the rAAV-based gene therapy can be diluted between daughter cells, which potentially decreases efficacy in rapidly growing organs such as the liver of neonates. Importantly, the use of rAAV-based therapeutics has been associated with several development challenges (Table 1), including an evolving regulatory agency environment, immunogenicity, hepatotoxicity, toxicity to the dorsal root ganglia (DRG), the potential for insertional mutagenesis. These are discussed in more detail below.
| Immunogenicity | • Pre-existing neutralizing antibodies to capsid • Innate immune response stimulates inflammation • T cell response that eliminates transduced cells • Humoral response against transgene product • Toxicity associated with complement activation observed clinically |
| Hepatotoxicity | • Hepatic necrosis observed in monkeys and piglets within 5 days after relatively high dose of AAV-based vector • Acute liver failure observed clinically |
| Dorsal Root Ganglia Toxicity | • Axonal degeneration and swelling of DRG, spinal cord, and/or peripheral nerve observed in monkeys and mice given AAV-based gene therapies • Human relevance unclear |
| Insertional Mutagenesis | • AAV integration associated with hepatocellular carcinoma observed in neonatal mice but not other species |
| Evolving Regulatory Environment | • Different regions (each with their own guidance documents) can have different requirements on nonclinical endpoints/studies needed to support clinical trials |
Guidance documents with information relevant for the nonclinical development of rAAV-based gene therapies have been provided by the US Food and Drug Administration (FDA), the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) and the Pharmaceuticals and Medical Devices Agency (PMDA) of Japan, some of which are listed in Table 2.
| US FDA | CHMP / EMA | PMDA |
|---|---|---|
| Guidance for Industry: Preclinical Assessment of Investigational Cellular and Gene Therapy Products (2013) | Draft Guideline on the quality, nonclinical, and clinical requirements for investigational advanced therapy medicinal products in clinical trials (Jan 2019) | Guideline for Assuring the Quality and Safety of the Gene Therapy Products (2019) under The Pharmaceuticals, Medical Devices, and Other Therapeutic Products Act (2014) |
| Design and Analysis of Shedding Studies for Virus or Bacteria-Based Gene Therapy and Oncolytic Products (2015) | Guideline on the non-clinical studies required before first clinical use of gene therapy medicinal products (2008) | Guidelines for Clinical Research such as Gene Therapy (2015) |
| Guidance for Industry. Gene Therapy Clinical Trials – Observing Subjects for Delayed Adverse Events (2006) | Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products (2018) | The Act on the Safety of Regenerative Medicine (2014) |
| Long Term Follow-Up After Administration of Human Gene Therapy Products (2020) | Guideline on non-clinical testing for inadvertent germline transmission of gene transfer vectors (2007) | |
| Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs) (2020) | Reflection paper on management of clinical risks deriving from insertional mutagenesis (2013) | |
| Reflection paper on quality, nonclinical and clinical issues related to the development of recombinant adeno-associated viral vectors (2010) |
With these guidance documents in mind, nonclinical toxicity study packages for rAAV-based gene therapies are typically designed on a case-by-case basis. For example, assuming the vector had human-relevant tropism in at least 1 nonclinical species and contained a well-characterized AAV serotype that used the same route of administration as previously tested, and assuming that peak transgene expression occurred at approximately 1 month post-dose and it was associated with minimal toxicity, then, a nonclinical toxicity package in this scenario could be a single-dose study, in a single species, with necropsy at 1-month and 3-months post-dose. This study would include standard toxicology endpoints plus measures that address safety concerns related to the specific disease state, transduced off target tissues and the extent of promoter organ transduction. Biodistribution assessments should be conducted at each timepoint, and in tissues that are predicted to have the greatest transduction (based on the properties of the capsid and expression cassette and the route of administration) as well as those recommended in available guidance documents (US FDA, 2020; IPRP, 2018). Germline transmission and vector shedding may also need to be assessed, often via biodistribution to the gonads and excretion in biofluids. Integration endpoints may be necessary to address regulatory concerns.
This minimal nonclinical toxicity study package described above may be modified based on the patient population, the properties of the rAAV-based gene therapy, and the effects observed with transgene overexpression. For example, studies in more than one species may be necessary, or studies may be conducted in healthy or diseased animals. The age of the patient population will impact the age of the animals. Studies longer than 3 months in duration may be needed to characterize durability of expression, immune response, long-term safety, and/or reversal of test article-related effects, particularly if a novel capsid is used. These variables further emphasize the need to develop nonclinical strategies on a case-by-case basis. Additional information on parameters included in nonclinical studies conducted to support the development of rAAV-based gene therapies are discussed elsewhere (Bolt et al., 2020).
Currently, harmonized guidance documents for the development of rAAV-based gene therapies are not available. In the absence of internationally harmonized documents, different geographic regions can have different requirements for nonclinical studies that support clinical trials and marketing authorizations. In addition, differences between gene therapy products (such as the capsid, promoter or transgene product strength and distribution) and indications (such as risk: benefit profile, patient population/age, and currently available therapies) can also increase uncertainty of the required nonclinical studies. Furthermore, as nonclinical toxicity data about immunogenicity, toxicity to the liver or DRG or tumorgenicity emerges, some regulatory agencies are more concerned about one or more of these potential rAAV-based toxicities than others, which influences the studies or endpoints that they are requiring. Together, these factors can make it difficult to design a nonclinical toxicity study package that is acceptable to all the different regions, and Sponsors often have to consult with multiple regulatory agencies to agree and align on the necessary studies and parameters.
Harmonized guidance documents are of significant benefit. In fact, efforts by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) to create a guidance on the nonclinical biodistribution studies for gene therapy products (S12) are ongoing. However, it is uncertain if now is the right time to create additional ICH guidance documents to support other aspects of gene therapy development because the field is evolving so rapidly.
Immune responses to rAAV can be separated into innate and adaptive immune response and the latter can be further separated into humoral and cell-mediated response. Both pre-existing and post-treatment adaptive immune responses influence the development of safe and effective gene therapies, as does the innate immune response that can occur shortly after administration of a gene therapy vector.
rAAV gene therapy vectors are derived from widely disseminated nonpathogenic AAV viruses that infect numerous species, including humans. Following infection, humans and animals develop antibodies to these wild-type AAV serotypes, and these antibodies will cross-react and prevent transduction of tissues by therapeutic rAAV vectors. Consequently, an important consideration in the design of nonclinical animal studies (as well as patient eligibility for treatment) is the identification of individuals that have a pre-existing AAV antibody titer that is permissive to tissue transduction by the rAAV serotype that is being investigated. As such, an AAV serotype that has a high prevalence of pre-existing antibody in a patient population of interest, may be deprioritized for therapeutic development because an insufficient proportion of the population will be eligible for treatment. In nonclinical studies, screening for pre-existing antibodies to select animals for a study is typically done for nonrodents, but not rodents. Rodents are not pre-screened because they generally do not have antibodies that neutralize rAAVs that are being evaluated for therapeutic development.
The post-treatment adaptive humoral immune response to the rAAV vector serotype results in high titer antibody responses that prevent subsequent administration of the same AAV vector. Approaches to induce immune tolerance to inhibit antibody production and approaches to eliminating pre-existing antibodies is an active area of investigation, that in the future, may lead to wider application of gene therapy as a therapeutic platform. However, the post-treatment humoral response to the transgene protein produced by the vector should also be considered. In animal studies, the antibody response to a foreign human protein may result in neutralization of the pharmacologic activity of the protein, and prevent the assessment of efficacy in a disease model or the potential for pharmacologically mediated toxicity. Antigen-antibody complex disease is also a consideration, although this has not been reported in gene therapy studies. If immune response to the foreign human protein impacts the nonclinical development of a gene therapy, then an animal specific transgene surrogate or immune suppression may be considered.
Another emerging consideration for pre-existing antibody responses is understanding the impact of binding antibodies that do not prevent (i.e. neutralize) transduction. Neutralizing antibodies are typically evaluated in cell-based assays that evaluate the ability of a serum sample to prevent a rAAV vector from expressing its transgene in a cell. Binding antibody assays look at total antibody that binds to the capsid in an assay similar to an ELISA antibody assay. Binding antibodies do not necessarily prevent transduction and there is some evidence that they may enhance transduction of the liver (Fitzpatrick et al., 2018).
In contrast to pre-existing antibodies to an AAV vector serotype, animals and humans are not typically screened for pre-existing cell-mediated immunity prior to selection for treatment. This is because the prevalence of pre-existing cell-mediated immunity in humans and animals, as detected by gamma-interferon ELISPOT, is very low, and does not impact the initial transduction of tissue by a therapeutic vector.
Post-treatment cell-mediated immune responses to the vector are an important consideration in both nonclinical studies and in patients. A cell-mediated response to either the vector capsid or transgene protein may cause cytotoxic T-cells and/or NK cells to attack transduced cells, which results in cell death and loss of expression of the therapeutic protein (Kuranda et al., 2018; Ertl and High, 2017; Nathwani et al., 2014). In some instances, this elimination of transduced cells may not be a safety concern and only lead to loss of efficacy. However, if a large proportion of cells in an organ are transduced and the organ has limited reserve capacity to replace lost cells, then the cell-mediated immune response-induced cell death may be a safety concern. Consequently, when treating human subjects, the potential for cell-mediated immune response is addressed by the application of prophylactic immune suppression and / or frequent monitoring for organ injury (e.g. liver enzymes) and cell-mediated immune response. If prophylactic immune suppression is not used, then administration of immunosuppressants (e.g. corticosteroids) is used in response to tissue injury markers and/or decreased transgene protein expression. Animals appear to be less prone to develop cell-mediated immune responses that result in tissue injury and have not predicted the cell-mediated response seen in human hemophilia gene therapy clinical trials (Ertl and High, 2017). However, there are examples in animal studies reported in both the literature (Hadaczek et al., 2009; Gao et al., 2009) and conducted at Pfizer (Fig. 1) where cell-mediated immune responses occur.
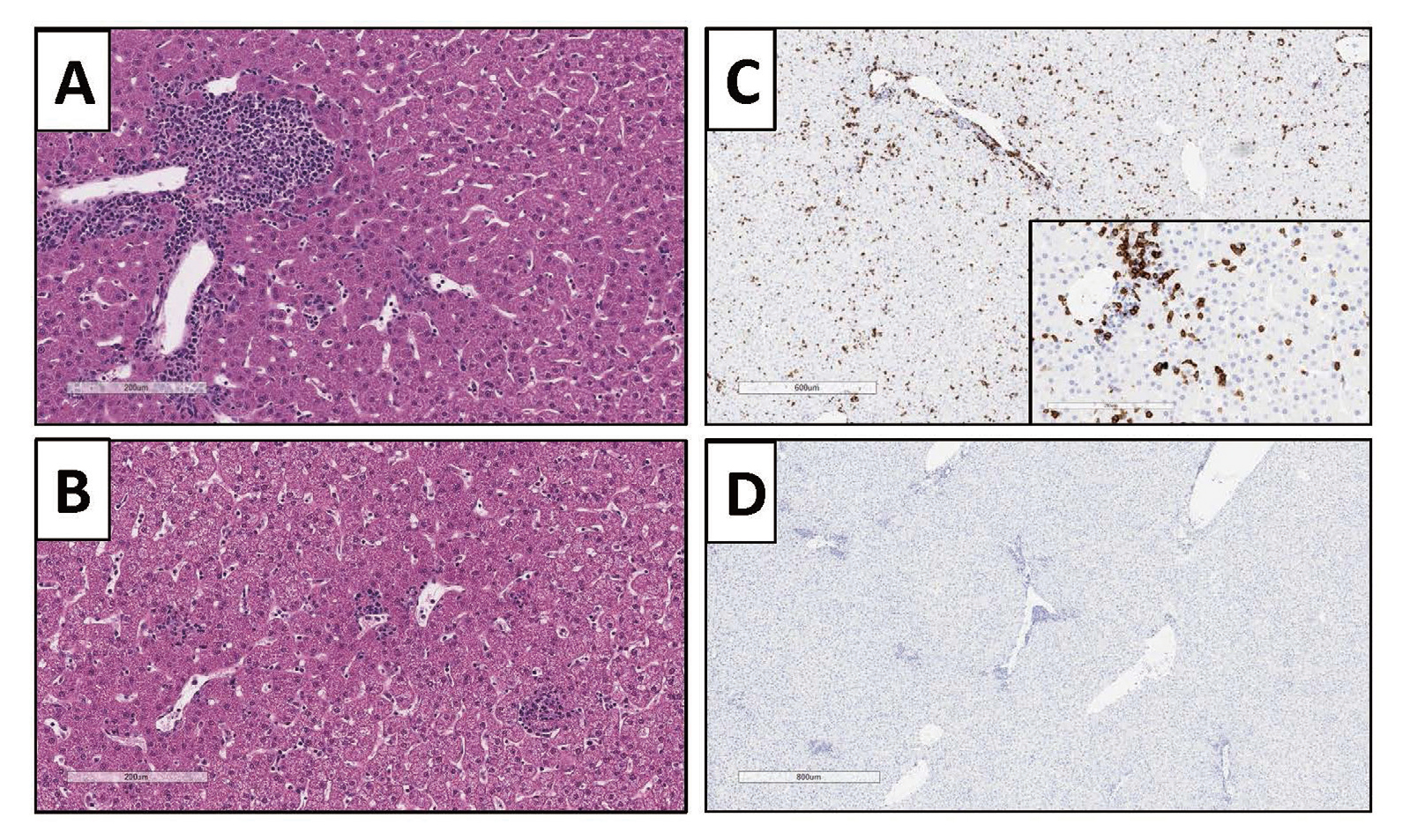
Suspected adaptive cell-mediated immune response to a rAAV vector in a cynomolgus monkey liver 4 weeks post treatment with 1E14 vg/kg of a rAAV vector that expresses a human protein. A and B: H& E stain showing periportal and intraparenchymal mononuclear cell infiltrate. C: Immunohistochemical stain for CD8 indicating mononuclear cell infiltrate is CD8 positive. D: IgG negative control for immunohistochemical stain.
The innate immune response involves the interaction of the vector with mediator systems designed to respond rapidly to foreign pathogens. These mediator systems include complement and foreign nucleic acid or protein recognition receptors, such as toll-like receptors and other foreign pathogen recognition pathways (Schlee and Hartmann, 2016; Herzog, 2019; Mingozzi and High, 2017). The activation of these innate systems has an important role in promoting the initiation of an adaptive immune response. They can also potentially lead to adverse consequences. For example, thrombocytopenia and renal compromise that was associated with concurrent complement activation has been observed within the first 2 weeks after rAAV administration in gene therapy clinical trials (Solid Biosciences, 2019). However, it should be noted that the causal link of these findings to complement activation, has not been proven (Ronzitti et al., 2020).
In nonclinical biodistribution studies with rAAV, the liver often has the greatest quantity of viral genomes. Consequently, one may anticipate that hepatoxicity would be associated with rAAV administration. Two recent publications in nonhuman primates (NHP) demonstrated that 2 different rAAV vectors (administered at doses ≥ 7.5E13 vg/kg) induced severe hepatotoxicity and morbidity within 4-5 days post vector administration (Hinderer et al., 2018; Hordeaux et al., 2018a). Given the acute time course of the events and the severe liver injury that is reported, the mechanism of the injury is unlikely to be related to an adaptive immune response, but is either secondary to initiation of innate immune pathways and /or caused by direct cellular injury to hepatocytes. In these studies, NHP were administered AAVhu68-CB7-hSMN1 (2E14 GC/kg) or AAVPHP.B-CB7-eGFP-WPRE-rBG (7.5E13 GC/kg) by IV injection. Common findings in both studies include morbidity and euthanasia on days 4 to 5 with peritoneal fluid transudate observed at necropsy. There was a marked increase in liver enzymes (ALT, AST and ALP) and bilirubin along with hypoproteinemia, consistent with the marked hepatocyte necrosis that was observed in the histologic sections of liver. Additional observations common to both studies included thrombocytopenia and decreased white blood cell counts. In the study conducted by Hordeaux et al. (2018a), but not in the study by Hinderer et al. (2018), D-dimer and coagulation time (PT/PTT) were evaluated and found to be increased, which is consistent with disseminated intravascular coagulation. Cytokines were measured in both studies and IL-6 was found to be markedly elevated in the Hinderer study but not the study reported by Hordeaux. However, in the Hordeaux study the animal was treated with corticosteroids which may have reduced cytokine release. A key finding in both studies was the very high vector genome copy number found in the liver (> 1000 viral genomes per host cell genome). We have also observed similar hepatocellular injury in one of our internal studies in cynomolgus monkeys dosed a rAAV gene therapy at 1E14 vg/kg (Fig. 2).
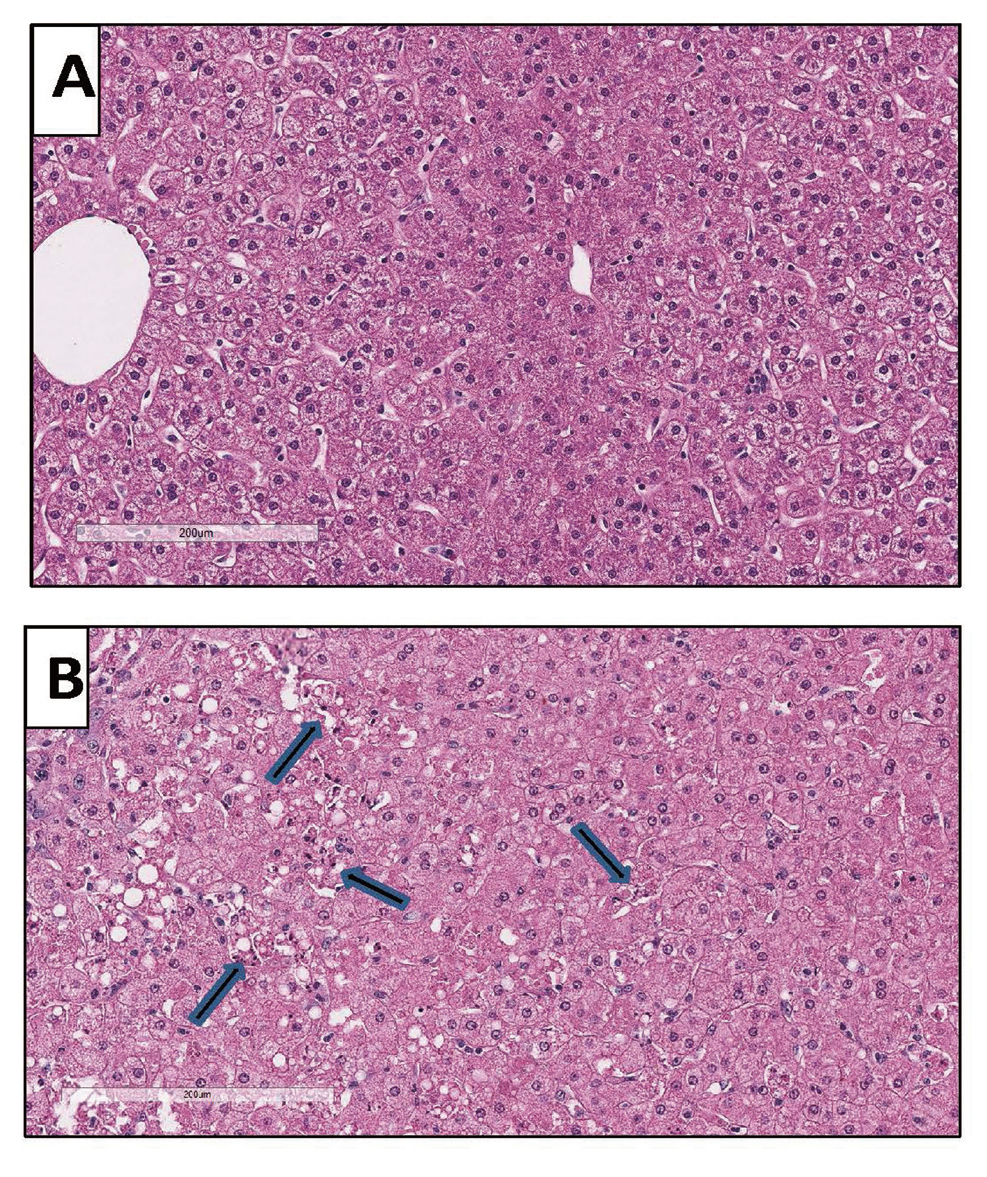
Livers from cynomolgus monkeys: A is from a nontreated control monkey and B is from a monkey necropsied 3 days after treatment with 1E14 vg/kg of a rAAV vector that expressed a human protein. In B there is degeneration and necrosis of hepatocytes (arrows) without a cellular infiltrate, suggesting direct injury to hepatocytes by the rAAV vector.
Hepatotoxicity has also recently been observed clinically following administration of rAAV gene therapies. Specifically, in a clinical trial, hepatobiliary disease occurred in 3 young boys approximately 4-6 weeks after administered AT-132, a rAAV8-based gene therapy for the treatment of X-linked myotubular myopathy, at 3E14 vg/kg by intravenous administration (Audentes press release, 2020; Ronzitti et al., 2020; Morales et al., 2020). However, it should be noted that all 3 of these boys had evidence of pre-existing hepatobiliary disease. ZOLGENSMA is a rAAV9-based gene therapy that contains the human survival motor neuron gene (SMN1) under the control of the cytomegalovirus (CMV) enhancer/ chicken-β-actin-hybrid promotor (CB), which is currently marketed for the treatment of spinal muscular atrophy (SMA) type 1. Two patients (4-20 months old) have been reported to have transient acute liver failure approximately 7-8 weeks after administration of ZOLGENSMA (1.1E14 vg/kg IV), which was characterized by jaundice, and increased serum hepatic enzymes and high international normalized ratios (INRs) indicating prolongation of coagulation times. In addition, liver biopsies showed hepatocellular swelling or hepatic ballooning, inflammation of periportal areas, bile duct reactive changes, and fibrosis in the portal, periportal or central vein areas in these patients (Feldman et al., 2020). The mechanism of the clinical hepatotoxicity is not known, but the onset of findings, several weeks after vector administration, indicates that the mechanism of liver injury is different than the acute liver injury in NHP described above. The most likely explanation is an adaptive immune response because of the anti-transgene and anti-capsid immune response that was observed (Ronzitti et al., 2020). Regardless, these nonclinical and clinical findings indicate that high doses of rAAV gene therapies can result in toxicity in the liver.
DRG are a cluster of cell bodies for sensory nerves that carry sensory information, such as pain or temperature, to the spinal cord. They form the sensory arm of the reflex arc that is required for sensory action and are located in the dorsal root of the spinal nerve, which is outside the blood brain barrier (Krames, 2014). Following administration of rAAV-based gene therapies, toxicity in the DRG have been reported in NHP and piglets (Hinderer et al., 2018; Hordeaux et al., 2018b; Hordeaux et al., 2019; EMA, 2020; US Securities and Exchange Commission, 2020). Toxicity is characterized by neuronal degeneration and loss, sattelitosis and mononuclear cell infiltration, and associated secondary axonal degeneration and swelling in the spinal cord and peripheral nerve. The microscopic findings in the DRG, spinal cord, and peripheral nerve were associated with proprioceptive deficiencies in piglets, but in NHP, CNS clinical signs are very rarely observed. Using aggregate data from 33 monkey studies that evaluated five different capsids, five different promoters, and 20 different transgenes, Hordeaux et al. (2020a) were able to demonstrate that following rAAV-based gene therapies, DRG toxicity occurred more frequently when given via the cerebral spinal fluid (vs IV route), and severity increased with IV dose, and was more severe when administered to adult (vs juvenile) animals.
DRG toxicity matching that observed in monkeys has recently been observed in mice administered a relatively high dose (1E14 vg/kg) of a rAAV vector with DRG tropism and a promoter that is active in the CNS. Specifically, DRG in these mice had multifocal areas of neuronal degeneration, satellitosis, and mononuclear cell infiltration, which was associated with a secondary axonopathy in the spinal cord and peripheral nerve (Fig. 3). These findings demonstrate that mice are a potential model for evaluating if a gene therapy molecule can cause DRG toxicity.
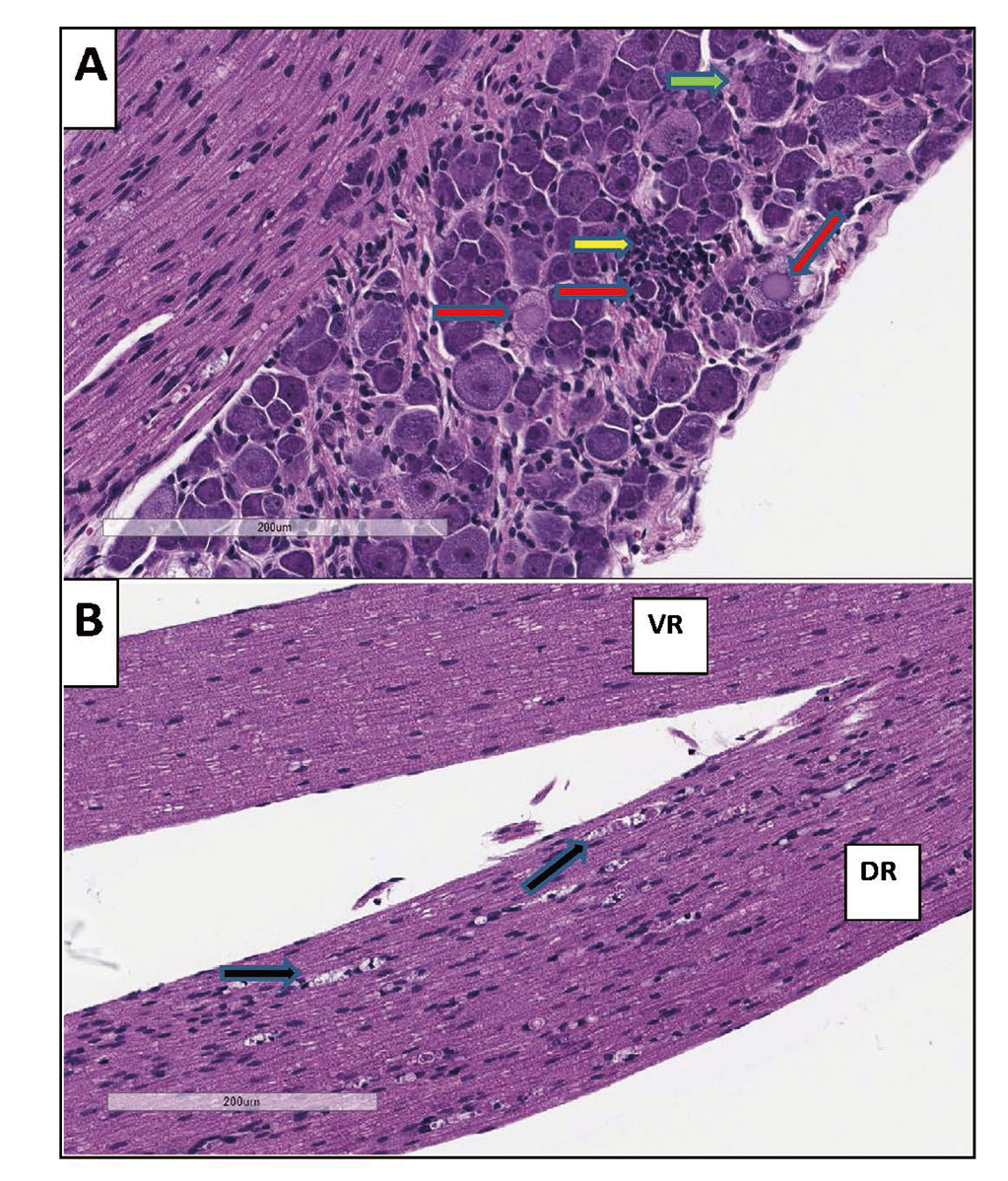
Dorsal Root Ganglia of Mice. A: Neuronal degeneration (red arrows), satellitosis (green arrow), and mononuclear cell infiltration (yellow arrow) in the dorsal root ganglia of mice administered an AAV-based viral vector at a dose of 1E14 vg/kg. B: Secondary axon degeneration with multiple axonal digestion chambers (black arrows) in dorsal nerve root (DR) while ventral nerve root (VR) is not affected.
The clinical translation of DRG toxicity seen in animals is unclear, and more clinical data and scholarship to understand the potential mechanisms of toxicity are needed. Despite many ongoing clinical trials with rAAV-based gene therapies, little evidence of CNS effects have been reported in humans. DRG have been reported as a target organ in NHP administered ZOLGENSMA by the intrathecal route (IT), but not when administered by the IV route. This difference in response may be due to the relatively high local vector concentrations in DRG neurons following IT administration, when compared to those achieved with IV dosing. Two patients that were administered ZOLGENSMA IV, and subsequently died, were examined for lesions in DRG. One deceased patient showed no evidence of effects in the DRG. The second deceased patient showed DRG ganglion cell loss and an excess of satellite cells. However, it could not be determined if the findings in the DRG were caused by the SMA pathology, were secondary to the hypoxic/ischemic injury associated with the terminal illness of this patient, or due to ZOLGENSMA administration (EMA, 2020). Regardless, further studies are needed to understand the clinical relevance of DRG toxicity observed in animals. More recently, Mueller et al. (2020), reported neuronal depletion in the DRG of a patient with familial amyotrophic lateral sclerosis (ALS) and mutations in the gene encoding superoxide dismutase 1 (SOD1) that was intrathecally infused with 4.2E14 vg of AAVrh10 containing an anti-SOD1 microRNA (AAV-miR-SOD1). Interestingly, this patient reported having transient tingling in both hands, and a feeling of “electric shocks” in his left foot. The onset of symptoms at 4 weeks post dose was coincident with a positive ELISPOT for capsid and laboratory measures at 8 weeks (CSF white cell count and protein elevations), which implicated an immune-mediated mechanism. However, no data was collected that could confirm/refute the possibility that a primary DRG toxicity related to the microRNA was also present with the immune-mediated mechanism. No clinical signs indicative of sensory dysfunction were observed in a second patient administered the same dose of 4.2E14 vg AAV-miR-SOD1 by intrathecal infusion. Unlike the patient above, this patient was pre-treated with rituximab and methylprednisolone for 3 weeks prior to treatment, which further indicates that DRG toxicity may involve an immune component.
The mechanism of DRG toxicity is not well understood and active investigations are ongoing. Given that toxicity has been observed with different rAAVs (including rAAV1, rAAV5, rAAV9, rAAV9-CRB3 and rAAVhu68) that delivered different transgenes, by different routes, DRG toxicity observed in animals is unlikely to be capsid specific. It has been hypothesized that overproduction of the transgene product leads to cellular stress which ultimately leads to neuronal loss. In support of this hypothesis, NHP were administered an intracisternal magna injection of a rAAV vector that expresses a transgene mRNA containing a nucleotide sequence that is recognized by endogenous miRNA expressed in DRG and leads to repression of vector transgene expression. NHP administered this vector had a reduction of transgene expression, as well as toxicity, in the DRG (Hordeaux et al., 2020b). Based on this finding, employing techniques that reduce transgene expression in DRG (such as incorporating miRNA target sequences into the expression cassette or using non-DRG tissue-specific promoters) could potentially minimize the risk for DRG toxicity.
Since ITR-flanked transgenes delivered by rAAV vectors form circular, episomal concatemers in the nucleus of transduced cells (Choi et al., 2006), rAAV vectors were initially considered to be non-integrating. However, hepatocellular carcinoma (HCC) that was associated with rAAV insertions into the RNA imprinted and accumulated in nucleus (Rian) locus, and subsequent overexpression of the adjacent retrotransposon like-1(Rtl1) gene have been reported following administration of rAAV-based viral vectors to neonatal mice (Chandler et al., 2015; Donsante et al., 2007). The increased susceptibility of neonatal mice to develop HCC is likely due to the higher expression of Rian in livers of neonatal mice, relative to adult mice (Li et al., 2012) and the preference for AAV to integrate into actively transcribed genes (Nakai et al., 2003). Other factors that increase susceptibility for neonatal mice to develop HCC included higher doses and the use of stronger promotor/enhancers (such as CB or thyroxin binding globulin [TGB]). However, it should be noted that HCC was not observed after administration to mice 6-8 weeks of age (Bell et al., 2005) or older (Li et al., 2011).
Following administration of rAAV-based vectors, low AAV integration have been reported in neonatal Wistar rats (Gauttier et al., 2013), NHP (Pañeda et al., 2013) and humans (Gil-Farina et al., 2016). However, the integrations appeared to be random, and were not associated with HCC. Nguyen et al. (2020), reported results from hemophilia A dogs that were treated with an AAV-canine FVIII therapy and followed for up to 10 years. These dogs had numerous integration events and evidence of clonal expansion near genes previously associated with growth control, but examination of the liver at necropsy did not show evidence of neoplasia in any dog. Collectively these data indicate that in most nonclinical species (excluding neonatal mice), rAAV vectors can integrate into the host DNA, randomly, and at a low rate, but without tumorgenicity.
The clinical relevance of integration events remains to be elucidated, and additional data is needed to ascertain the correlation between insertions and tumorgenicity. The Rian locus is not present in humans. Humans become infected with wild-type AAV at a young age without clear evidence of an association with carcinogenesis (Büning and Schmidt, 2015; Calcedo et al., 2011). Furthermore, gene therapy trials have been conducted for more than 20 years, but there has been minimal evidence HCC occurring in patients receiving gene therapies, and the cause of the HCC in these patients has not been clearly determined. Nevertheless, the potential for integration assessments should be considered, and based on the risk:benefit profile for rAAV-based gene therapies in development, nonclinical studies may be necessary to support some clinical trials.
Four major categories for potential toxicities associated with rAAV-based gene therapies have emerged, and involve the hepatic, immune, and nervous systems and theoretical risk of insertional mutagenesis. Although researchers continue to respond to new data and collectively work through these emerging safety issues, the field of gene therapy is rapidly evolving, and there is still much to learn. For example, it is not known how long efficacy will last, or what the long-term safety profile is in a patient administered a gene therapy. This is particularly important for some gene therapies administered to the very young, because following cell division, episomal vector is distributed to some daughter cells leading to dilution and eventual loss of transgene expression and efficacy in rapidly growing/dividing organs like the livers of pediatric patients. Understanding durability of efficacy is critical for establishing the risk:benefit profile for both pediatric and adult patients, and is something that Regulatory Authorities appear to be concerned about. In fact, marketing approval for valoctocogene roxaparvovec, a gene therapy in development for the treatment of hemophilia A, was recently delayed so that additional clinical data could be obtained to support its durability of effect (BioMarin, 2020).
We also have much to learn about modifying / dampening the immune response to rAAV-based gene therapies, to possibly allow for dosing to patients with pre-existing nAbs or to re-dose patients that have developed nAbs after the first administration of a gene therapy. Immunosuppression strategies that overcome AAV antibodies are currently being investigated (Meliani et al., 2018). For example, monkeys with pre-existing nAbs to AAV8 and pretreated with imlifidase, an IgG-degrading enzyme derived from Streptococcus pyogenes, demonstrated lower anti-AAV8 IgG titers and nAbs, and successful liver transduction of the transgene (Leborgne et al., 2020). However, use of this technology is limited by pre-existing humoral immunity to imlifidase, caused by exposure to S. pyogenes. Furthermore, imlifidase does not target transgene- or vector-induced B or T cell responses, including memory responses (Herzog and Biswas, 2020).
Nonclinical studies involving the administration of a gene therapy to embryonic cells (Ma et al., 2017) or in utero (Chan et al., 2019) have been reported. Germline or in utero gene therapy can correct disease-causing mutations early in development (i.e. when present in only a few embryonic cells), prevent the passage of the genetic defect to dividing daughter cells, and provide curative treatment before birth. In addition, the immune system of the fetus may tolerate gene therapies to a greater extent. However, there are numerous challenges that need to be resolved before extensive clinical trials can be conducted. These include ensuring the safety of both the mother and fetus, understanding the optimal timing for gene therapy administration, controlling the increased risk for infection, preterm labor and fetal loss, as well as the multitude of ethical concerns with dosing embryos or fetuses (Peranteau and Flake, 2020).
In summary, rAAV-based gene therapies have tremendous promise, but as with any new modality, they have some development challenges. Nonclinical studies in disease models and/or regulatory toxicology species provide important information on the safety of rAAV-based gene therapies, as well as for determining the relative risk for toxicity to occur in patients. Future scholarship in nonclinical models may be warranted to further investigate toxicities related to high doses, determine optimal human doses, and understand the potential mechanisms of liver, immune, and DRG effects. Furthermore, additional experience may help to determine if insertional mutagenesis and subsequent tumorigenicity is more than a theoretical risk for patients. Knowledge sharing across industry and academia, in close collaboration with regulatory authorities, can accelerate learnings in this field and increase the availability of safe rAAV-based gene therapies.
Note from AuthorsFor nonclinical studies that resulted in data shown in Figs. 1-3, all procedures performed on animals were in accordance with regulations and established guidelines and were reviewed and approved by the Institutional Animal Care and Use Committee.
The authors wish to thank Basel Assaf for his assistance with the histopathology evaluation of mouse dorsal root ganglion.
Conflict of interestMichael Bolt, Joseph Brady, Lawrence Whiteley and Nasir Khan are all paid employees of Pfizer, and have stock interests in these companies.