Abstract
Vascular endothelial cell growth is essential for the repair of intimal injury. Perlecan, a large heparan sulfate proteoglycan, intensifies fibroblast growth factor-2 (FGF-2) signaling as a co-receptor for FGF-2 and its receptor, and promotes the proliferation of vascular endothelial cells. Previously, we reported that 2 µM of lead, a toxic heavy metal, downregulated perlecan core protein expression and then suppressed the growth of vascular endothelial cells. However, since the mechanisms involved in the repression of perlecan by lead remains unclear, we analyzed its detailed signaling pathway using cultured bovine aortic endothelial cells. Our findings indicate that 2 µM of lead inhibited protein tyrosine phosphatase (PTP) activity and induced cyclooxygenase-2 (COX-2) via phosphorylation of the epidermal growth factor receptor (EGFR) and its downstream extracellular signal-regulated kinases (ERK1/2). In addition, among the prostanoids regulated by COX-2, prostaglandin I2 (PGI2) specifically contributes to the downregulation of perlecan expression by lead. This study revealed an intracellular pathway—the EGFR-ERK1/2-COX-2-PGI2 pathway activated by inhibition of PTP by lead—as a pathway that downregulates endothelial perlecan synthesis. The pathway is suggested to serve as a mechanism for the repression of perlecan expression, which leads to a delay in cell proliferation by lead.
INTRODUCTION
Lead is a heavy metal that is widely distributed in the environment and humans are exposed to lead from water, food, soil, dust, and occupational environment. The half-life of lead in vivo is approximately 20-30 days in the blood and soft tissue, approximately 2 years in the brain, and several decades in the bone (Schütz et al., 1987). Gastrointestinal absorption of lead is only 10-15% in adults, but as high as 50% in infants (Patrick, 2006). Epidemiological studies have revealed that lead is associated with the development of vascular lesion (Poręba et al., 2011). In addition, animal studies have shown that lead induces vascular lesions such as arteriosclerosis, because oral intake of lead promotes the proliferation of vascular smooth muscle cells and hypertension (Revis et al., 1981). Since lead induces organ toxicity, it must enter the blood vessels, pass across the endothelial cell layer, and reach the parenchymal cells of the target organ. Therefore, clarification of the mechanisms of lead toxicity in vascular endothelial cells is essential for understanding the organ toxicity as well as vascular toxicity of lead.
We have reported that although lead has low toxicity to vascular endothelial cells (Kaji et al., 1995b), it reduces fibrinolytic activity by inhibiting the synthesis of tissue-type plasminogen activator (Kaji et al., 1992) and induces endoplasmic reticulum stress via the JNK-AP1 pathway (Shinkai et al., 2010) in the cells. Additionally, we found that lead enhances the growth of vascular smooth muscle cells (Fujiwara et al., 1995) but inhibits the growth of vascular endothelial cells (Kaji et al., 1995a; Fujiwara et al., 1997). This indicates that lead can promote the progression of atherosclerosis by both stimulation and inhibition of the proliferation of vascular smooth muscle cells and endothelial cells, respectively. Specifically, stimulation of vascular smooth muscle cells would contribute to the intimal thickening and inhibition of vascular endothelial cells would be a factor for delay of the repair of the damaged endothelial monolayer, which is involved in the initiation and progression of atherosclerosis (Ross et al., 1977).
Perlecan, a large heparan sulfate proteoglycan, is present in the basement membrane and is essential for the maintenance of blood vessels. Perlecan on vascular endothelium has two important physiological roles in the endothelium: one is to regulate the anticoagulant property of endothelium by exhibiting a heparin-like activity and the other is to regulate the stimulation of vascular endothelial cell proliferation. Perlecan is secreted by vascular endothelial cells, which enhances the binding of fibroblast growth factor-2 (FGF-2) to its receptor and promotes cell growth (Aviezer et al., 1994). In fact, perlecan null mice die between E10–E12 and perinatally (Costell et al., 1999). We also reported that the inhibition of perlecan synthesis contributes to the suppression of bovine vascular endothelial cell growth by 2 µM of lead (Fujiwara and Kaji, 1999a; Fujiwara and Kaji, 1999b).
Previously, we reported that forskolin-mediated cyclic AMP (cAMP) pathway activation suppresses the synthesis of heparan sulfate proteoglycan, in fact, perlecan, in bovine vascular endothelial cells (Kaji et al., 1996). Additionally, we found that methylmercury inhibits the activity of protein tyrosine phosphatase 1B (PTP1B) and increases the phosphorylation of epidermal growth factor receptor (EGFR) that induces the phosphorylation of p38 mitogen-activated protein kinase (p38 MAPK) in human brain microvascular endothelial cells. The p38 MAPK pathway mediates the methylmercury-induced expression of cyclooxygenase-2 (COX-2) that synthesizes prostaglandin I2 (PGI2), an activator of the cAMP pathway (Yoshida et al., 2017). However, the molecular mechanisms for the repression of perlecan expression by lead remain unknown. We hypothesized that lead also may activate the EGFR-MAPK-COX-2 pathway that activates cAMP pathways and suppresses the synthesis of perlecan in vascular endothelial cells. This study aimed to elucidate this mechanism using bovine aortic endothelial cells in a culture system.
MATERIALS AND METHODS
Materials
Bovine aortic endothelial cells were purchased from Cell Applications (San Diego, CA, USA). Tissue culture dishes were obtained from AGC Techno Glass (Shizuoka, Japan). Dulbecco’s modified Eagle’s medium (DMEM) and Ca2+- and Mg2+-free phosphate-buffered saline (CMF-PBS) were obtained from Nissui Pharmaceutical (Tokyo, Japan). Fetal bovine serum (FBS), a bicinchoninic acid (BCA) protein assay kit, and a High-Capacity cDNA Reverse Transcription kit were purchased from Thermo Fisher Scientific (Waltham, MA, USA). ISOGEN-II and GeneAce SYBR qPCR mix α were purchased from Nippon Gene (Tokyo, Japan). Anti-EGFR (MI-12-1) was purchased from MBL (Tokyo, Japan). Anti-phospho-EGFR (#3777), anti-p44/42 MAPK (ERK1/2) (#9102), anti-phospho-ERK1/2 (#9101), anti-p38 MAPK (#9212), anti-phospho-p38 MAPK (#9211), anti-JNK (#9252), anti-phospho-JNK (#9255) antibodies, and HRP-conjugated anti-rabbit and mouse IgG secondary antibodies (#7074 and #7076, respectively) were obtained from Cell Signaling Technology (Beverly, MA, USA). Anti-COX-2 antibody (A1253) was obtained from ABclonal (Woburn, MA, USA). Anti-β-actin antibody (60008-1-Ig) was purchased from Proteintech (Rosemont, IL, USA). Amersham™ Hybond™ P 0.2 PVDF was obtained from Cytiva (Marlborough, MA, USA). EGFR inhibitor PD153035 was purchased from MedChemExpress (Monmouth Junction, NJ, USA). p38 MAPK inhibitor SB203580 was obtained from Enzo Life Sciences (Farmingdale, NY, USA). ERK1/2 inhibitor SCH772984, COX-2 inhibitor NS-398, PGI2 analogue beraprost, and 6-keto-PGF1α ELISA Kit were purchased from Cayman Chemical (Ann Arbor, MI, USA). PGE2 was purchased from Tokyo Chemical Industry (Tokyo, Japan). Thromboxane A2 (TXA2) receptor analog U-46619 was obtained from Abcam (Cambridge, UK). Disodium p-nitrophenyl phosphate hexahydrate (pNPP) was purchased from Fujifilm Wako Pure Chemical Industries (Osaka, Japan). Lead (II) nitrate, Chemi-Lumi One Super, and other reagents of the highest grade available were obtained from Nacalai Tesque (Kyoto, Japan).
Cell culture and treatments
Bovine aortic endothelial cells were cultured in a humidified atmosphere containing 5% CO2 at 37°C in DMEM supplemented with 10% FBS until confluence. Then, the cells were transferred into 100 mm (for ELISA) or 60 mm (for the other experiments) dishes at 2 × 104 cells/cm2 and cultured for 24 hr (“glowing culture”). The medium was discarded, and the cells were washed twice with serum-free DMEM. The cells were then exposed to lead nitrite, beraprost, PGE2, or U-46619. In some experiments, PD153035, SCH772984, SB203580, and SP600125 were treated prior to lead exposure. On the other hand, NS-398 was coexposed to lead. After incubation, the following experiments were performed.
Total RNA extraction and quantitative RT-PCR
Total RNA was extracted according to the manufacturer’s instructions. After incubation, the conditioned medium was discarded, and 300 µL of cold ISOGEN II was added to each well. The cells were then homogenized by pipetting. Next, 120 µL of ultrapure water was added to the collected homogenate and incubated for 5 min. The samples were centrifuged at 15,000 × g at 4°C for 15 min with 300 µL of the collected supernatant and mixed with 300 µL of 2-propanol. After incubation for 5 min, the samples were centrifuged at 15,000 × g at 4°C for 15 min. The supernatant was discarded and the RNA pellet was washed with 500 µL of 70% ethanol. Finally, the RNA pellets were dissolved in ultrapure water. cDNA was synthesized from the total RNA using a High-Capacity cDNA Reverse Transcription kit. Quantitative PCR was performed using GeneAce SYBR qPCR mix α with 1 ng of cDNA and 0.2 µM primers (Table 1) on a CFX Connect Real-Time PCR Detection System (BioRad, Hercules, CA, USA). The levels of perlecan, COX-2, and beta 2 microglobulin (B2M) transcripts were quantified using the relative standard curve method. The fold-change in the intensity value of the target gene was normalized to that of B2M.
Table 1. Gene-specific primers for quantitative RT-PCR
| Gene |
Forward primer (5′ – 3′) |
Reverse primer (5′ – 3′) |
Amplicon |
| Perlecan |
ATGGCAGCGATGAAGCGGAC |
TTGTGGACACGCAGCGGAAC |
201 bp |
| COX-2 |
AAGTCTTTGGTCTGGTGCCTGGTC |
GTCTGGAACAACTGCTCATCGCC |
124 bp |
| B2M |
CCATCCAGCGTCCTCCAAAGA |
TTCAATCTGGGGTGGATGGAA |
110 bp |
Western blotting
EGFR, ERK1/2, p38 MAPK, JNK, COX-2, and β-actin proteins were detected using western blot analysis. After incubation, the conditioned medium was discarded, and the cell layer was washed twice with CMF-PBS. Next, the cells were lysed with 100 µL of 50 mM Tris-HCl buffer (pH 6.8) containing 2% SDS and 10% glycerol. The cells were then lysed by incubation at 95°C for 10 min, and a portion of each lysate was analyzed for protein content using a BCA protein assay kit. The samples were separated by SDS-PAGE, and transferred onto an Amersham Hybond P 0.2 PVDF membrane at 2 mA/cm2 for 1 hr. Membranes were blocked for 1 hr with 5% skim milk in 20 mM Tris-HCl buffer solution (pH 7.5) containing 150 mM NaCl and 0.1% Tween 20 (TTBS) or 2% bovine serum albumin-TTBS solution, and incubated overnight with a primary antibody at room temperature. The membranes were washed and incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hr. Immunoreactive bands were visualized using Chemi-Lumi One Super and scanned using Amersham Imager 600 (GE Healthcare). Representative blots are shown from three independent experiments.
6-keto-PGF1α ELISA
After incubation, the conditioned medium was harvested and used for the determination of PGI2, which was measured as 6-keto PGF1α, using ELISA kits. The cell layer was washed twice with CMF-PBS and lysed with 100 µL of 50 mM Tris-HCl buffer (pH 6.8) containing 2% SDS and 10% glycerol. The cells were then lysed by incubation at 95°C for 10 min, and 90 µL of the lysate was used for the determination of DNA content using the fluorometric method (Kissane and Robins, 1958) to express the content of 6-keto PGF1α as ng/µg DNA.
Phosphatase activity assay
After incubation, the conditioned medium was discarded, and the cell layer was washed twice with CMF-PBS and harvested in 1.5-mL tubes with a cell scraper in the presence of 1 mL CMF-PBS. The well was then washed with 0.3 mL CMF-PBS and the wash was combined with the corresponding harvested cell suspension. The combined cell suspension was then centrifuged at 15,000 × g for 5 min at 4°C, and the supernatant was discarded. Next, 100 µL of assay buffer (25 mM HEPES-NaOH [pH 7.5] containing 100 mM NaCl) was added to each sample and sonicated for five cycles (1-sec pulse and 1-sec rest, on ice) using a homogenizer (UR-20, TOMY, Tokyo, Japan). The samples were centrifuged at 4°C at 3,000 × g for 3 min to remove cell debris, and 90 µL of the supernatant was transferred to another 1.5-mL tube. A portion of the homogenates was analyzed for protein content using a BCA protein assay kit. Then, 30 µg of the homogenates in 1.5-mL tubes were added to 360 µL of assay buffer. After that, 40 µL of pNPP solution [0.1 M glycine-NaOH (pH 10.4) containing 1 mM MgCl2, 1 mM ZnCl2, and 40 mM pNPP] was added to each sample, mixed well, and then incubated at 37°C for 60 min. The absorbance of the samples was measured at 405 nm to determine phosphatase activity.
Statistical analysis
Data were analyzed for statistical significance using Student’s t-test or analysis of variance and Dunnett’s multiple t-tests, when applicable. Statistical significance was set at p < 0.05.
RESULTS
Perlecan expression is suppressed via EGFR/ERK1/2/ pathway by lead exposure in vascular endothelial cells
We first examined whether lead suppresses the expression of perlecan in vascular endothelial cells.
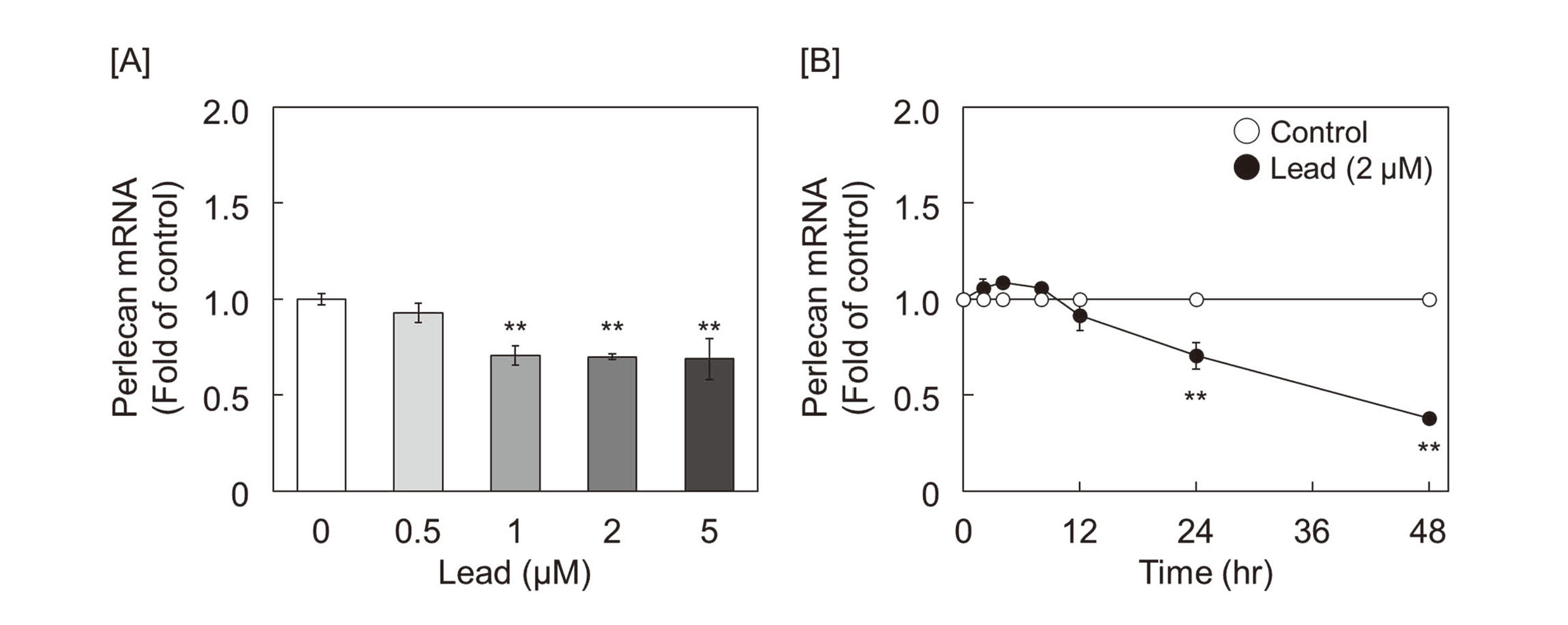
As shown in Fig. 1, perlecan mRNA expression was suppressed by lead at 1 µM and more (Fig. 1A). The suppression by lead at 2 µM was in a time-dependent manner (Fig. 1B). These results showed that the decrease in perlecan core protein expression by lead, which we have previously reported (Fujiwara and Kaji, 1999b) is caused at the gene expression level.
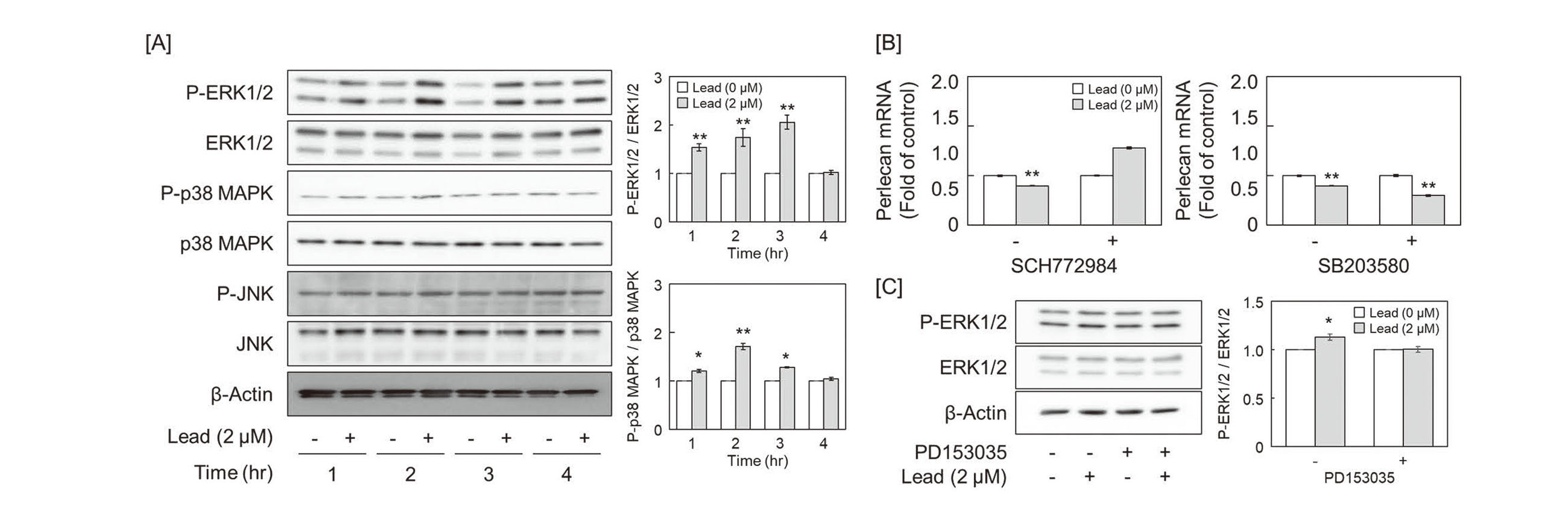
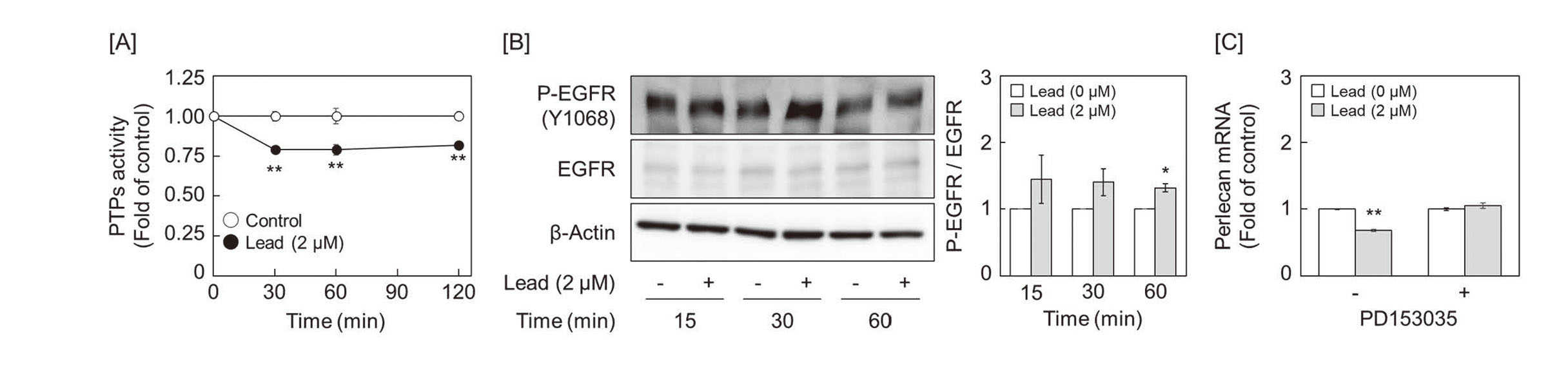
Since it has been reported that heavy metals, such as zinc, cadmium, and methylmercury, inhibit the activity of protein tyrosine phosphatases (PTPs) and promote autophosphorylation of EGFR, a receptor-type tyrosine kinase (Yoshida et al., 2017; Bellomo et al., 2014; Zhang et al., 2021), we tested whether lead inhibits PTPs. The activity of PTPs was suppressed from 30 to 120 min after lead exposure (Fig. 2A), and autophosphorylation of EGFR, which is dephosphorylated by PTPs, such as PTP1B, was persistently enhanced up to 60 min after lead exposure (Fig. 2B). In addition, pretreatment with EGFR inhibitor PD153035 rescued the lead-induced suppression of perlecan mRNA expression (Fig. 2C). These results suggest that suppression of perlecan expression by lead is mediated by downstream signaling from EGFR activated by autophosphorylation that was not inhibited by PTPs.
Next, we investigated the effect of MAPKs (ERK1/2, p38 MAPK, and JNK), which are generally located downstream of EGFR, on the repression of perlecan expression by lead. ERK1/2 and p38 MAPK were activated in vascular endothelial cells after 1–4 hr and 1–2 hr of lead exposure, respectively. However, no significant activation of JNK was observed (Fig. 3A). Although p38 MAPK inhibitor SB203580 did not affect perlecan mRNA suppression by lead, ERK1/2 inhibitor SCH772984 hindered the suppression in vascular endothelial cells (Fig. 3B). Furthermore, pretreatment with EGFR inhibitor PD153035 suppressed lead-induced ERK1/2 activation (Fig. 3C), suggesting that ERK1/2 is a downstream signal molecule of EGFR that mediates the repression of perlecan by lead.
Lead upregulates COX-2 expression while PGI2 suppresses mRNA level of perlecan in vascular endothelial cells
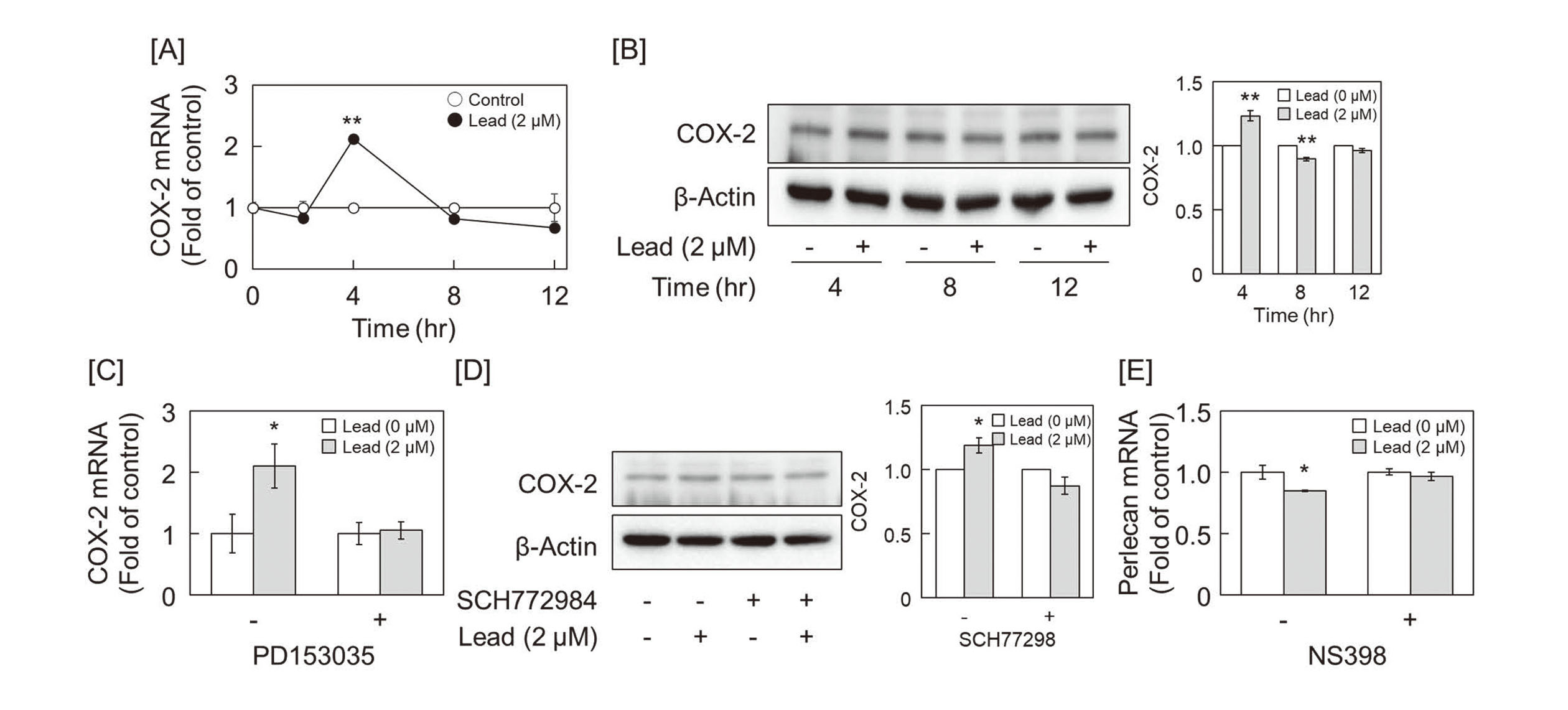
We have previously reported that methylmercury activates EGFR and upregulates the expression of COX-2, which is involved in prostanoid synthesis, in human brain microvascular endothelial cells and pericytes (Yoshida et al., 2017). Lead also transiently induced COX-2 mRNA and protein expression in growing vascular endothelial cells at 4 hr after exposure (Fig. 4A and 4B), and the lead-induced-COX-2 upregulation was markedly suppressed by pretreatment with EGFR inhibitor PD153035 and ERK1/2 inhibitor SCH772984 (Fig. 4C and 4D). The results showed that the suppression of PTP activity by lead-induced COX-2 expression via activation of the EGFR-ERK1/2 pathway. In addition, as shown in Fig. 4E, co-treatment with the COX-2 inhibitor NS398 recovered the lead-induced downregulation of perlecan, suggesting that perlecan expression is suppressed by a series of these mechanisms. COX-2 converts arachidonic acid to PGH2, which is subsequently metabolized to various prostanoids such as PGI2 and PGE2 (Brock et al., 1999). We investigated which prostanoids contributed to the repression of perlecan mRNA expression. Although PGE2 and TXA2 analog U-46619 failed to suppress perlecan mRNA expression, the PGI2 analog beraprost reduced its expression in a dose-dependent manner (Fig. 5A), and 6-keto-PGF1α, a stable metabolite of PGI2 was increased in the conditioned medium of vascular endothelial cells 12 hr after lead exposure (Fig. 5B).

DISCUSSION
We previously reported that lead inhibits the repair of wounded monolayers of vascular endothelial cells (Fujiwara et al., 1997; Fujiwara et al., 1998) and that this is due to the suppression of perlecan core protein synthesis by lead without changing the length of the heparan sulfate chain (Fujiwara and Kaji, 1999b). This study aimed to elucidate the mechanism of perlecan suppression by lead, and the following results were obtained using a culture of vascular endothelial cells.
(1) Lead suppressed perlecan expression at the gene expression level.
(2) Lead inhibited PTP activity and enhanced EGFR and ERK1/2 activation.
(3) Lead induces COX-2 expression via activation of the EGFR-ERK1/2 pathway.
(4) Inhibition of EGFR, ERK1/2, and COX-2 hindered the lead-induced repression of perlecan.
(5) PGI2 production was increased by lead and perlecan expression was suppressed by the PGI2 analog beraprost.
These results indicate that lead suppresses perlecan mRNA expression via the EGFR-ERK1/2-COX-2-PGI2 pathway in bovine vascular endothelial cells. The PGI2 receptor is a G protein-coupled IP (Ushikubi et al., 1995; Narumiya et al., 1999); activation of the IP receptor increases the intracellular cAMP level via activation of adenylate cyclase, and increased cAMP leads to activation of protein kinase A (PKA) (Steinert et al., 2009; El-Haroun et al., 2008). Therefore, the cAMP-PKA pathway may serve as the downstream signal pathway of PGI2 that mediates endothelial perlecan expression. Our previous study (Kaji et al., 1996) partly supports this assumption.
This study not only presented the molecular mechanism mediating the inhibition of vascular endothelial cell proliferation by lead in terms of the suppression of perlecan expression, but also revealed for the first time that EGFR and PGI2 are involved in the regulation of perlecan expression. Additionally, as shown in Fig. 5A, vascular endothelial cells have a mechanism to induce perlecan via PGE2 and perlecan may be bidirectionally modulated by prostanoids. On the other hand, considering the events under lead treatment, it is expected that PGI2 has stronger effects on endothelial cells than PGE2, or that lead would cause inhibition of PGE2 synthase (PGES) activity, loss of PGE2 receptor (EP1-4) function, or inhibition of downstream PGE2 receptor signaling. As a result, the phenotype may have resulted from suppression of perlecan expression by PGI2, rather than induction of perlecan expression by PGE2. The synthesis of perlecan and its heparan sulfate chains in vascular endothelial cells is regulated by growth factors and cytokines. We have reported that transforming growth factor-β1 (TGF-β1) (Kaji et al., 2000) and vascular endothelial growth factor (VEGF) (Kaji et al., 2006) modulate perlecan expression in the cells. Namely, TGF-β1 increased perlecan synthesis in confluent cultures of cells with a high cell density but not in cultures with a low cell density. Similarly, VEGF, which is expressed in capillary endothelial cells, enhanced perlecan synthesis in confluent endothelial cells at a high cell density. However, VEGF expression in aortic endothelial cells is low (Ladoux and Frelin, 1993). Since we used vascular endothelial cells from the aorta at a low cell density in this study, it is unlikely that the conclusion of this study is secondary to the decreased expression of TGF-β1 or VEGF.
Comparing the effects of lead on aortic endothelial cells shown in this study with those of methylmercury on brain microvascular endothelial cells (Yoshida et al., 2017), both inhibited PTP activity, phosphorylated EGFR, and induced COX-2 via downstream signaling. However, the MAPKs that contribute to the induction of COX-2 expression were different. In brain microvascular endothelial cells, methylmercury induced COX-2 expression via p38 MAPK activation. On the other hand, lead activated ERK1/2 and p38 MAPK, but only ERK1/2 contributed to the induction of COX-2 expression. There have been many reports of MAPK-mediated induction of COX-2 expression, and it is also known that the critical MAPK depends on the compound (Cui et al., 2004; Elder et al., 2002; Tsatsanis et al., 2006; Ki et al., 2013; Zhou et al., 2005). In addition, the affinity of methylmercury for the thiol group (-SH, -S−) of the cysteine residue located in the catalytic center of PTP is higher than that of lead (Simpson, 1961; Sangvanich et al., 2014), which may be one of the reasons why the induction manner of COX-2 is different between methylmercury and lead. Moreover, the activation of ERK1/2 is observed at 2~3 hours after lead treatment, while activation of ERK1/2 returns to steady-state levels at 4 hr after treatment, which may explain why the induction of COX-2 expression by lead is transient.
EGF has been known to exhibit proliferative activity against vascular endothelial cells (Carpenter and Cohen, 1979) and induces PGI2 (Ristimäki et al., 1988). However, this study showed that lead can inhibit vascular endothelial cell proliferation by inhibiting the synthesis of perlecan through the phosphorylation of EGFR. Although this result is inconsistent with the proliferative activity of EGFR, this inconsistency can be explained by the unique characteristics of vascular endothelial cells, which require perlecan and endogenous FGF-2 for proliferation. The phosphorylation of EGFR by lead may temporarily promote endothelial cell proliferation, especially by activating ERK1/2, whereas the effect of inhibiting perlecan synthesis gradually increases, probably resulting in a decline in cell proliferation. The disturbance of intracellular signaling and its outcomes is a typical example of signal toxicology (Kanno, 2016). In other words, the present study provided evidence that inhibition of vascular endothelial cell proliferation by lead results from a signal toxicity of lead.
ACKNOWLDGMENTS
This work was supported by JSPS KAKENHI Grant Numbers JP 19K19418 (to T.H.) and JP 18K06638 (to C.Y.).
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Aviezer, D., Hecht, D., Safran, M., Eisinger, M., David, G. and Yayon, A. (1994): Perlecan, basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis. Cell, 79, 1005-1013.
- Bellomo, E., Massarotti, A., Hogstrand, C. and Maret, W. (2014): Zinc ions modulate protein tyrosine phosphatase 1B activity. Metallomics, 6, 1229-1239.
- Brock, T.G., McNish, R.W. and Peters-Golden, M. (1999): Arachidonic acid is preferentially metabolized by cyclooxygenase-2 to prostacyclin and prostaglandin E2. J. Biol. Chem., 274, 11660-11666.
- Carpenter, G. and Cohen, S. (1979): Epidermal growth factor. Annu. Rev. Biochem., 48, 193-216.
- Costell, M., Gustafsson, E., Aszódi, A., Mörgelin, M., Bloch, W., Hunziker, E., Addicks, K., Timpl, R. and Fässler, R. (1999): Perlecan maintains the integrity of cartilage and some basement membranes. J. Cell Biol., 147, 1109-1122.
- Cui, Y., Kim, D.S., Park, S.H., Yoon, J.A., Kim, S.K., Kwon, S.B. and Park, K.C. (2004): Involvement of ERK AND p38 MAP kinase in AAPH-induced COX-2 expression in HaCaT cells. Chem. Phys. Lipids, 129, 43-52.
- El-Haroun, H., Clarke, D.L., Deacon, K., Bradbury, D., Clayton, A., Sutcliffe, A. and Knox, A.J. (2008): IL-1β, BK, and TGF-β1 attenuate PGI2-mediated cAMP formation in human pulmonary artery smooth muscle cells by multiple mechanisms involving p38 MAP kinase and PKA. Am. J. Physiol. Lung Cell. Mol. Physiol., 294, L553-L562.
- Elder, D.J., Halton, D.E., Playle, L.C. and Paraskeva, C. (2002): The MEK/ERK pathway mediates COX-2-selective NSAID-induced apoptosis and induced COX-2 protein expression in colorectal carcinoma cells. Int. J. Cancer, 99, 323-327.
- Fujiwara, Y., Kaji, T., Yamamoto, C., Sakamoto, M. and Kozuka, H. (1995): Stimulatory effect of lead on the proliferation of cultured vascular smooth-muscle cells. Toxicology, 98, 105-110.
- Fujiwara, Y., Kaji, T., Sakurai, S., Sakamoto, M. and Kozuka, H. (1997): Inhibitory effect of lead on the repair of wounded monolayers of cultured vascular endothelial cells. Toxicology, 117, 193-198.
- Fujiwara, Y., Watanabe, S., Sakamoto, M. and Kaji, T. (1998): Repair of wounded monolayers of cultured vascular endothelial cells after simultaneous exposure to lead and zinc. Toxicol. Lett., 94, 181-188.
- Fujiwara, Y. and Kaji, T. (1999a): Possible mechanism for lead inhibition of vascular endothelial cell proliferation: a lower response to basic fibroblast growth factor through inhibition of heparan sulfate synthesis. Toxicology, 133, 147-157.
- Fujiwara, Y. and Kaji, T. (1999b): Lead inhibits the core protein synthesis of a large heparan sulfate proteoglycan perlecan by proliferating vascular endothelial cells in culture. Toxicology, 133, 159-169.
- Kaji, T., Yamamoto, C., Sakamoto, M. and Kozuka, H. (1992): Inhibitory effect of lead on the release of tissue plasminogen activator from human vascular endothelial cells in culture. Toxicology, 73, 219-227.
- Kaji, T., Fujiwara, Y., Hoshino, M., Yamamoto, C., Sakamoto, M. and Kozuka, H. (1995a): Inhibitory effect of lead on the proliferation of cultured vascular endothelial cells. Toxicology, 95, 87-92.
- Kaji, T., Suzuki, M., Yamamoto, C., Mishima, A., Sakamoto, M. and Kozuka, H. (1995b): Severe damage of cultured vascular endothelial cell monolayer after simultaneous exposure to cadmium and lead. Arch. Environ. Contam. Toxicol., 28, 168-172.
- Kaji, T., Inada, M., Yamamoto, C., Fujiwara, Y. and Koizumi, F. (1996): Cyclic AMP-dependent pathway that mediates suppressive regulation of glycosaminoglycan production in cultured vascular endothelial cells. Thromb. Res., 82, 389-397.
- Kaji, T., Yamada, A., Miyajima, S., Yamamoto, C., Fujiwara, Y., Wight, T.N. and Kinsella, M.G. (2000): Cell density-dependent regulation of proteoglycan synthesis by transforming growth factor-beta(1) in cultured bovine aortic endothelial cells. J. Biol. Chem., 275, 1463-1470.
- Kaji, T., Yamamoto, C., Oh-i, M., Fujiwara, Y., Yamazaki, Y., Morita, T., Plaas, A.H. and Wight, T.N. (2006): The vascular endothelial growth factor VEGF165 induces perlecan synthesis via VEGF receptor-2 in cultured human brain microvascular endothelial cells. Biochim. Biophys. Acta, 1760, 1465-1474.
- Kanno, J. (2016): Introduction to the concept of signal toxicity. J. Toxicol. Sci., 41 (Special), SP105-SP109.
- Ki, Y.W., Park, J.H., Lee, J.E., Shin, I.C. and Koh, H.C. (2013): JNK and p38 MAPK regulate oxidative stress and the inflammatory response in chlorpyrifos-induced apoptosis. Toxicol. Lett., 218, 235-245.
- Kissane, J.M. and Robins, E. (1958): The fluorometric measurement of deoxyribonucleic acid in animal tissues with special reference to the central nervous system. J. Biol. Chem., 233, 184-188.
- Ladoux, A. and Frelin, C. (1993): Expression of vascular endothelial growth factor by cultured endothelial cells from brain microvessels. Biochem. Biophys. Res. Commun., 194, 799-803.
- Narumiya, S., Sugimoto, Y. and Ushikubi, F. (1999): Prostanoid receptors: structures, properties, and functions. Physiol. Rev., 79, 1193-1226.
- Patrick, L. (2006): Lead toxicity, a review of the literature. Part 1: Exposure, evaluation, and treatment. Altern. Med. Rev., 11, 2-22.
- Poręba, R., Gać, P., Poręba, M. and Andrzejak, R. (2011): Environmental and occupational exposure to lead as a potential risk factor for cardiovascular disease. Environ. Toxicol. Pharmacol., 31, 267-277.
- Revis, N.W., Zinsmeister, A.R. and Bull, R. (1981): Atherosclerosis and hypertension induction by lead and cadmium ions: an effect prevented by calcium ion. Proc. Natl. Acad. Sci. USA, 78, 6494-6498.
- Ristimäki, A., Ylikorkala, O., Perheentupa, J. and Viinikka, L. (1988): Epidermal growth factor stimulates prostacyclin production by cultured human vascular endothelial cells. Thromb. Haemost., 59, 248-250.
- Ross, R., Glomset, J. and Harker, L. (1977): Response to injury and atherogenesis. Am. J. Pathol., 86, 675-684.
- Sangvanich, T., Morry, J., Fox, C., Ngamcherdtrakul, W., Goodyear, S., Castro, D., Fryxell, G.E., Addleman, R.S., Summers, A.O. and Yantasee, W. (2014): Novel oral detoxification of mercury, cadmium, and lead with thiol-modified nanoporous silica. ACS Appl. Mater. Interfaces, 6, 5483-5493.
- Schütz, A., Skerfving, S., Ranstam, J. and Christoffersson, J.O. (1987): Kinetics of lead in blood after the end of occupational exposure. Scand. J. Work Environ. Health, 13, 221-231.
- Shinkai, Y., Yamamoto, C. and Kaji, T. (2010): Lead induces the expression of endoplasmic reticulum chaperones GRP78 and GRP94 in vascular endothelial cells via the JNK-AP-1 pathway. Toxicol. Sci., 114, 378-386.
- Simpson, R.B. (1961): Association constants of methylmercury with sulfhydryl and other bases. J. Am. Chem. Soc., 83, 4711-4717.
- Steinert, D., Küper, C., Bartels, H., Beck, F.X. and Neuhofer, W. (2009): PGE2 potentiates tonicity-induced COX-2 expression in renal medullary cells in a positive feedback loop involving EP2-cAMP-PKA signaling. Am. J. Physiol. Cell Physiol., 296, C75-C87.
- Tsatsanis, C., Androulidaki, A., Venihaki, M. and Margioris, A.N. (2006): Signalling networks regulating cyclooxygenase-2. Int. J. Biochem. Cell Biol., 38, 1654-1661.
- Ushikubi, F., Hirata, M. and Narumiya, S. (1995): Molecular biology of prostanoid receptors; an overview. J. Lipid Mediat. Cell Signal., 12, 343-359.
- Yoshida, E., Kurita, M., Eto, K., Kumagai, Y. and Kaji, T. (2017): Methylmercury promotes prostacyclin release from cultured human brain microvascular endothelial cells via induction of cyclooxygenase-2 through activation of the EGFR-p38 MAPK pathway by inhibiting protein tyrosine phosphatase 1B activity. Toxicology, 392, 40-46.
- Zhang, T., Xu, Z., Wen, L., Lei, D., Li, S., Wang, J., Huang, J., Wang, N., Durkan, C., Liao, X. and Wang, G. (2021): Cadmium-induced dysfunction of the blood-brain barrier depends on ROS-mediated inhibition of PTPase activity in zebrafish. J. Hazard. Mater., 412, 125198.
- Zhou, H., Ivanov, V.N., Gillespie, J., Geard, C.R., Amundson, S.A., Brenner, D.J., Yu, Z., Lieberman, H.B. and Hei, T.K. (2005): Mechanism of radiation-induced bystander effect: role of the cyclooxygenase-2 signaling pathway. Proc. Natl. Acad. Sci. USA, 102, 14641-14646.