Abstract
Hepatotoxicity is one of the most common toxicities observed in non-clinical safety studies of drug candidates, and it is important to understand the hepatotoxicity mechanism to assess the risk of drug-induced liver injury in humans. In this study, we investigated the mechanism of hepatotoxicity caused by 2-[2-Methyl-1-(oxan-4-yl)-1H-benzimidazol-5-yl]-1,3-benzoxazole (DSP-0640), a drug candidate that showed hepatotoxicity characterized by centrilobular hypertrophy and vacuolation of hepatocytes in a 4-week oral repeated-dose toxicity study in male rats. In the liver of rats treated with DSP-0640, the expression of aryl hydrocarbon receptor (AHR) target genes, including Cyp1a1, was upregulated. In in vitro reporter assays, however, DSP-0640 showed only minimal AHR-activating potency. Therefore, we investigated the possibility that DSP-0640 indirectly activated AHR by inhibiting the CYP1 enzyme-dependent clearance of endogenous AHR agonists. In in vitro assays, DSP-0640 showed inhibitory effects on both rat and human CYP1A1 and enhanced rat and human AHR-mediated reporter gene expression induced by 6-formylindolo[3,2-b]carbazole, a well-known endogenous AHR agonist. The possible involvement of CYP1A1 inhibition in AHR activation was also demonstrated with other hepatotoxic compounds tacrine and albendazole. These results suggest that CYP1A1 inhibition-mediated AHR activation is involved in the hepatotoxicity caused by DSP-0640 and that DSP-0640 might induce hepatotoxicity in humans as well. We propose that CYP1A1 inhibition-mediated AHR activation is a novel mechanism for drug-induced hepatotoxicity.
INTRODUCTION
Drug-induced liver injury (DILI) is one of the most frequent drug-induced adverse events in humans that can be responsible for drug withdrawal from the market or discontinuation of drug development (Hornberg et al., 2014; Chen et al., 2011). In preclinical safety studies, hepatotoxicity is one of the most frequently observed toxicities in animals and is a finding that may lead to the discontinuation of further development of candidate compounds (McGill and Jaeschke, 2019). Therefore, it is important to understand the mechanism of hepatotoxicity observed in preclinical safety studies in order to predict the risk of hepatotoxicity in humans or to identify compounds that have less potential for hepatotoxicity.
Aryl hydrocarbon receptor (AHR) is known to be associated with hepatotoxicity and tumor promotion in the liver (Fernandez-Salguero et al., 1996; Harrill et al., 2016; Moennikes et al., 2004; Lu et al., 2019). It is a ligand-activated transcription factor classified as a member of the basic helix-loop-helix/Per-Arnt-Sim (bHLH-PAS) family and is generally known as a physiological sensor for xenobiotics in a wide range of species (Chopra and Schrenk, 2011; Denison and Nagy, 2003; Denison et al., 2011). The prototypical and most potent AHR ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) causes hepatic alterations, including cellular hypertrophy, lipid accumulation within hepatocytes, and inflammatory cell infiltration in rodents in an AHR-dependent manner (Fernandez-Salguero et al., 1996; Harrill et al., 2016). In addition, in mice expressing constitutively activated AHR in the liver, an increase in liver weight and accumulation of lipid droplets in hepatocytes were observed (Lee et al., 2010). In humans, although the route of exposure was not clarified in most reports, hepatotoxic effects such as slight and transient alterations in blood levels of hepatic enzymes, including alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (Pocchiari et al., 1979; Mocarelli et al., 1991), histopathological changes, including steatosis, periportal fibrosis or activation of Kupffer cells (Pazderova-Vejlupková et al., 1981), and alterations in lipid profiles (Pazderova-Vejlupková et al., 1981) have been reported in people accidentally and occupationally exposed to TCDD and its related compounds. Although limited information is available on AHR-dependent toxicity in humans, AHR activation may have toxicological effects on the liver in humans as well as in rodents.
AHR regulates the expression of a large battery of genes and causes biological and toxicological alterations in systemic organs, including the liver. The most representative AHR responsive genes are those encoding cytochrome P450 family 1 (CYP1) members, including CYP1A1, CYP1A2, and CYP1B1. In particular, CYP1A1 mRNA levels and/or CYP1A1 activity have long been regarded as the most sensitive indicators of AHR activation because they are rapidly and significantly induced by AHR activation. In addition, AHR has been reported to regulate genes related to oxidative stress responses and involved in lipid metabolism, transport, and processing in the liver of rodents (Boverhof et al., 2006; Dere et al., 2011). These studies suggest that AHR activation causes dynamic transcriptional changes and induces hepatotoxicity via transcriptional regulation of AHR target genes.
In recent years, it has become clear that not only xenobiotics such as TCDD but also endogenous substances play important roles in the control of AHR functions (Kou and Dai, 2021). Endogenous AHR ligands, including various indoles, tryptophan metabolites, heme metabolites, flavonoids, and arachidonic acid, have been identified so far (Murray and Perdew, 2017; Nguyen and Bradfield, 2008). Among them, 6-formylindolo[3,2-b]carbazole (FICZ), a tryptophan-derived metabolite formed by a light-dependent or -independent pathway (Wincent et al., 2009; Smirnova et al., 2016), is considered to be the most potent AHR ligand with high affinity for AHR, comparable to TCDD (Oberg et al., 2005; Wei et al., 1998; Rannug and Rannug, 2018). Several FICZ-derived metabolites were detected in human urine samples, indicating that FICZ is present in humans (Wincent et al., 2009). These reports implicate the contribution of endogenous ligands, including FICZ, to the regulation of AHR signaling in vivo.
Recent studies have suggested that AHR signaling in vivo is strictly regulated not only by AHR itself and its ligands but also by the modulation of ligand availability through the clearance of endogenous AHR ligands (Stockinger et al., 2014; Lamas et al., 2018). These ligands induce the expression of CYP1 family enzymes through AHR activation but are also efficiently metabolized by the same enzymes (Nguyen and Bradfield, 2008; Wincent et al., 2009; Ito et al., 2007). For example, FICZ is a strong inducer of CYP1A1 but also an excellent substrate for CYP1 family members, particularly CYP1A1. These facts suggest that autoregulatory feedback by CYP1 family members plays a central role in the regulation of AHR signaling by endogenous ligands (Rannug and Rannug, 2018). These facts also indicate that compounds that inhibit CYP1 enzymes can suppress the metabolic clearance of FICZ, resulting in increased levels of FICZ and prolonged activation of AHR (Wincent et al., 2009). Indeed, AHR activation by this mechanism has been reported for a variety of CYP1-inhibiting compounds (Wincent et al., 2012; Manzella et al., 2020; Mohammadi-Bardbori et al., 2012) and for the conditions where functional CYP1A1 is deficient (Wei et al., 2000). However, the toxicological relevance of the disruption of AHR signaling through interference with CYP1-dependent metabolism has been unclear.
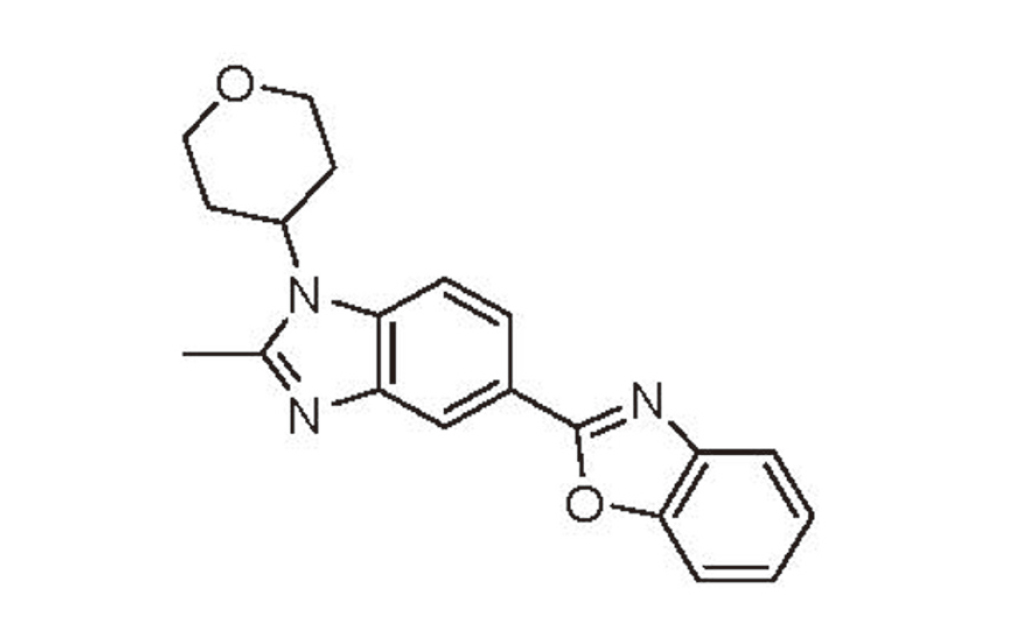
2-[2-Methyl-1-(oxan-4-yl)-1H-benzimidazol-5-yl]-1,3-benzoxazole (DSP-0640) (Fig. 1), a candidate phosphodiesterase 4 inhibitor indicated for Alzheimer’s disease (Kuroda et al., 2016), showed hepatotoxicity characterized by cellular hypertrophy and vacuolation in hepatocytes and marked Cyp1a1 induction in the liver in a 4-week repeated oral dose toxicity study in male rats. Here, we demonstrate the mechanism of Cyp1a1 induction by DSP-0640 and two other hepatotoxic compounds with CYP1A1-inhibitory effects (tacrine and albendazole) and propose the possible involvement of CYP1A1 inhibition-mediated AHR activation in their hepatotoxicity.
MATERIALS AND METHODS
Chemicals
The methanesulfonate and hemifumarate of DSP-0640 were synthesized at Sumitomo Chemical Co., Ltd. and Sumitomo Pharma Co., Ltd., respectively (Kuroda et al., 2016). FICZ, CH223191, and albendazole was purchased from Sigma-Aldrich Co. LLC (Saint Louis, MO, USA). Dimethyl sulfoxide (DMSO) was purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). 3-methylcholanthrene (3-MC) was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Tacrine hydrochloride was purchased from Cayman Chemical Company (Ann Arbor, MI, USA).
Animals
Five-week-old male Crl:CD (SD) rats were purchased from The Jackson Laboratory Japan, Inc. (Yokohama, Japan). The animals were housed in a room with a 12-hr light/dark cycle (lighting from 8:00 a.m. to 8:00 p.m.), temperature (20–26°C), relative humidity (40–70%), and ventilation exchange rate (at least 10 times per hour). Animals were given food and water ad libitum but were fasted approximately 15–20 hr before necropsy. All animal experiments were approved by the Institutional Animal Care and Use Committee of the test sites (Sumitomo Chemical Co., Ltd. for the 4-week repeated oral dose toxicity study of DSP-0640 or Sumitomo Pharma Co., Ltd. for the 2-week repeated oral dose toxicity study of DSP-0640).
The 4-week repeated oral dose toxicity study of DSP-0640 in male rats was performed as follows: The methanesulfonate of DSP-0640 was suspended in distilled water for injection. The rats, 6-week-old at the start of dosing, were orally dosed with DSP-0640 once daily at a volume of 5 mL/kg for 28 days. Dose levels of the methanesulfonate of DSP-0640 were 0, 100, 300, and 1000 mg/kg per day (5 animals/group). During the dosing period, clinical signs, body weights, and food consumption were evaluated regularly. On the day of necropsy, blood was collected from the abdominal aorta under ether anesthesia and anticoagulated with heparin sodium, and livers were removed, weighed, and fixed in 10% neutral buffered formalin. The trimmed livers were embedded in paraffin, and sections were prepared and stained with hematoxylin and eosin for histopathology.
The 2-week repeated oral dose toxicity study of DSP-0640 in male rats was performed as follows: The hemifumarate of DSP-0640 was suspended in 0.5% methylcellulose solution. The rats, 6-week-old at the start of dosing, were orally dosed with DSP-0640 once daily at a volume of 5 mL/kg for 14 days. Dose levels of the hemifumarate of DSP-0640 were 0, 150, and 300 mg/kg per day (5 animals/group). During the dosing period, clinical signs, body weights, and food consumption were monitored. On the day of necropsy, the rats were exsanguinated under isoflurane anesthesia, and livers were removed, weighed, and fixed in 10% neutral buffered formalin. The trimmed livers were embedded in paraffin, and sections were prepared and stained with hematoxylin and eosin for histopathology. For Oil Red O staining, the fixed specimens were frozen and sections were prepared as described previously (Tochitani et al., 2011).
Blood chemistry
Plasma collected by centrifugation was used for the measurement of total protein (TP), albumin (Alb), albumin/globulin ratio (A/G ratio), glucose (Glu), total cholesterol (T-Cho), triglycerides (TG), phospholipids (PL), total bilirubin (T-Bil), blood urea nitrogen (BUN), creatinine (Cre), AST, ALT, alkaline phosphatase (ALP), γ-glutamyl transpeptidase (γ-GTP), lactate dehydrogenase (LDH), creatine kinase (CK), sodium (Na), potassium (K), chloride (Cl), calcium (Ca) and inorganic phosphorus (P) using commercial kits (information will be provided upon request) and a Clinical Biochemistry Analyzer JCA-BM1250 (JEOL Ltd., Tokyo, Japan).
Microarray analysis
The livers of 3 animals in the 0 and 300 mg/kg groups of the 4-week study were used for microarray analysis. The livers were submerged in RNAlater RNA Stabilization Reagent (Qiagen N.V., Venlo, Netherlands) for 24 hr at room temperature and the stabilized tissues were stored at −20°C until use. Up to 30 mg of liver samples were homogenized in 100 μL of diethylpyrocarbonate-treated water and 350 μL of Buffer RLT containing 1% 2-mercaptoethanol by a Mixer Mill MM300 (Qiagen N.V.) at 25 Hz, for 4 min, twice. Then total RNA was extracted using an RNeasy Mini Kit (Qiagen N.V.) according to the manufacturer’s protocol. The quantity of isolated total RNA was determined using an Ultrospec 3100 pro Spectrophotometer (GE Healthcare, Chicago, IL, USA) and the quality was assessed with a Bioanalyzer 2100 using an RNA 6000 Nano kit (Agilent Technologies, Santa Clara, CA, USA). Sample preparation for gene expression analysis was performed using kits obtained from Thermo Fisher Scientific (Waltham, MA, USA) according to the manufacturer’s instructions and recommendations. Briefly, 5 μg of total RNA from each sample was converted into double-stranded cDNA using a One-Cycle cDNA Synthesis Kit (Thermo Fisher Scientific). After cleaning-up, biotin-labeling was performed by in vitro transcription using an IVT labeling Kit (Thermo Fisher Scientific). The 20 μg of amplified cRNA was fragmented using 5 × fragmentation buffer (Sample Cleanup Module). The cRNA was then hybridized to GeneChip Rat Genome 230 2.0 Array (Thermo Fisher Scientific). After hybridization, the chips were washed and stained in a GeneChip Fluidics Station (Thermo Fisher Scientific) and scanned with a GeneChip Scanner 3000 (Thermo Fisher Scientific). The data obtained were normalized by the MAS5 algorithm and analyzed by the empirical Bayes method, using Transcriptome Analysis Console (TAC) Software (Thermo Fisher Scientific).
Cell culture
A human hepatoma cell line HepG2 and a rat hepatoma cell line H-4-II-E (H4IIE) were purchased from American Type Culture Collection (Manassas, VA, USA) and the European Collection of Authenticated Cell Cultures (Salisbury, UK), respectively. They were maintained in D-MEM (FUJIFILM Wako Pure Chemical Corporation) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 1% MEM Non-essential Amino Acids at 37°C in a humidified incubator with 5% CO2/95% air. Rat primary hepatocytes were isolated from 10-week-old male Sprague-Dawley rats (The Jackson Laboratory Japan, Inc.), as described previously (Goto et al., 2015) with some modifications. The animals were handled by specialized personnel of the SUMIKA TECHNOSERVICE CORPORATION and the handling procedures were approved by its Institutional Animal Care and Use Committee. The liver was perfused with Hank’s balanced salt solution (HBSS), supplemented with ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’-tetraacetic acid (EGTA), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and sodium bicarbonate. Subsequently, the liver was digested by perfusion with collagenase solution. After digestion, the liver was excised and dispersed in Williams’ medium E supplemented with 5% FBS, 2 mM L-glutamine, and 1% penicillin/streptomycin (Cell washing solution). The cell suspension was centrifuged (50 × g for 2 min at 4°C), and the supernatant was removed. Then, the washing step, including the addition of 40 mL of Cell washing solution, filtration of the cell suspension through a cell strainer, centrifugation, and supernatant removal, was repeated 3 times. To obtain hepatocytes, the precipitate was suspended in Williams’ medium E supplemented with 5% FBS, 2 mM L-glutamine, 1 μM dexamethasone, 100 nM insulin, 10 mM nicotinamide, 200 μM L-ascorbic acid, and 1% penicillin/streptomycin. Cells were seeded on collagen-coated plates and cultured at 37°C in a humidified incubator with 5% CO2/95% air.
Quantitative reverse transcription PCR
Livers of the 2-week study rats were immediately submerged in RNAlater RNA Stabilization Reagent and incubated overnight at 4°C, and the stabilized tissues were stored at −80°C until use. Total RNA was extracted by the TRIzol-chloroform method and purified using RNeasy spin columns (Qiagen N.V.). cDNA was synthesized using a QuantiTect Reverse Transcription Kit (Qiagen N.V.) according to the manufacturer’s instructions.
For cDNA preparation from cultured cells, samples were prepared using a TaqMan Gene Expression Cells-to-CT Kit (Thermo Fisher Scientific) in accordance with the manufacturer’s instructions. Briefly, cultured cells were washed with phosphate-buffered saline (PBS), mixed with Lysis Solution, and incubated at room temperature. After 5 min, Stop Solution was added to the lysate and the mixture was incubated for 2 min and then reverse-transcribed to synthesize cDNA using 20 × RT Enzyme Mix and 2 × RT Buffer.
cDNA samples were used for TaqMan probe-based semi-quantitative real-time PCR. The mRNA levels of rat Cyp1a1, Cd36, and Abcc3 were measured in triplicate on a 7300 Real-Time PCR System using TaqMan Gene Expression Master Mix (Thermo Fisher Scientific). The mRNA levels were normalized to those of rat Gapdh. The following TaqMan probes were used: Rn00487218_m1 for Cyp1a1, Rn00580728_m1 for Cd36, Rn01452854_m1 for Abcc3, and Rn01775763_g1 for Gapdh.
AHR reporter assay
To examine CYP1A1 inhibition-dependent AHR activation in reporter assays, it is better to use cell lines in which CYP1A1 can be induced but CYP1A1 expression is low as in the liver in vivo. In this study, we thus used HepG2 and H4IIE cells as human- and rat-derived cells, respectively, because it is reported in these cells that CYP1A1 is enzymatically active although the basal activity is low and that it is inducible by FICZ treatment (Mohammadi-Bardbori et al., 2017; Rannug and Rannug, 1989; Wei et al., 1999).
In AHR reporter assays, 3×XRE-TK-pGL4.10, containing 3 copies of XRE followed by TK promoter (Matsuzaka et al., 2020) and pGL4.74[hRluc/TK] control plasmid (Promega Corporation, Madison, WI, USA) were used. Transfection of plasmids and detection of firefly and Renilla luciferase activity were performed using Lipofectamine 3000 reagent (Thermo Fisher Scientific) and the Dual-Glo Luciferase Assay System (Promega Corporation), respectively, in accordance with the manufacturers’ instructions. Briefly, the transfection reagent was prepared by adding solution A (Opti-MEM I reduced Serum Medium [Thermo Fisher Scientific], containing pGL4.74[hRluc/TK], 3 × XRE-TK-pGL4.10, and P3000 reagent) to Solution B (Opti-MEM I reduced Serum Medium containing Lipofectamine 3000), and then incubating the mixture for 10 min at room temperature. The transfection reagent was added to cell suspension (HepG2 or H4IIE), and the mixture was seeded on LumiNunc 96-well solid plates (Thermo Fisher Scientific). After 24 hr, serum-free medium containing each test compound was added to the cells after removing the medium. Following additional incubation for 24 hr, the cells were washed with PBS, and then luminescence was determined using an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA, USA).
Cytochrome P450 (P450) inhibition assay
P450 inhibition assay was conducted using the P450-Glo CYP1A1 Assay, P450-Glo CYP1A2 Induction/Inhibition Assay, and P450-Glo CYP1B1 Assay kits (Promega Corporation) as described previously (Shimizu et al., 2021; Watanabe et al., 2020) with some modifications. The enzyme sources included human CYP1A1R EasyCYP Bactosomes, human CYP1A2R EasyCYP Bactosomes, human CYP1B1LR EasyCYP Bactosomes, rat CYP1A2LR Bactosomes, and rat CYP1B1LR Bactosomes (Cypex, Dundee, Scotland, UK). Due to the unavailability of rat CYP1A1 recombinant protein, β-naphthoflavone-treated SD rat liver microsomes (SEKISUI XenoTech, LLC., Kansas City, KS, USA) were used as a rat CYP1A1-predominant enzyme source. Briefly, 25 μL of substrate mixture containing a P450-Glo substrate, enzyme source, and each test compound in potassium phosphate buffer (pH 7.4) was added to LumiNunc 96-well solid plates, and the mixture was incubated for 10 min at the assay temperature indicated (Supplemental Table 1). Reactions were then initiated by adding 25 μL of the 2 × NADPH regeneration system (Promega Corporation), which was prepared according to the manufacturer’s instructions. The final concentrations of each component and conditions in each assay are shown in Supplemental Table 1. After incubation, the addition of 25 μL of Luciferin Detection Reagent to the reaction mixture was followed by incubation at room temperature for 20 min, and luminescence was determined using an EnSpire Multimode Plate Reader. The half-maximal inhibitory concentration (IC50) values were calculated by fitting the data to the sigmoidal dose-response equation using Origin Pro 2021b (OriginLab Corporation, Northampton, MA, USA). Typical P450 inhibitors (Supplemental Table 1) were used to confirm assay conditions (Shimizu et al., 2021; Watanabe et al., 2020).
Statistical analysis
Results are presented as the means ± standard deviation (S.D.). Comparisons between groups were performed by Dunnett’s multiple comparison test using SAS version 9.4 software (SAS Institute Inc., Cary, NC, USA). A p-value less than 0.05 was considered statistically significant.
RESULTS
Four-week repeated oral dose toxicity study of DSP-0640 in male rats
To understand the toxicological properties of DSP-0640, a 4-week repeated oral dose toxicity study was performed using male rats and the results are summarized in Table 1. In this study, no death or moribundity was observed at any of the tested dose levels (i.e., 100, 300, and 1000 mg/kg). In the blood chemistry, increases in T-Bil, ALT and γ-GTP at 1000 mg/kg were noted. Apart from the increases in typical hepatic parameters, increases in Alb at 300 mg/kg and above and in TP, A/G ratio, T-Cho, PL and Ca at 1000 mg/kg, and a decrease in TG at 1000 mg/kg were observed, among the parameters evaluated. Dose-dependent increases in relative liver weight at 100 mg/kg and above were observed. Histopathological examination of the liver revealed centrilobular hypertrophy of hepatocytes at 100 mg/kg and above, centrilobular single cell necrosis of hepatocytes and yellowish-brown pigments in hepatocytes or Kupffer cells at 300 mg/kg and above, and eosinophilic droplet in hepatocytes at 1000 mg/kg. In addition, centrilobular or mid-zonal vacuolation of hepatocytes was noted in all the DSP-0640 dose groups. Although there was no clear dose-dependency for the vacuolation, this finding was considered DSP-0640-related because it was not observed in the 0 mg/kg group.
Table 1. Findings in the 4-week repeated oral dose toxicity study of DSP-0640 in rats.
 Microarray analysis using the liver of DSP-0640-treated rats
Microarray analysis using the liver of DSP-0640-treated rats
To investigate the alteration of gene expression in the livers of DSP-0640-treated rats, a microarray analysis was conducted using liver samples from the 0 and 300 mg/kg groups in the 4-week study. Among 31,042 genes represented on the rat Affymetrix GeneChip 230 2.0, the expression of 6 genes were upregulated in the 300 mg/kg group compared to the 0 mg/kg group by empirical Bayes analysis (fold change, > 1.5 or < –1.5; p-value, < 0.05; and false discovery rate [FDR], < 0.05; Table 2). Among the genes listed, Cyp1a1, a representative AHR target gene, was upregulated remarkably. The upregulation of Abcc3 and Akr7a3, which are known as oxidative stress response-related genes (Maher et al., 2007; Dewa et al., 2008), was also prominent. In addition, the expression of the Cd36 gene, which encodes a fatty acid transport protein involved in hepatic steatosis caused by AHR activation (Lee et al., 2010), was also upregulated. These results suggest the activation of AHR signaling in the liver of DSP-0640-treated rats.
Table 2. Genes upregulated after 4-week treatment with DSP-0640 (300 mg/kg) in rat livers.
| Gene symbol |
Description |
Fold change |
p-value |
FDR |
| Abcc3 |
ATP-binding cassette, subfamily C (CFTR/MRP), member 3 |
139 |
7.25E-07 |
0.0225 |
| Akr7a3 |
Aldo-keto reductase family 7, member A3 (aflatoxin aldehyde reductase) |
11.5 |
1.88E-06 |
0.0292 |
| Cd36 |
CD36 molecule (thrombospondin receptor); scavenger receptor class B, member 2-like; platelet glycoprotein 4-like |
5.47 |
9.77E-06 |
0.0448 |
| Cyp1a1 |
Cytochrome P450, family 1, subfamily a, polypeptide 1 |
371 |
9.82E-06 |
0.0448 |
| Gsta3 |
Glutathione S-transferase alpha 3 |
88.1 |
1.11E-05 |
0.0448 |
| Aldh1a1 |
Aldehyde dehydrogenase 1 family, member A1 |
7.77 |
1.15E-05 |
0.0448 |
FDR, false discovery rate.
DSP-0640-induced hepatotoxic phenotypes and changes in hepatic gene expression
To investigate a possible cause of the centrilobular or mid-zonal vacuolation of hepatocytes and its association with gene expression changes in AHR signaling, male rats were orally treated with DSP-0640 for 2 weeks at 150 and 300 mg/kg. In the histopathological examination, centrilobular hypertrophy and mid-zonal vacuolation of hepatocytes but not single cell necrosis was observed at 150 and 300 mg/kg. The frequency and severity of vacuolation were dose-dependent, and the vacuolation was confirmed to be lipid accumulation by Oil Red O staining (Fig. 2A and 2B). In addition, the dose-dependent upregulation of Cyp1a1, Cd36, and Abcc3 mRNA levels in the liver was confirmed (Fig. 2C). These results suggest that DSP-0640 treatment activates AHR signaling and oxidative stress responses at earlier time points than single cell necrosis is observed.
Mechanism of CYP1A1 induction by DSP-0640
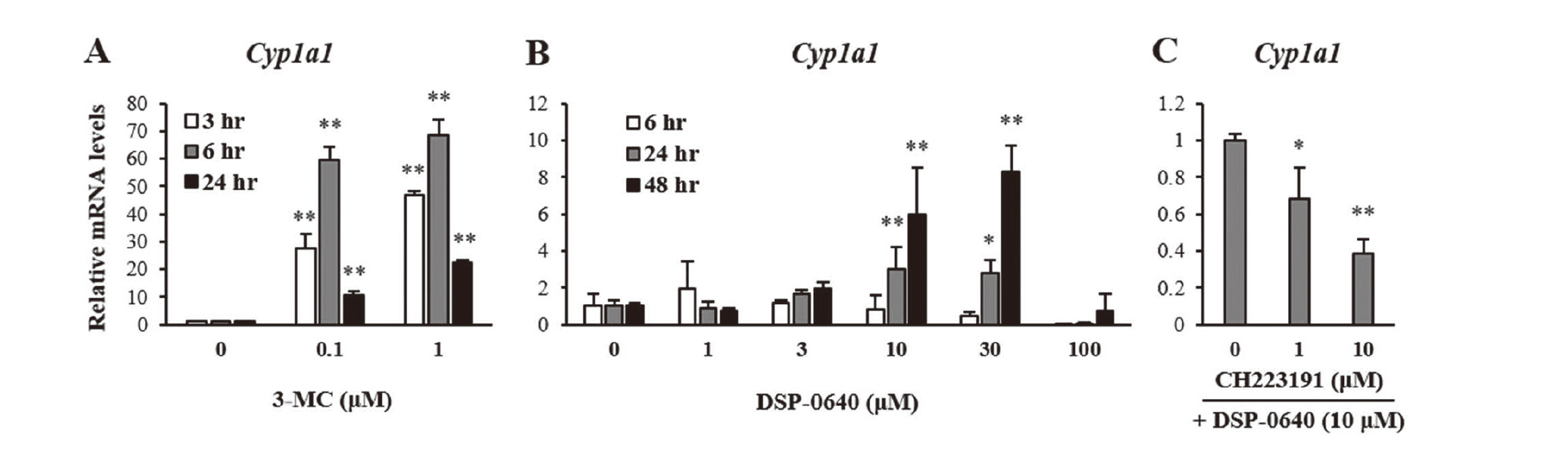
Next, we evaluated the Cyp1a1 induction by DSP-0640 in vitro using rat primary hepatocytes. In these cells, 3-MC, a typical AHR ligand, increased Cyp1a1 mRNA levels drastically at 3 and 6 hr after treatment (Fig. 3A). On the other hand, DSP-0640 increased the mRNA levels only at 24 and 48 hr but not at 6 hr after treatment (Fig. 3B). The induction was concentration-dependent except for 100 μM, at which the viability of rat hepatocytes was reduced (data not shown). In addition, the induction by DSP-0640 was considered AHR-dependent because it was suppressed by co-treatment with the AHR antagonist CH223191 at 1 μM or more (Fig. 3C), concentrations that did not affect the cell viability (data not shown).
To investigate the AHR agonism of DSP-0640, reporter gene assays were conducted using rat hepatoma-derived H4IIE cells. In this assay system, AHR-dependent reporter activity was induced more than 10-fold by treatment with FICZ, a well-known endogenous AHR agonist (Fig. 4A). DSP-0640 also increased the reporter activity, but its degree was less than 3-fold even at the highest concentration of 100 μM (Fig. 4B, open bars).
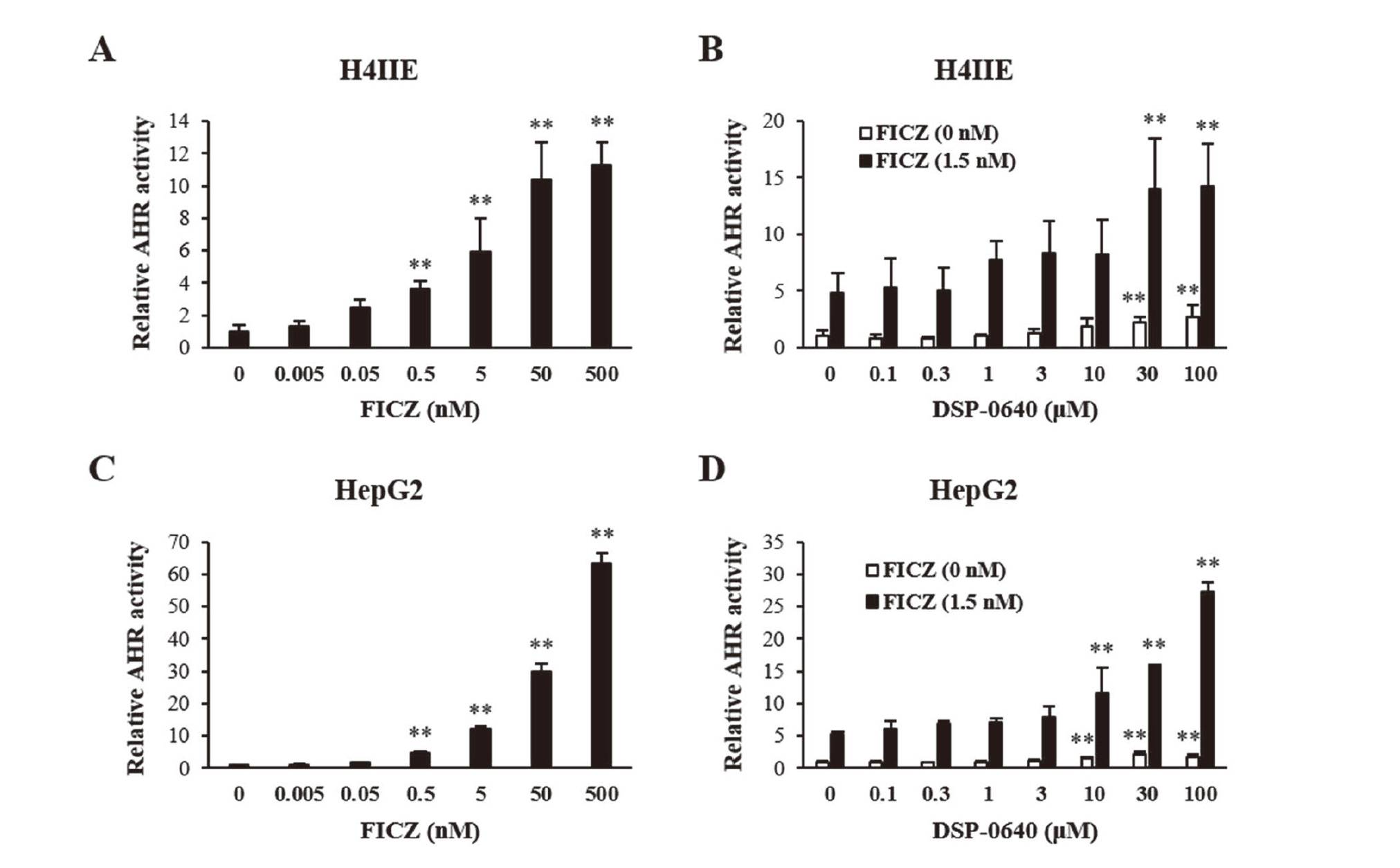
Since the AHR-activating potency of DSP-0640 was considered to be minimal up to 100 μM, a concentration about 16 times the maximum serum concentration (Cmax) on day 28 when 300 mg/kg of DSP-0640 was administered to rats for 4 weeks (data not shown), we focused on the mechanism by which CYP1-inhibiting compounds indirectly activate AHR through inhibiting metabolic clearance of endogenous AHR ligands (Wincent et al., 2012; Manzella et al., 2020; Mohammadi-Bardbori et al., 2012). To assess the involvement of this mechanism in DSP-0640-dependent CYP1A1 induction, we conducted reporter gene assays by co-treatment with DSP-0640 and FICZ (Fig. 4B). In this assay, FICZ concentration was set at 1.5 nM, where FICZ was expected to moderately activate AHR in the reporter assay (Fig. 4A). Compared to the reporter activity in the group treated with DSP-0640 alone, reporter activity in each FICZ co-treated group was augmented (Fig. 4B, closed bars). The augmentation was especially marked in the FICZ-treated groups receiving 30 or 100 μM of DSP-0640.
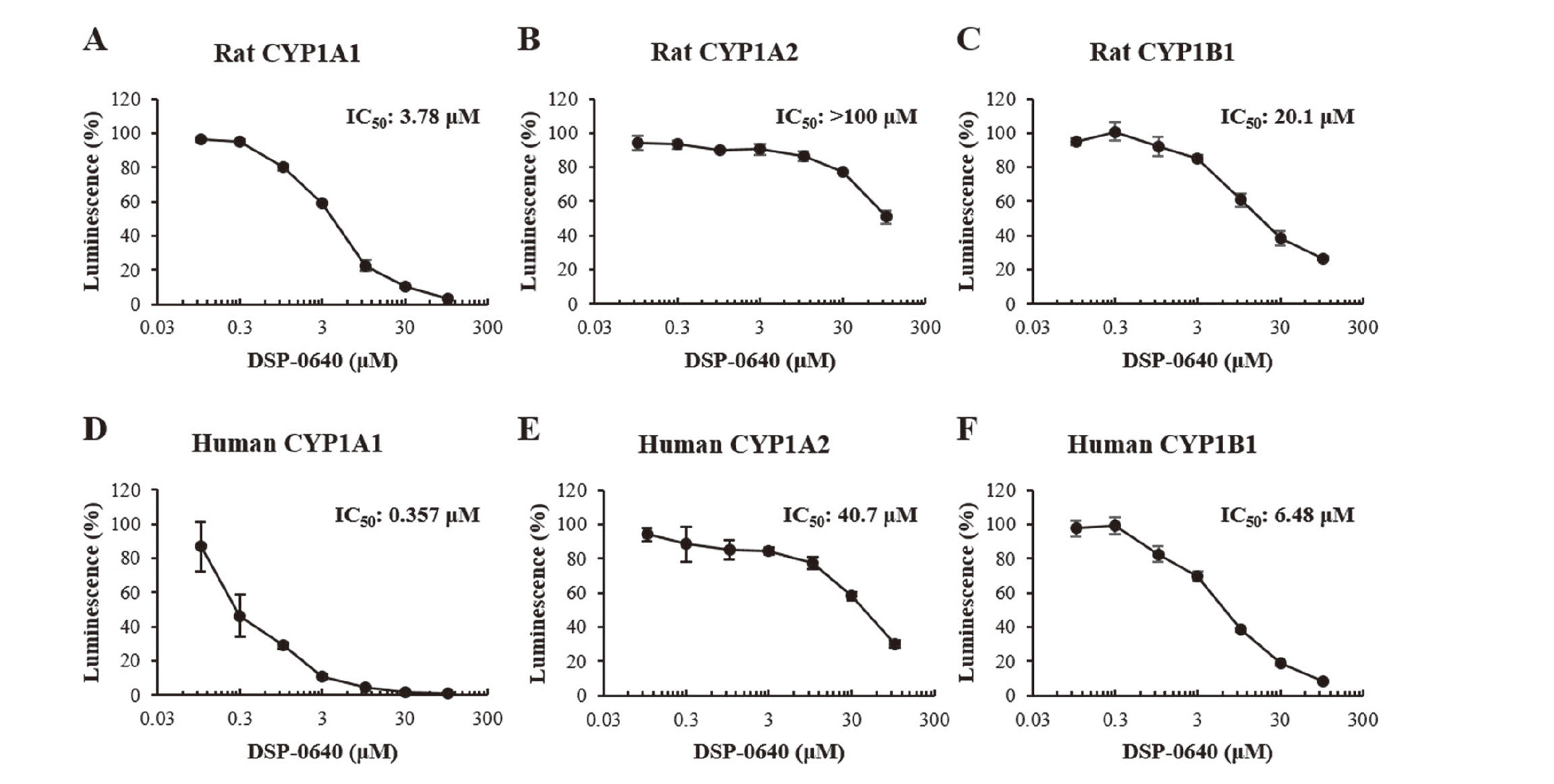
To investigate whether DSP-0640 had inhibitory effects on rat CYP1 family enzymes, P450 inhibition assays were conducted using recombinant rat CYP1A2 and CYP1B1, and β-naphthoflavone-treated rat liver microsomes as the CYP1A1-enriched enzyme source. Under the conditions used (Supplemental Table 1), the IC50 values of the DSP-0640-mediated inhibition for the reactions mediated by rat CYP1A1-enriched microsomes, CYP1A2, and CYP1B1 were 3.78, > 100, and 20.1 μM, respectively (Fig. 5A–5C). Although β-naphthoflavone induces not only CYP1A1 but also CYP1A2 and the IC50 values for rat CYP1A2 metabolism was > 100 μM, the IC50 value determined for rat CYP1A1 using the microsomes was considerably low, indicating that the contribution of CYP1A2 to the microsome-mediated reaction was minimal. These results suggest that DSP-0640 shows strong inhibitory effects on CYP1A1 among CYP1 family enzymes in rats.
CYP1 inhibition-mediated AHR activation by DSP-0640 in humans
To investigate whether DSP-0640 could activate AHR by inhibiting CYP1 enzymes in humans, AHR reporter assays were conducted using human-derived HepG2 cells. In this assay system, FICZ increased the reporter activity more than 60-fold (Fig. 4C). DSP-0640 also increased the activity, but its degree was minimal (up to around 2-fold at 100 μM) (Fig. 4D, open bars) as in rat H4IIE cells. On the other hand, in the presence of FICZ (1.5 nM), the reporter activity was greatly increased by DSP-0640 at 10 μM or more (Fig. 4D, closed bars), suggesting that DSP-0640 inhibits the CYP1-dependent metabolism of FICZ in humans as well.
To evaluate the inhibitory effects of DSP-0640 on human CYP1 family enzymes, P450 inhibition assays were conducted using human recombinant proteins. Under the conditions used, the IC50 values for the inhibition by DSP-0640 of CYP1A1-, CYP1A2-, and CYP1B1-mediated reactions were 0.357, 40.7, and 6.48 μM, respectively (Fig. 5D–5F). These results indicate that DSP-0640 inhibits human CYP1 family enzymes with a preference for CYP1A1 as in rats.
CYP1A1 inhibition-mediated AHR activation by other hepatotoxic drugs
To understand the toxicological significance of the CYP1A1 inhibition-associated AHR activation observed with DSP-0640, we investigated whether this mechanism could occur for other hepatotoxic compounds, namely tacrine and albendazole, which have been used in clinical practice. The former has been reported to show CYP1A1 inhibitory effects in rats and humans (Westerink et al., 2008), and the latter is known to induce CYP1A1 extensively in rat livers in vivo without direct AHR binding in vitro (Hu et al., 2007). It is reported that CYP1A1-inducing ability in vivo does not necessarily correlate with the ability to bind to AHR and activate it in vitro for a number of compounds, including albendazole (Hu et al., 2007), and we thus thought that the involvement of the CYP1A1 inhibition-associated mechanism might explain this discrepancy.
Using these two drugs, P450 inhibition assays and AHR reporter assays were performed with rat- and human-derived samples. Tacrine showed a relatively weak inhibitory effect against rat CYP1A1 (IC50: 24.7 μM, Fig. 6A) and strong inhibitory effect against human CYP1A1 (IC50: 0.142 μM, Fig. 6B). In reporter gene assays, as expected, tacrine only slightly augmented the FICZ-mediated increase in reporter activity in rat H4IIE cells (Fig. 6C, closed bars), while it drastically increased reporter activity in human cells in the presence of FICZ (Fig. 6D, closed bars). Tacrine alone activated AHR less than 5-fold in humans (Fig. 6D, open bars) and did not activate AHR in rats (Fig. 6C, open bars).

Albendazole showed a strong inhibitory effect against CYP1A1-dependent metabolism in rats (IC50: 0.106 μM, Fig. 6E) and humans (IC50: 0.108 μM, Fig. 6F), and it clearly augmented reporter activity in the presence of FICZ in both species (Fig. 6G and 6H, closed bars). Albendazole alone increased reporter activity less than 3-fold and up to 15-fold in rat- and human-derived cells (Fig. 6G and 6H, open bars), respectively.
DISCUSSION
In this study, DSP-0640, which caused liver toxicity in the rat 4-week study, enhanced AHR signaling, and mechanistic analyses suggest that the DSP-0640-mediated AHR activation is associated with its inhibitory effects against CYP1A1. Furthermore, CYP1A1 inhibition-mediated AHR activation was also observed with other hepatotoxic drugs tacrine and albendazole. These results suggest that this indirect AHR activation mechanism plays a role in hepatotoxicity caused by compounds with CYP1A1 inhibitory potential.
In DSP-0640-treated rats, centrilobular hypertrophy and vacuolation of hepatocytes were observed in the liver. These histopathological changes in the liver have been also noted after TCDD administration in rodents (Fernandez-Salguero et al., 1996; Harrill et al., 2016). The hepatocyte hypertrophy is known to be associated with enzyme induction (Maronpot et al., 2010), and the significant upregulation of Cyp1a1, a representative AHR responsive gene, was noted in the livers of DSP-0640-treated rats. The vacuolation, which was confirmed to be lipid accumulation in hepatocytes, was also observed in mice expressing constitutively activated AHR in the liver, and the involvement of CD36, a fatty acid transport protein, in lipid accumulation in this model has been demonstrated (Lee et al., 2010). In the present study, we confirmed the upregulation of Cd36 expression in the liver of DSP-0640-treated rats (Fig. 2C). These results suggest that the alterations of gene expressions following AHR activation contribute to the abovementioned histopathological findings observed after treatment with DSP-0640.
In the histopathological examination of the 4-week study of DSP-0640 in male rats, the yellowish-brown pigment in hepatocytes or Kupffer cells was observed at 300 mg/kg and above and was considered to be lipofuscin pigmentation, suggestive of oxidative stress. Correspondingly, the expression of oxidative stress response-related genes, including Abcc3 (Maher et al., 2007) and Akr7a3 (Dewa et al., 2008), was increased in the livers of DSP-0640-treated rats. AHR activation is generally considered to be associated with a cellular oxidative stress response (Dalton et al., 2002). In addition, during the monooxygenase reaction by P450s including CYP1 family members, reactive oxygen species (ROS) are released in the course of the electron transfer from NADPH-cytochrome P450 oxidoreductase to bound oxygen (Hrycay and Bandiera, 2015). ROS produced by this mechanism is considered to contribute significantly to the total cellular production of ROS in rat livers, even in the absence of enzyme induction (Bondy and Naderi, 1994). Based on these reports, it is proposed that DSP-0640-induced AHR activation and/or CYP1A1 induction may have induced oxidative stress and enhanced hepatotoxicity, as indicated by single cell necrosis.
In the present study, DSP-0640 augmented AHR activity under cotreatment with an endogenous AHR agonist, even though DSP-0640 itself showed only weak AHR-activating potency, suggesting the involvement of CYP1A1 inhibition-mediated mechanism in DSP-0640-dependent AHR activation. In the 4-week repeated oral dose toxicity study in rats, remarkable upregulation of Cyp1a1 was noted at 300 mg/kg, and the Cmax on day 28 at this dose level was 2080 ng/mL (6.2 μM, data not shown). On the other hand, in the AHR reporter assay using rat hepatoma-derived H4IIE cells, AHR-activating potency of DSP-0640 was minimal up to 100 μM, a concentration about 16 times the Cmax on day 28 at 300 mg/kg in the 4-week study. In addition, in rat primary hepatocytes, DSP-0640 increased the Cyp1a1 mRNA levels only at 24 and 48 hr but not at 6 hr after treatment, even though typical AHR ligand 3-MC increased Cyp1a1 mRNA levels drastically at 3 and 6 hr after treatment, suggesting involvement of indirect mechanism in DSP-0640-dependent AHR activation. Based on these results, AHR agonism of DSP-0640 was considered to be minimal in rats. Instead, DSP-0640 showed CYP1A1 inhibiting potency and enhanced FICZ-dependent AHR reporter activity in rat-derived in vitro assay systems. Given that compounds with CYP1-inhibiting potency have been reported to enhance FICZ-driven AHR activation (Wincent et al., 2012; Manzella et al., 2020; Mohammadi-Bardbori et al., 2012), DSP-0640 was considered to enhance AHR activity in vivo by this mechanism.
Although the number of reports on the toxicity caused by the CYP1 inhibition-mediated AHR activation mechanism is limited, several studies using FICZ as an endogenous AHR ligand have revealed the enhancement of FICZ-induced toxicity by CYP1 inhibition. In chicken and Japanese quail embryos, FICZ caused liver toxicity and embryo mortality with an increase in CYP1A4 and CYP1A5 expression levels. In addition, when chick embryos were treated with FICZ and the CYP1 inhibitor ketoconazole, mortality and increase in CYP1A4 expression by FICZ were enhanced (Jönsson et al., 2016). Furthermore, CYP1A knockdown or treatment with α-naphthoflavone (a CYP1 inhibitor) increased the mortality of zebrafish embryos caused by FICZ (Wincent et al., 2016). Although the information on rodents and humans is limited, it is possible that CYP1-inhibiting compounds indirectly activate AHR by increasing the levels of endogenous AHR ligands to dysregulate AHR functions and thereby cause toxicological changes.
In this study, we investigated the CYP1A1 inhibition-mediated AHR activation mechanism using tacrine and albendazole, to test whether this mechanism could be commonly observed with hepatotoxic drugs. Tacrine is a cholinesterase inhibitor indicated for Alzheimer’s disease. In rat toxicity studies of tacrine for up to 52 weeks, rats were exposed to concentrations up to 10 times higher than human blood levels (Cmax), but there were no histological findings suggestive of hepatotoxicity (Woolf et al., 1996). However, in clinical practice, elevated blood ALT levels were observed in ~50% of patients, and the compound was withdrawn from the market in many countries (Woolf et al., 1996). In the present study, tacrine inhibited human CYP1A1 (IC50: 0.142 μM) more potently than rat CYP1A1 (IC50: 24.7 μM), and it markedly enhanced FICZ-induced AHR activity in human-derived HepG2 cells. Although the involvement of reactive metabolites has been suggested in tacrine-induced DILI (Usui et al., 2009), the AHR activation by a CYP1A1 inhibition-mediated mechanism may contribute to the ALT elevation in humans.
Albendazole is a drug for parasitic infections. In its rat toxicity study, the enlargement of centrilobular hepatocytes was noted (Dayan, 2003). In clinical practice, after a prolonged period of treatment, the asymptomatic elevation of ALT was observed in up to 50% of patients (Piloiu and Dumitrascu, 2021). In the present study, albendazole strongly inhibited both rat and human CYP1A1 to a similar extent, suggesting that it could cause CYP1A1 inhibition-mediated AHR activation in both species. In fact, albendazole augmented the FICZ-dependent AHR activation in both rat and human-derived cells. In addition, albendazole is reported to significantly induce Cyp1a1 in rat livers in vivo but not to be a classic agonist of rat AHR (Hu et al., 2007). Based on our present findings, we suggest that the CYP1A1 inhibition-mediated AHR activation may explain the strong CYP1A1 induction by albendazole in vivo.
In both rats and humans, the inhibitory potential of DSP-0640 was strongest against CYP1A1 among the CYP1 family members tested, and this tendency was also noted for tacrine and albendazole (Supplemental Fig. 1), but tacrine similarly inhibited human CYP1B1. This is noteworthy, as it may explain in part our recent findings on the significant association between human CYP1A1 inhibition and DILI risk in humans (Shimizu et al., 2021). In their study, a total of 358 drugs were examined at a concentration of 10 μM, and approximately 10%, 1% and 6% of drugs showed inhibitory activity by 90% or more against human CYP1A1, CYP1A2 and CYP1B1, respectively. Similarly, our previous study using 148 industrial chemicals demonstrated that 11%, 1%, 4%, 11%, and 5% of them showed inhibitory activity by 80% or more against human CYP1A1, human CYP1A2, human CYP1B1, rat CY1A1 and rat CYP1A2, respectively (Watanabe, et al., 2020). In general, polyaromatic hydrocarbons (PAHs) are considered typical substrates of CYP1A1 and CYP1B1, however, the compounds demonstrated to inhibit those enzymes in these studies do not necessarily have PAH-like structures, as with the compounds examined in our present study. In addition, CYP1A1 substrates can take various conformations due to their flexibility (Yamazoe and Yoshinari, 2020). Further study may clarify common physicochemical characteristics among CYP1A1-inhibiting compounds and its relation to DILI risk in humans.
Generally, the basal expression level of CYP1A1 in the liver is considerably low in both rats and humans, even with high interindividual variability in humans (Lang et al., 2019), and therefore the effect of CYP1A1 inhibition in the intact liver on the amount of AHR endogenous ligands is considered to be minimal in these species. However, CYP1A1 at an induced expression level may interact with drugs and then change the clearance of AHR endogenous ligands in the liver. In addition, it is possible that CYP1A1 inhibition in extrahepatic tissues, where CYP1A1 is highly expressed compared to the liver tissue (Lindell et al., 2003), may increase the systemic amounts of AHR endogenous ligands and contribute to the enhancement of AHR activity in the liver. In accordance with this possibility, recent studies suggest that the CYP1A1 expression level in the gastrointestinal tract affects the systemic exposure of AHR ligands (Ito et al., 2007; Schiering et al., 2017). In oral dose toxicity studies, the gastrointestinal tract is exposed to high concentrations of a test compound, and thus CYP1A1 in the gastrointestinal tract may be inhibited by the test compound. The contribution of CYP1A1 inhibition in extrahepatic tissues, including the gastrointestinal tract, to the activation of AHR in the liver is worth studying in the future.
DSP-0640 and tacrine showed species differences in CYP1A1 inhibition and subsequent AHR activation in this study. They inhibited human CYP1A1 more than rat CYP1A1. Some compounds are also reported to show species differences in CYP1A1 inhibition (Westerink et al., 2008; Watanabe et al., 2020). On the other hand, the AHR-activating potency of FICZ is reported to be similar in rats and humans (Doan et al., 2020), and the AHR response to exogenous ligands is generally considered to vary little between species. From these facts, it will be of great interest to further investigate whether a compound showing species differences in CYP1A1 inhibition may show species differences in hepatotoxicity caused by this mechanism.
In conclusion, we have shown that DSP-0640 and the hepatotoxic compounds tacrine and albendazole could activate AHR through a CYP1A1 inhibition-mediated mechanism. Given the relatively high incidence of DILI for tacrine and albendazole in humans, the hepatotoxic effects of these compounds may be explained at least in part by CYP1A1 inhibition-mediated AHR activation, and interindividual variations of hepatotoxicity might be dependent on the interindividual variations of endogenous AHR ligand levels. Although its hepatotoxic potential in humans has not been evaluated, DSP-0640 may have the potential to cause hepatotoxicity in humans, given that it has a greater inhibitory effect on human CYP1A1 than rat CYP1A1. We believe that our present findings are helpful in understanding hepatotoxicity observed in preclinical studies and in developing a strategy for compound screening to avoid hepatotoxicity in humans.
ACKNOWLEDGMENTS
We thank Yuka Fujii for her contributions to confirming the quality of DSP-0640. We thank members in Toxicology Group I and II, Preclinical Research Unit, Sumitomo Pharma Co., Ltd. for help with the animal studies and histotechnical works.
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Bondy, S.C. and Naderi, S. (1994): Contribution of hepatic cytochrome P450 systems to the generation of reactive oxygen species. Biochem. Pharmacol., 48, 155-159.
- Boverhof, D.R., Burgoon, L.D., Tashiro, C., Sharratt, B., Chittim, B., Harkema, J.R., Mendrick, D.L. and Zacharewski, T.R. (2006): Comparative toxicogenomic analysis of the hepatotoxic effects of TCDD in Sprague Dawley rats and C57BL/6 mice. Toxicol. Sci., 94, 398-416.
- Chen, M., Vijay, V., Shi, Q., Liu, Z., Fang, H. and Tong, W. (2011): FDA-approved drug labeling for the study of drug-induced liver injury. Drug Discov. Today, 16, 697-703.
- Chopra, M. and Schrenk, D. (2011): Dioxin toxicity, aryl hydrocarbon receptor signaling, and apoptosis-persistent pollutants affect programmed cell death. Crit. Rev. Toxicol., 41, 292-320.
- Dalton, T.P., Puga, A. and Shertzer, H.G. (2002): Induction of cellular oxidative stress by aryl hydrocarbon receptor activation. Chem. Biol. Interact., 141, 77-95.
- Dayan, A.D. (2003): Albendazole, mebendazole and praziquantel. Review of non-clinical toxicity and pharmacokinetics. Acta Trop., 86, 141-159.
- Denison, M.S. and Nagy, S.R. (2003): Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol., 43, 309-334.
- Denison, M.S., Soshilov, A.A., He, G., DeGroot, D.E. and Zhao, B. (2011): Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci., 124, 1-22.
- Dere, E., Lo, R., Celius, T., Matthews, J. and Zacharewski, T.R. (2011): Integration of genome-wide computation DRE search, AhR ChIP-chip and gene expression analyses of TCDD-elicited responses in the mouse liver. BMC Genomics, 12, 365.
- Dewa, Y., Nishimura, J., Muguruma, M., Jin, M., Saegusa, Y., Okamura, T., Tasaki, M., Umemura, T. and Mitsumori, K. (2008): beta-Naphthoflavone enhances oxidative stress responses and the induction of preneoplastic lesions in a diethylnitrosamine-initiated hepatocarcinogenesis model in partially hepatectomized rats. Toxicology, 244, 179-189.
- Doan, T.Q., Connolly, L., Igout, A., Muller, M. and Scippo, M.L. (2020): In vitro differential responses of rat and human aryl hydrocarbon receptor to two distinct ligands and to different polyphenols. Environ. Pollut., 265 (Pt B), 114966.
- Fernandez-Salguero, P.M., Hilbert, D.M., Rudikoff, S., Ward, J.M. and Gonzalez, F.J. (1996): Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol., 140, 173-179.
- Goto, S., Deguchi, J., Nishio, N., Nomura, N. and Funabashi, H. (2015): Hepatotoxicants induce cytokine imbalance in response to innate immune system. J. Toxicol. Sci., 40, 389-404.
- Harrill, J.A., Layko, D., Nyska, A., Hukkanen, R.R., Manno, R.A., Grassetti, A., Lawson, M., Martin, G., Budinsky, R.A., Rowlands, J.C. and Thomas, R.S. (2016): Aryl hydrocarbon receptor knockout rats are insensitive to the pathological effects of repeated oral exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Appl. Toxicol., 36, 802-814.
- Hornberg, J.J., Laursen, M., Brenden, N., Persson, M., Thougaard, A.V., Toft, D.B. and Mow, T. (2014): Exploratory toxicology as an integrated part of drug discovery. Part I: why and how. Drug Discov. Today, 19, 1131-1136.
- Hrycay, E.G. and Bandiera, S.M. (2015): Involvement of Cytochrome P450 in Reactive Oxygen Species Formation and Cancer. Adv. Pharmacol., 74, 35-84.
- Hu, W., Sorrentino, C., Denison, M.S., Kolaja, K. and Fielden, M.R. (2007): Induction of cyp1a1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro. Mol. Pharmacol., 71, 1475-1486.
- Ito, S., Chen, C., Satoh, J., Yim, S. and Gonzalez, F.J. (2007): Dietary phytochemicals regulate whole-body CYP1A1 expression through an arylhydrocarbon receptor nuclear translocator-dependent system in gut. J. Clin. Invest., 117, 1940-1950.
- Jönsson, M.E., Mattsson, A., Shaik, S. and Brunström, B. (2016): Toxicity and cytochrome P450 1A mRNA induction by 6-formylindolo[3,2-b]carbazole (FICZ) in chicken and Japanese quail embryos. Comp. Biochem. Physiol. C Toxicol. Pharmacol., 179, 125-136.
- Kou, Z. and Dai, W. (2021): Aryl hydrocarbon receptor: its roles in physiology. Biochem. Pharmacol., 185, 114428.
- Kuroda, K., Tsuyumine, S. and Kodama, T. (2016): Direct Synthesis of a PDE4 Inhibitor by Using Pd–Cu-Catalyzed C–H/C–Br Coupling of Benzoxazole with a Heteroaryl Bromide. Org. Process Res. Dev., 20, 1053-1058.
- Lamas, B., Natividad, J.M. and Sokol, H. (2018): Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol., 11, 1024-1038.
- Lang, D., Radtke, M. and Bairlein, M. (2019): Highly Variable Expression of CYP1A1 in Human Liver and Impact on Pharmacokinetics of Riociguat and Granisetron in Humans. Chem. Res. Toxicol., 32, 1115-1122.
- Lee, J.H., Wada, T., Febbraio, M., He, J., Matsubara, T., Lee, M.J., Gonzalez, F.J. and Xie, W. (2010): A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology, 139, 653-663.
- Lindell, M., Lang, M. and Lennernäs, H. (2003): Expression of genes encoding for drug metabolising cytochrome P450 enzymes and P-glycoprotein in the rat small intestine; comparison to the liver. Eur. J. Drug Metab. Pharmacokinet., 28, 41-48.
- Lu, P., Cai, X., Guo, Y., Xu, M., Tian, J., Locker, J. and Xie, W. (2019): Constitutive Activation of the Human Aryl Hydrocarbon Receptor in Mice Promotes Hepatocarcinogenesis Independent of Its Coactivator Gadd45b. Toxicol. Sci., 167, 581-592.
- Maher, J.M., Dieter, M.Z., Aleksunes, L.M., Slitt, A.L., Guo, G., Tanaka, Y., Scheffer, G.L., Chan, J.Y., Manautou, J.E., Chen, Y., Dalton, T.P., Yamamoto, M. and Klaassen, C.D. (2007): Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology, 46, 1597-1610.
- Manzella, C.R., Ackerman, M., Singhal, M., Ticho, A.L., Ceh, J., Alrefai, W.A., Saksena, S., Dudeja, P.K. and Gill, R.K. (2020): Serotonin Modulates AhR Activation by Interfering with CYP1A1-Mediated Clearance of AhR Ligands. Cell. Physiol. Biochem., 54, 126-141.
- Maronpot, R.R., Yoshizawa, K., Nyska, A., Harada, T., Flake, G., Mueller, G., Singh, B. and Ward, J.M. (2010): Hepatic enzyme induction: histopathology. Toxicol. Pathol., 38, 776-795.
- Matsuzaka, Y., Hosaka, T., Ogaito, A., Yoshinari, K. and Uesawa, Y. (2020): Prediction Model of Aryl Hydrocarbon Receptor Activation by a Novel QSAR Approach, DeepSnap-Deep Learning. Molecules, 25.
- McGill, M.R. and Jaeschke, H. (2019): Animal models of drug-induced liver injury. Biochim. Biophys. Acta Mol. Basis Dis., 1865, 1031-1039.
- Mocarelli, P., Needham, L.L., Marocchi, A., Patterson, D.G. Jr., Brambilla, P., Gerthoux, P.M., Meazza, L. and Carreri, V. (1991): Serum concentrations of 2,3,7,8-tetrachlorodibenzo-p-dioxin and test results from selected residents of Seveso, Italy. J. Toxicol. Environ. Health, 32, 357-366.
- Moennikes, O., Loeppen, S., Buchmann, A., Andersson, P., Ittrich, C., Poellinger, L. and Schwarz, M. (2004): A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res., 64, 4707-4710.
- Mohammadi-Bardbori, A., Bengtsson, J., Rannug, U., Rannug, A. and Wincent, E. (2012): Quercetin, resveratrol, and curcumin are indirect activators of the aryl hydrocarbon receptor (AHR). Chem. Res. Toxicol., 25, 1878-1884.
- Mohammadi-Bardbori, A., Bastan, F. and Akbarizadeh, A.R. (2017): The highly bioactive molecule and signal substance 6-formylindolo[3,2-b]carbazole (FICZ) plays bi-functional roles in cell growth and apoptosis in vitro. Arch. Toxicol., 91, 3365-3372.
- Murray, I.A. and Perdew, G.H. (2017): Ligand activation of the Ah receptor contributes to gastrointestinal homeostasis. Curr. Opin. Toxicol., 2, 15-23.
- Nguyen, L.P. and Bradfield, C.A. (2008): The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol., 21, 102-116.
- Oberg, M., Bergander, L., Håkansson, H., Rannug, U. and Rannug, A. (2005): Identification of the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole, in cell culture medium, as a factor that controls the background aryl hydrocarbon receptor activity. Toxicol. Sci., 85, 935-943.
- Pazderova-Vejlupková, J., Lukás, E., Nĕmcova, M., Pícková, J. and Jirásek, L. (1981): The development and prognosis of chronic intoxication by tetrachlordibenzo-p-dioxin in men. Arch. Environ. Health, 36, 5-11.
- Piloiu, C. and Dumitrascu, D.L. (2021): Albendazole-Induced Liver Injury. Am. J. Ther., 28, e335-e340.
- Pocchiari, F., Silano, V. and Zampieri, A. (1979): Human health effects from accidental release of tetrachlorodibenzo-p-dioxin (TCDD) at Seveso, Italy. Ann. N. Y. Acad. Sci., 320, 311-320.
- Rannug, A. and Rannug, U. (1989): UV-irradiation of tryptophan gives rise to AHH-inducing compounds with very high affinity for the Ah receptor. Chemosphere, 18, 1101-1106.
- Rannug, A. and Rannug, U. (2018): The tryptophan derivative 6-formylindolo[3,2-b]carbazole, FICZ, a dynamic mediator of endogenous aryl hydrocarbon receptor signaling, balances cell growth and differentiation. Crit. Rev. Toxicol., 48, 555-574.
- Schiering, C., Wincent, E., Metidji, A., Iseppon, A., Li, Y., Potocnik, A.J., Omenetti, S., Henderson, C.J., Wolf, C.R., Nebert, D.W. and Stockinger, B. (2017): Feedback control of AHR signalling regulates intestinal immunity. Nature, 542, 242-245.
- Shimizu, Y., Sasaki, T., Yonekawa, E., Yamazaki, H., Ogura, R., Watanabe, M., Hosaka, T., Shizu, R., Takeshita, J.I. and Yoshinari, K. (2021): Association of CYP1A1 and CYP1B1 inhibition in in vitro assays with drug-induced liver injury. J. Toxicol. Sci., 46, 167-176.
- Smirnova, A., Wincent, E., Vikström Bergander, L., Alsberg, T., Bergman, J., Rannug, A. and Rannug, U. (2016): Evidence for New Light-Independent Pathways for Generation of the Endogenous Aryl Hydrocarbon Receptor Agonist FICZ. Chem. Res. Toxicol., 29, 75-86.
- Stockinger, B., Di Meglio, P., Gialitakis, M. and Duarte, J.H. (2014): The aryl hydrocarbon receptor: multitasking in the immune system. Annu. Rev. Immunol., 32, 403-432.
- Tochitani, T., Toyosawa, K., Matsumoto, I., Kouchi, M., Michimae, Y., Koujitani, T., Funabashi, H. and Seki, T. (2011): Immunohistochemical and ultrastructural analyses of cytoplasmic blood plasma inclusions of rat hepatocytes. J. Toxicol. Pathol., 24, 245-249.
- Usui, T., Mise, M., Hashizume, T., Yabuki, M. and Komuro, S. (2009): Evaluation of the potential for drug-induced liver injury based on in vitro covalent binding to human liver proteins. Drug Metab. Dispos., 37, 2383-2392.
- Watanabe, M., Sasaki, T., Takeshita, J.I., Kushida, M., Shimizu, Y., Oki, H., Kitsunai, Y., Nakayama, H., Saruhashi, H., Ogura, R., Shizu, R., Hosaka, T. and Yoshinari, K. (2020): Application of cytochrome P450 reactivity on the characterization of chemical compounds and its association with repeated-dose toxicity. Toxicol. Appl. Pharmacol., 388, 114854.
- Wei, Y.-D., Helleberg, H., Rannug, U. and Rannug, A. (1998): Rapid and transient induction of CYP1A1 gene expression in human cells by the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole. Chem. Biol. Interact., 110, 39-55.
- Wei, Y.-D., Rannug, U. and Rannug, A. (1999): UV-induced CYP1A1 gene expression in human cells is mediated by tryptophan. Chem. Biol. Interact., 118, 127-140.
- Wei, Y.D., Bergander, L., Rannug, U. and Rannug, A. (2000): Regulation of CYP1A1 transcription via the metabolism of the tryptophan-derived 6-formylindolo[3,2-b]carbazole. Arch. Biochem. Biophys., 383, 99-107.
- Westerink, W.M., Stevenson, J.C. and Schoonen, W.G. (2008): Pharmacologic profiling of human and rat cytochrome P450 1A1 and 1A2 induction and competition. Arch. Toxicol., 82, 909-921.
- Wincent, E., Amini, N., Luecke, S., Glatt, H., Bergman, J., Crescenzi, C., Rannug, A. and Rannug, U. (2009): The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo[3,2-b]carbazole is present in humans. J. Biol. Chem., 284, 2690-2696.
- Wincent, E., Bengtsson, J., Mohammadi Bardbori, A., Alsberg, T., Luecke, S., Rannug, U. and Rannug, A. (2012): Inhibition of cytochrome P4501-dependent clearance of the endogenous agonist FICZ as a mechanism for activation of the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA, 109, 4479-4484.
- Wincent, E., Kubota, A., Timme-Laragy, A., Jönsson, M.E., Hahn, M.E. and Stegeman, J.J. (2016): Biological effects of 6-formylindolo[3,2-b]carbazole (FICZ) in vivo are enhanced by loss of CYP1A function in an Ahr2-dependent manner. Biochem. Pharmacol., 110-111, 117-129.
- Woolf, T.F., Pool, W.F., Walker, R.M. and Monteith, D.K. (1996): Liver reactions to tacrine. In: Cameron, R.G., Feuer, G., de la Iglesia, F.A. (eds) Drug-Induced Hepatotoxicity. Handbook of Experimental Pharmacology, vol 121. Springer, Berlin, Heidelberg.
- Yamazoe, Y. and Yoshinari, K. (2020): Prediction of regioselectivity and preferred order of CYP1A1-mediated metabolism: solving the interaction of human and rat CYP1A1 forms with ligands on the template system. Drug Metab. Pharmacokinet., 35, 165-185.
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/47_359-t1.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)