Original Article
Dihydropyrazine suppresses TLR4-dependent inflammatory responses by blocking MAPK signaling in human hepatoma HepG2 cells
2022 年 47 巻 9 号 p. 381-387
詳細
2022 年 47 巻 9 号 p. 381-387
Dihydropyrazines (DHPs), including 3-hydro-2,2,5,6-tetramethylpyrazine (DHP-3), are glycation products generated through non-enzymatic reactions in vivo and in food. They are recognized as compounds that are toxic to organisms as they produce radicals. However, our previous study indicated that DHP-3 suppressed Toll-like receptor 4 (TLR4) expression and decreased the phosphorylation of nuclear factor-κB (NF-κB) in lipopolysaccharide (LPS)-treated HepG2 cells. TLR4 signaling is involved in the onset of various inflammatory diseases, and NF-κB and mitogen-activated protein kinase (MAPK) play important roles in TLR4 signaling. Thus, we aimed to elucidate the effects of DHP-3 on MAPK signaling and in turn on the activated TLR4 signaling pathway. In LPS-stimulated HepG2 cells, DHP-3 reduced the phosphorylation of MAPK, extracellular signal-regulated kinase, c-Jun NH2-terminal kinase, and p38. The expression of c-jun, a subunit of activator protein-1, was decreased by DHP-3 treatment. Furthermore, DHP-3-induced suppression of MAPK signaling resulted in a decrease in various inflammatory regulators, such as interleukin-6, CC-chemokine ligand 2, and cyclooxygenase-2. These results suggest that DHP-3 exerts an inhibitory effect on TLR4-dependent inflammatory response by suppressing MAPK signaling.
Glycation products are generated by the Maillard reaction, which progresses non-enzymatic reactions between reducing sugars and an amino group of biological molecules (Glomb and Monnier, 1995). Glycation products have complex molecular structures because the biological molecules responsible for the reaction are divergent. Therefore, their biological effects are not yet fully understood. Most glycation products are known to accumulate in the living body. As a result, these glycation products are suggested to contribute to various diseases, such as diabetes (Aragno and Mastrocola, 2017), atherosclerosis (Wang et al., 2012), and Alzheimer’s and Parkinson’s disease (Koriyama et al., 2015). However, it was recently reported that some glycation products have antioxidant and antimutagenic effects that are beneficial to human health (Del Pino-García et al., 2012; Ohtani et al., 2007). Therefore, it is necessary to elucidate the detailed molecular mechanisms of each glycation product to estimate the accurate biological effects of each compound. Dihydropyrazines (DHPs), which have a chemical skeleton formed by the dimerization of 5-aminolevulinic acid (Teixeira et al., 2001) or D-glucosamine (Kashige et al., 1995), are a type of glycation product. Among DHPs, methyl-substituted DHPs generate carbon-centered radicals and hydroxyl radicals as well as other glycation products (Yamaguchi et al., 2012; Kashige et al., 2000; Ishida et al., 2012). In addition, 3-hydro-2,2,5,6-tetramethylpyrazine (DHP-3; Fig. 1) is one of the most toxic compounds to cells. DHP-3 activates the nuclear factor erythroid 2-related factor 2 pathway (Ishida et al., 2014), which is a protective mechanism against oxidative stress (Kobayashi et al., 2016). Pyrazine is one of the metabolites of DHP-3. Pyrazine has been detected in human urine (Zlatkis et al., 1973). Thus, DHP-3 can be generated and elicit some biological effects in vivo as occurs in vitro.
Chemical structure of 3-hydro-2,2,5,6-tetramethylpyrazine (DHP-3).
The Toll-like receptor 4 (TLR4) pathway is widely accepted as an important contributor to inflammatory responses, and two downstream signaling pathways, including myeloid differentiation primary response gene 88 (MyD88)-dependent and MyD88-independent pathways (Zughaier et al., 2005). TLRs recognize various pathogens (Akira and Takeda, 2004), such as lipopolysaccharides (LPS) in gram-negative bacteria (Peri and Calabrese, 2014). Therefore, numerous studies have used LPS as an activator of TLR4 signaling. Treatment with LPS enhances the interaction between TLR4 and MyD88, an adaptor protein, and subsequently activates downstream signaling pathways. the signaling pathway include nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK). The progression of MAPK signaling is executed by the phosphorylation of MAPKs such as extracellular signal-regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK), and p38, which are known to strongly contribute to the production of inflammatory mediators (Qian et al., 2015). MAPK signaling activated by MyD88 transmits a signal to activator protein-1 (AP-1) in the nucleus. Activated AP-1 functions as a transcriptional factor regulating an inflammatory response (Plotnikov et al., 2011). Therefore, the activation of MAPKs leads to an inflammatory state (Johnson and Lapadat, 2002). In a previous study, we showed that the increase in TLR4 and MyD88 protein levels in LPS-stimulated HepG2 cells were alleviated by treatment with DHP-3 (Esaki et al., 2020). Additionally, DHP-3 decreases LPS-induced phosphorylation of inhibitor of NF-κB (IκB) and NF-κB. These results led us to hypothesize that DHP-3 could suppress the TLR4 signaling pathway activated by LPS through the inhibition of NF-κB signaling. However, it remains unclear whether DHP-3 affects MAPK signal transduction, starting with TLR4. Therefore, we investigated the effects of DHP-3 on MAPK phosphorylation and production of inflammatory mediators in LPS-stimulated human HepG2 cells.
DHP-3 was synthesized according to a previously described method (Yamaguchi et al., 1996). LPS was purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against ERK (sc-135900), phospho-ERK (sc-7383), JNK (sc-7345), phospho-JNK (sc-6254), p38 (sc-81621), phospho-p38 (sc-166182), c-jun (sc-74543), cyclooxygenase-2 (COX-2) (sc-514489), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (sc-32233) were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Horseradish peroxidase (HRP)-conjugated goat anti-mouse immunoglobulin G antibody (A4416) was obtained from Sigma-Aldrich. All other reagents and chemicals used were of the highest grade of commercially available products.
Cell cultureHepG2 cells (JCRB1054) were obtained from the Human Science Research Resources Bank (Osaka, Japan). Dulbecco’s modified Eagle medium (DMEM) was purchased from FUJIFILM Wako Pure Chemical Corp. (Osaka, Japan). Cells were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum, in a humidified atmosphere with 5% CO2 at 37°C. The cells were grown in 60-mm culture dishes to 80%–90% confluence before being subjected to LPS or DHP-3 treatments.
Immunoblotting assayHepG2 cells were exposed to various concentrations of DHP-3 (50–400 µM) after treatment with 1 μg/mL LPS in a serum-free medium. The cells were then lysed with a sample buffer containing 50 mM Tris-HCl (pH 6.8), 2% sodium dodecyl sulfate (SDS), 10% glycerol, 5% 2-mercaptoethanol, and 0.1% bromophenol blue. Cell lysates were separated by 10% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The membrane was blocked with PVDF Blocking Reagent (TOYOBO Co. Ltd., Tokyo, Japan) and then incubated with primary antibodies. After washing with phosphate-buffered saline with 0.1% Tween20, the membranes were treated with HRP–conjugated secondary antibodies. SuperSignal™ West Dura Extended Duration Substrate (Thermo Fisher Scientific, Waltham, MA, USA) was used as the HRP substrate. The signals were detected using the VersaDoc™ Imaging System (Bio-Rad Laboratories, Hercules, CA, USA) and iBright system (Thermo Fisher Scientific).
Quantitative real-time PCRAccording to the manufacturer’s instructions, HepG2 cell lysates were subjected to reverse transcription to synthesize cDNA using Power SYBR® Green Cells-to-CT™ (Thermo Fisher Scientific). Real-time PCR was performed with each primer using the StepOnePlus™ Real-Time PCR System (Thermo Fisher Scientific). The primers used are listed in Table 1. The PCR conditions were: 95°C for 10 min, followed by 40 cycles of 15 sec at 95°C, and then 1 min at 60°C. The results were normalized by GAPDH mRNA expression and the fold change (2−ΔΔCt) was compared with that of the control group.
| Target Gene | Primer Sequence |
|---|---|
| IL-6 | Forward: 5’-CAGAACGAATTGACAAACAAATTC-3’ |
| Reverse: 5’-TTGTTACATGTCTCCTTTCTCAGG-3’ | |
| CCL2 | Forward: 5’-CTCAGCCAGATGCAATCAATG-3’ |
| Reverse: 5’-AGATCACAGCTTCTTTGGGACAC-3' | |
| GAPDH | Forward: 5’-GGACTCATGACCACAGTCCATGCC-3’ |
| Reverse: 5’-TCAGGGATGACCTTGCCCACA-3’ |
Data analysis was performed with KaleidaGraph 4.5 software (KaleidaGraph Software, Tokyo, Japan). Statistical significance was determined by ANOVA, followed by Dunnett’s or Tukey’s test.
To estimate the effect of DHP-3 on MAPK signaling in LPS-stimulated HepG2 cells, phosphorylation levels of kinases were analyzed by immunoblotting (Fig. 2A). The intensity of each band was standardized with that of GAPDH; the quantitative results of the three experiments are shown in Fig. 2B–G. Total ERK and p38 protein levels were unaffected by treatment with LPS and DHP-3 (Figs. 2B and F). In contrast, the total JNK protein level was increased by treatment with LPS and ≥ 100 µM DHP-3 (Fig. 2D). Although significant differences were not detected, the phosphorylation levels of ERK, JNK, and p38 showed a tendency to increase after LPS treatment in HepG2 cells (Figs. 2C, E, and G). In contrast, treatment with 50 µM or more of DHP-3 after LPS treatment demonstrated a significant reduction in phosphorylated ERK, JNK, and p38 levels to almost the control levels (Figs. 2C, E, and G). Additionally, DHP-3 significantly suppressed the induction of c-jun, a subunit of AP-1 complex, in LPS-stimulated HepG2 cells in a dose-dependent manner (Figs. 3A and B).

The effect of DHP-3 on activation of mitogen-activated protein kinases (MAPKs) in lipopolysaccharide (LPS)-stimulated HepG2 cells. After the treatment of 1 μg/mL LPS for 6 hr, the HepG2 cells were exposed to various concentrations of DHP-3 (50, 100, 200, 400 µM) for 1 hr. The control was an LPS-treated sample without DHP-3. (A) Immunoblot analysis of phospho-extracellular signal-regulated protein kinase (p-ERK), ERK, p-c-Jun N-terminal kinase (p-JNK), JNK, p-p38, and p38. (B, D, F) The band intensities of the total ERK, JNK, and p38 are normalized to the expression levels of GAPDH. (C, E, G) The relative ratios of p-ERK/ERK, p-JNK/JNK, and p-p38/p38. The values represent the mean ± S.E. of three independent experiments. (*p < 0.05, **p < 0.01, ***p < 0.001 indicate significant differences from each control.)

The effect of DHP-3 on protein expression of c-jun in LPS-stimulated HepG2 cells. After the treatment of 1 μg/mL LPS for 6 hr, the HepG2 cells were exposed to various concentrations of DHP-3 (50, 100, 200, 400 µM) for 1 hr. The control was an LPS-treated sample without DHP-3. (A) Immunoblot analysis of c-jun. (B) The band intensities of c-jun were normalized to the expression levels of GAPDH. The values represent the mean ± S.E. of three samples. (*p < 0.05, **p < 0.01, ***p < 0.001 indicate significant differences from control.)
Next, we analyzed the effects of DHP-3 on the expression of pro-inflammatory cytokine mRNAs regulated by the TLR4-MAPK axis. LPS stimulation of HepG2 cells resulted in the upregulation of interleukin-6 (IL-6) and CC-chemokine ligand 2 (CCL2) mRNAs (Figs. 4A and B). The upregulation of these mRNAs was inhibited by treatment with 200 µM DHP-3 after LPS stimulation (Figs. 4A and B, each third column from the left). The same phenomena was observed even if DHP-3 was treated before LPS stimulation (Figs. 4A and B, each fourth column from the left).

The effect of DHP-3 on the production of pro-inflammatory cytokines in LPS-stimulated HepG2 cells. The mRNA levels of IL-6 (A) and CCL2 (B) were analyzed using real-time PCR and normalization to GAPDH mRNA. After the treatment of 1 μg/mL LPS for 6 hr, the HepG2 cells were exposed to 200 µM DHP-3 for 1 hr (pre-treatment LPS). We also switched the order of LPS and DHP treatments as follows. After the treatment of 200 µM DHP-3 for 1 hr, the HepG2 cells were stimulated by 1 μg/mL LPS for 6 hr (post-treatment LPS). Values represent the mean ± S.E. of 3 samples. (##p < 0.01, ###p < 0.001 indicates significant differences from the untreated sample. ***p < 0.001 indicates significant differences from the LPS-treated sample without DHP-3.)
In inflammatory reactions, pro-inflammatory cytokines activate eicosanoid biosynthesis via COX-2 induction. We examined the effects of DHP-3 on COX-2 expression in LPS-stimulated HepG2 cells. In LPS-stimulated cells, COX-2 protein levels showed a tendency to increase compared to those in the control (Figs. 5A and B). In contrast, similar to other induces of inflammatory reactions, such as phosphorylated MAPKs and pro-cytokine mRNAs, the increase in COX-2 expression level showed a tendency toward suppression following DHP-3 treatment (Figs. 5A and B).
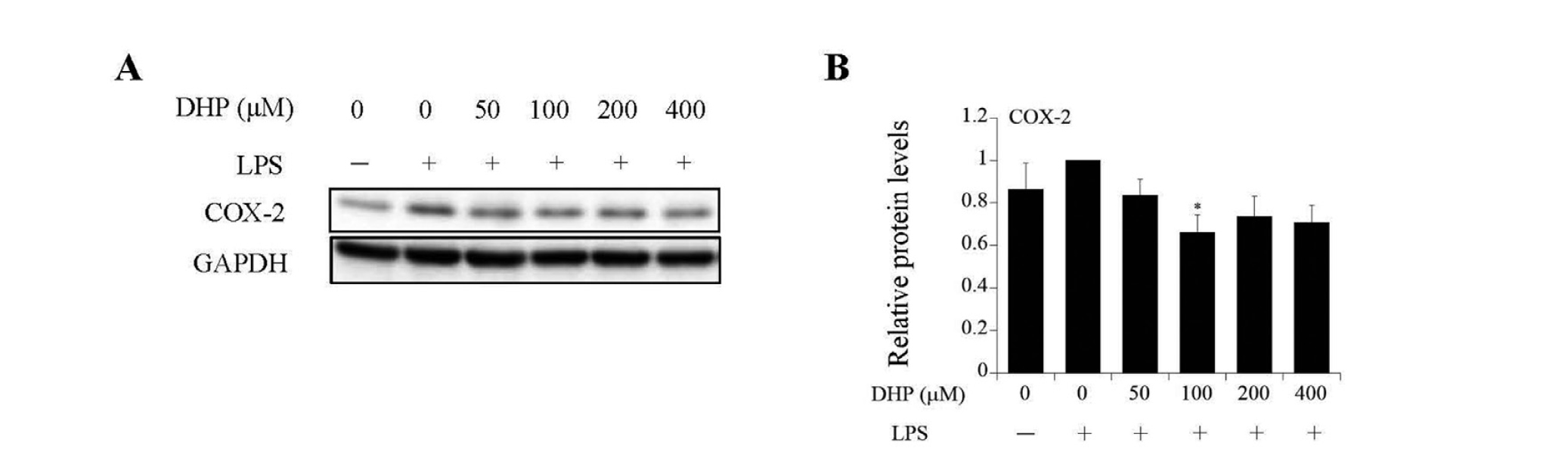
The effect of DHP-3 on the eicosanoid biosynthesis pathway in LPS-stimulated HepG2 cells. After the treatment of 1 μg/mL LPS for 6 hr, the HepG2 cells were exposed to various concentrations of DHP-3 (50, 100, 200, 400 µM) for 1 hr. The control was an LPS-treated sample without DHP-3. (A) Immunoblot analysis of cyclooxygenase-2 (COX-2). (B) The band intensities of COX-2 were normalized to the expression levels of GAPDH. The values represent the mean ± S.E. of three samples. (*p < 0.05 indicate significant differences from control.)
Although TLR4 elicits an inflammatory reaction through the MyD88-dependent or -independent pathways, MyD88-dependent signal transduction is known to contribute more than the MyD88-independent pathway (Pålsson-McDermott and O'Neill, 2004). After the binding of ligands, such as LPS to TLR4, the MyD88-dependent signal transduction is activated, and MAPKs, including ERK, JNK, and p38, are phosphorylated downstream. This activation of MAPKs results in the induction of several pro-inflammatory cytokines, such as IL-6, CCL2, and tumor necrosis factor α (TNFα) (McKeown-Longo and Higgins, 2017). Therefore, activation of MAPKs through MyD88-dependent signaling is considered to play a critical role in the progression of several inflammatory diseases (Kim and Choi, 2010). In the present study, LPS treatment induced phosphorylation of ERK, JNK, and p38 in HepG2 cells. The increase in the phosphorylation levels of these kinases was suppressed to almost control levels after treatment with DHP-3. In addition, DHP-3 decreased the expression of c-jun, which is located downstream of MAPK signaling. We further examined whether DHP-3 affects the expression level of pro-inflammatory cytokines. We found that the increase in IL-6 and CCL2 mRNA levels in LPS-stimulated HepG2 cells was downregulated by pre- and post-treatment with DHP-3. Although we were unable to identify the detailed mechanisms exerted by DHP-3, these results suggest that DHP-3 can suppress TLR4-dependent activation of inflammatory reactions by affecting downstream signal transduction.
Ligand binding to TLR4 also activates NF-κB signal transduction, which is a radical oxygen species-sensitive signaling pathway. We previously reported that DHP-3 inhibits NF-κB nuclear translocation in LPS-stimulated HepG2 cells (Esaki et al., 2020). In addition, the degradation of IκB, which is known to repress NF-κB signaling, was lowered by treatment with DHP-3. MAPKs and NF-κB are mainly activated by the MyD88-dependent TLR4 pathway (Zughaier et al., 2005), and the activation of both signaling cascades results in the induction of inflammatory cytokines such as IL-6, TNFα, and IL-1β. Therefore, DHP-3 may affect multiple signaling pathways downstream of TLR4.
Excessive activation of TLR4 is involved in the pathogenesis of various inflammatory diseases, such as hepatitis (Kanmani et al., 2019), sepsis (Leon et al., 2008), and cancer metastasis (Li et al., 2017). In addition, it has been reported that many patients transported to the hospital with a viral infection are in a state of TLR4 over-activation (Sohn et al., 2020). Effective inhibitors against excessive TLR4 activation have not been found until now. However, our study demonstrated the possibility that DHP-3 is an attractive candidate. Fermented foods, such as coffee and soy sauce, are enriched in glycation products. These products are also generated through the Maillard reaction during the aging process. The fermented food products have antihypertensive and antibacterial effects (Rufián-Henares and Morales, 2007), leading some people to actively consume these foods. In addition, pyrazines, which have a skeleton similar to that of DHP-3, are used in food flavoring. According to the United States Flavor and Extract Manufacturers Association, most pyrazines investigated are non-toxic but have anti-inflammatory and anti-tumor activities (Adams et al., 2002). In mice, some pyrazines reportedly reduce the mortality rate due to sepsis caused by overactivation of TLR4 signaling (Fu and Hu, 1999). Although glycation products are widely considered harmful to the body, some have the potential to be useful in treating inflammatory diseases. Further studies are necessary to determine the molecular mechanisms underlying the DHP-3-mediated anti-inflammatory effects.
Conflict of interestThe authors declare that there is no conflict of interest.