Original Article
CHAC1 exacerbates arsenite cytotoxicity by lowering intracellular glutathione levels
2023 年 48 巻 9 号 p. 487-494
詳細
2023 年 48 巻 9 号 p. 487-494
We here examined whether CHAC1 is implicated in arsenite (As(III))-induced cytotoxicity in HaCaT cells. We found that HaCaT cells in which the intracellular GSH levels were elevated by transfection with CHAC1 siRNA showed decreased sensitivity to As(III) compared to the control cells. Treatment with BSO (an inhibitor of GSH biosynthesis) abolished the decrease in sensitivity to As(III), suggesting that an increase in intracellular GSH levels was involved in the decrease in sensitivity to As(III) due to the decrease in the levels of CHAC1 expression. When we examined the expression of CHAC1 after exposure of HaCaT cells to As(III), the levels of CHAC1 were increased. Since CHAC1 is a proapoptotic factor, we examined appearance of apoptotic cells and cleavage of caspase-3 after exposure to As(III) to determine whether As(III)-induced CHAC1 up-regulation was involved in apoptosis induction. The results showed that induction of apoptosis by As(III) exposure was not detected in CHAC1 siRNA-transfected cells. Together, our findings indicate that CHAC1 is involved in the sensitivity of HaCaT cells to As(III) by regulating the intracellular GSH levels, and in particular, CHAC1 is involved in As(III)-induced apoptosis.
Glutathione (GSH) is a tripeptide of γ-glutamyl-cysteinyl-glycine and is the most abundant thiol compound in cells (Meister and Anderson, 1983). GSH is oxidized during the scavenging of reactive oxygen species (ROS) by antioxidant enzymes such as glutathione peroxidase, and contributes to maintaining cells in a reduced state. In addition, GSH acts as a donor in GSH conjugation of xenobiotics by glutathione S-transferase (GST), and promotes extracellular excretion of xenobiotics (Meister and Anderson, 1983). The homeostasis of GSH relies on biosynthesis, consumption and degradation to maintain the intracellular environment. Prior to 2003, GSH was thought to be degraded only by the extracellular space of γ-glutamyl-transpeptidase (GGT), but through experiments using a genetic-based analysis, Kumar et al. revealed the potential existence of GSH degradation independent of GGT (Kumar et al., 2003). Subsequent research revealed that cation transport regulator-like protein 1 (CHAC1), a member of the γ-glutamylcyclotransferase family, hydrolyzes GSH to Cys-Gly and 5-oxoproline in cells (Mungrue et al., 2009; Crawford et al., 2015; Kumar et al., 2012). Given that the Km of CHAC1 is 2–3 mM and the concentration of intracellular GSH is ~10 mM, CHAC1 contributes to the degradation of intracellular GSH under physiological conditions (Kumar et al., 2012).
Inorganic arsenic that has entered cells is converted into less toxic substances such as methylarsonic acid and dimethylarsinic acid through metabolic reactions such as oxidative methylation by arsenic (+3 oxidation state) methyltransferase (AS3MT), GSH conjugation by GST, and reduction by GSH (Aposhian and Aposhian, 2006; Hayakawa et al., 2005; Kumagai and Sumi, 2007; Kojima et al., 2005). Analyses of a survey of residents with chronic arsenic poisoning revealed the relationship between polymorphisms in the genes encoding these enzymes, their effects on metabolic capacity and symptoms of arsenic poisoning (Sumi and Himeno, 2012; Sarker et al., 2021). Furthermore, arsenic compounds are known to produce ROS through the activation of oxidase/inhibition of antioxidant enzyme activity, thereby causing cytotoxicity to cells (Lu et al., 2007; Lynn et al., 2000; Smith et al., 2001; Jing et al., 1999), and GSH thus contributes to a reduction in the ROS-mediated cytotoxicity of arsenic compounds. Many reports have shown that treatment with BSO (an inhibitor of GSH biosynthesis) increases arsenic toxicity, making intracellular GSH levels an important factor in determining arsenic toxicity (Sumi et al., 2011; Chen et al., 2006; Shen et al., 2012).
In this study, we investigated whether CHAC1 exacerbates arsenite (As(III))-induced cytotoxicity via GSH degradation using HaCaT cells. Furthermore, since CHAC1 is a proapoptotic factor, we examined whether the induction of apoptosis by As(III) would be affected in HaCaT cells treated with CHAC1 siRNA.
Sodium arsenite (As(III)) (90% purity), buthionine sulfoximine, and GAPDH antibody (#016-25523) were purchased from Wako Pure Chemicals (Osaka, Japan). 4-fluoro-7-sulfobenzofurazan was purchased from DOJINDO (Kumamoto, Japan). Tri-n-butylphosphine was purchased from KISHIDA Chemical (Osaka, Japan). Antibody for CHAC1 (#NBP1-57685) was purchased from Novus Biologicals (Centennial, CO, USA) and antibodies for caspase-3 (#9662) and cleaved caspase-3 (#9661) were purchased from Cell Signaling Technology (Beverly, MA, USA). All other reagents and chemicals used were of the highest grade available.
Cell cultureHaCaT cells were obtained from CLS (Eppelheim, Germany) and cultured at 37°C in a humidified atmosphere of 5% CO2 using Dulbecco’s modified Eagle’s medium (Wako Pure Chemicals) containing 10% fetal calf serum, penicillin (100 U/mL) and streptomycin (100 μg/mL).
siRNA transfectionThe sequences of the No. 1 and No. 2 siRNAs against the human CHAC1 gene are 5′-GACUUACUACUUGAAACUU-3′ (No. 1) and 5′-CCAAGGAGGUCACCUUCUA -3′ (No. 2) (Sigma-Aldrich, St. Louis, MO, USA). The CHAC1 siRNA was transfected transiently with Lipofectamine RNAiMAX (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions, and a negative siRNA was used as a control. Briefly, siRNA and Lipofectamine RNAiMAX were diluted in Opti-MEMI, respectively (ThermoFisher Scientific). Then, the diluted siRNA duplex and the diluted Lipofectamine RNAiMAX were combined for 20 min at room temperature to allow formation of transfection complexes. Complexes were then added to the HaCaT cells and all were swirled gently to ensure uniform distribution. The final concentration of siRNA in the culture media was 10 nM.
RT-qPCRTotal RNA was isolated from cells using ISOGEN (NIPPON GENE, Tokyo, Japan). For the conversion of total RNA to cDNA, PrimeScript RT Master Mix (TaKaRa Bio, Shiga, Japan) was used. The reaction mixture was incubated at 37°C for 15 min followed by inactivation of the enzyme at 85°C for 30 sec. For the qPCR, a 20 μL mixture containing SYBR Premix TaqII (TaKaRa Bio), 2 μL cDNA, the specific PCR primers, and ROX reference dye was prepared. The qPCR was carried out using a StepOnePlus system (ThermoFisher Scientific). The PCR reaction protocol was 40 cycles of 95°C for 5 sec and 60°C for 30 sec. The primer sequences are shown in the Table 1. The levels of each mRNA were normalized by both β-actin and GAPDH levels, and the data are presented as the relative values.
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/48_487-t1.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09) Western blotting
Western blotting
The proteins were separated by SDS-PAGE. Gels were transferred to an immunoblot PVDF membrane and then placed in a blocking solution consisting of TBST (10 mM Tris (pH 8.0), 150 mM NaCl, and 0.05% Tween 20) and 5% skim milk for 1 hr. The blotted membranes were incubated with the appropriate antibody, washed with TBST, and incubated with HRP-conjugated secondary antibody. Bound IgG was visualized with Western blotting detection reagents (ThermoFisher Scientific) according to the manufacturer’s protocol.
Intracellular GSH levelsCellular GSH concentrations were determined primarily according to the method of Tanaka-Kagawa et al. (1993) with a modification. Briefly, the precipitated cells were mixed with 5 mM EDTA containing 5% TCA. The homogenates were then centrifuged at 4°C for 10 min to remove proteins. To an aliquot (300 μL) of the supernatant was added 100 μL of 0.2% SBD-F, 100 μL of 1% tri-n-butylphosphine and 350 μL of 1 M borate buffer containing 5 mM EDTA. The mixture was incubated at 60°C for 30 min. SBD-SH was separated by HPLC on a 5C18-MS-II column (Nacalai Tesque, Kyoto, Japan) with a solvent system (75 mM citrate buffer (pH 2.75)-methanol, 97:3), and the fluorescence of the eluate was monitored with a NANOSPACE SI-2 fluorescence detector (Osaka Soda, Osaka, Japan) (Ex = 384 nm; Em = 516 nm). Flow velocity was 2.0 mL/min.
Cell viabilityAn alamarBlue assay (Invitrogen, Carlsbad, CA, USA) was used for the determination of cell viability. The alamarBlue solution (1/50 volume) was added to the cells in the 96-well plate, and the mixture was cultured for another 12 hr in a CO2 incubator. The conversion of oxidized alamarBlue (blue) to the reduced form (pink) by active cells was monitored by absorbance at 540 nm.
Detection of apoptosisDetection of apoptosis was examined with Annexin V-FITC Apoptosis Detection Kit. Cells were stained with Annexin V-FITC conjugate and propidium iodide in Annexin V Binding Buffer and apoptotic cells were observed under a fluorescence microscope (Keyence, Osaka, Japan).
Statistical analysisData were obtained from three separate experiments. The values are shown as means ± SD. Statistical significance was assessed with student t-tests (Fig. 1B) and single-factor ANOVA followed by a Dunnet (Figs. 1A and 5A) and followed by a Tukey-Kramer post-hoc testing (Figs. 2, 3B and 4). Differences between groups were considered statistically significant at p < 0.05.

Evaluation of the actions of CHAC1 siRNA at two different target sites on HaCaT cells. Cells were harvested 48 hr after transfection of CHAC1 siRNA into HaCaT cells. A: The mRNA levels of CHAC1 in HaCaT cells transfected with No. 1 and No. 2 CHAC1 siRNA. Data are presented as the percentages of CHAC1 mRNA levels relative to the control siRNA. The values are mean ± SD. *p < 0.05, **p < 0.01 vs. control siRNA. B: Intracellular GSH levels in HaCaT cells transfected with No. 2 CHAC1 siRNA. The values are mean ± SD. **p < 0.01 vs. control siRNA. Data were obtained from three separate experiments.

Effects of CHAC1 siRNA on As(III)-induced cytotoxicity. HaCaT cells transfected with Control (Cont.) or CHAC1 siRNA for 48 hr were exposed to the indicated concentrations of As(III) for 24 hr. The cell viability was assessed by alamarBlue assay. Data are presented as the percentages of viable cells relative to the control. The values are mean ± SD. **p < 0.01 vs. control siRNA. Data were obtained from three separate experiments.
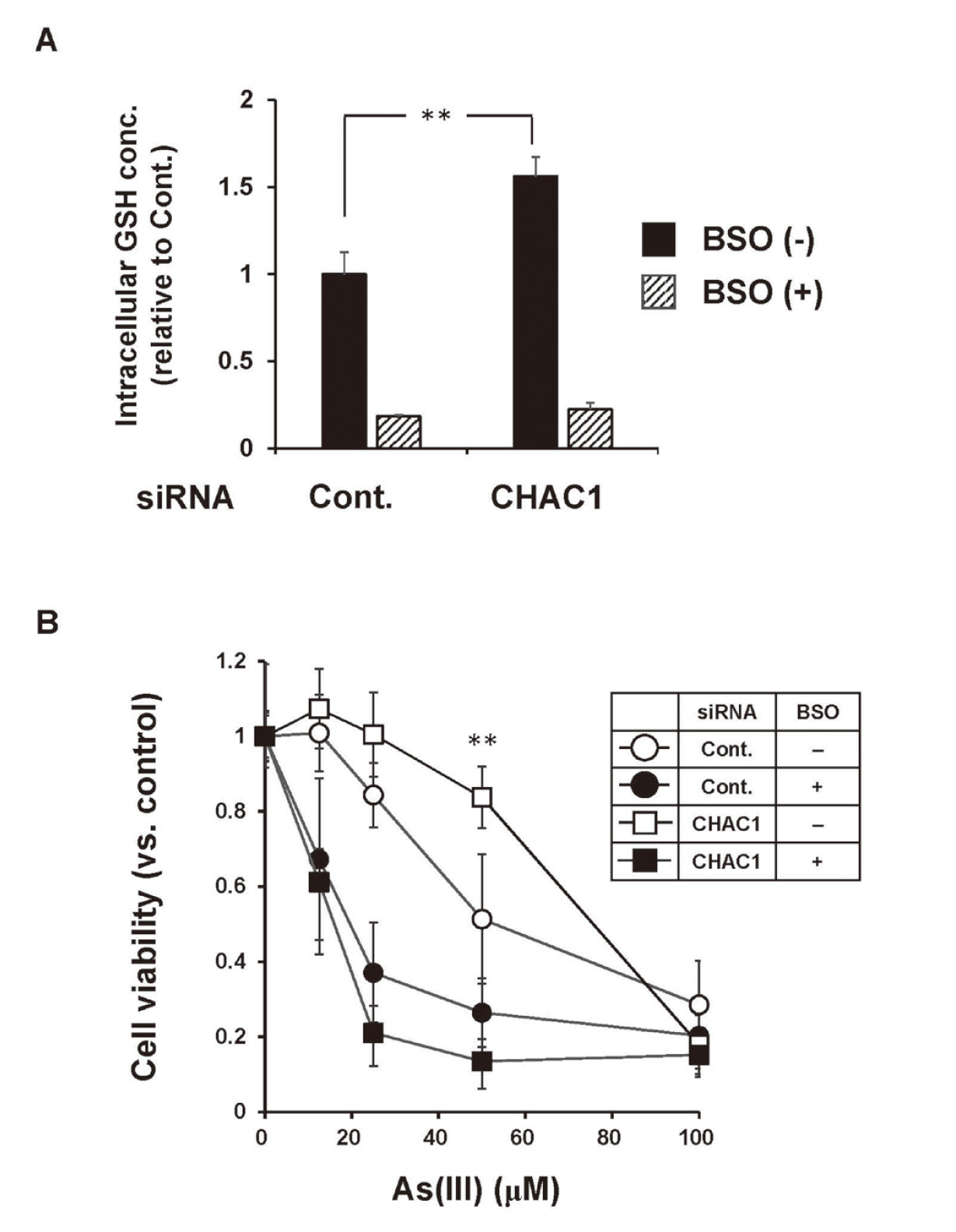
Effect of BSO on the reduction of As(III)-induced cytotoxicity by CHAC1 siRNA. HaCaT cells transfected with CHAC1 siRNA for 36 hr were treated with BSO for 12 hr followed by exposure to the indicated concentrations of As(III) for 24 hr. A: Intracellular GSH levels in CHAC1 siRNA HaCaT cells treated with BSO. The values are mean ± SD. **p < 0.01 vs. control siRNA. B: The cell viability was assessed by alamarBlue assay. Data are presented as the percentages of the viable cells relative to the control. The values are mean ± SD. **p < 0.01 vs. control siRNA. Data were obtained from three separate experiments.
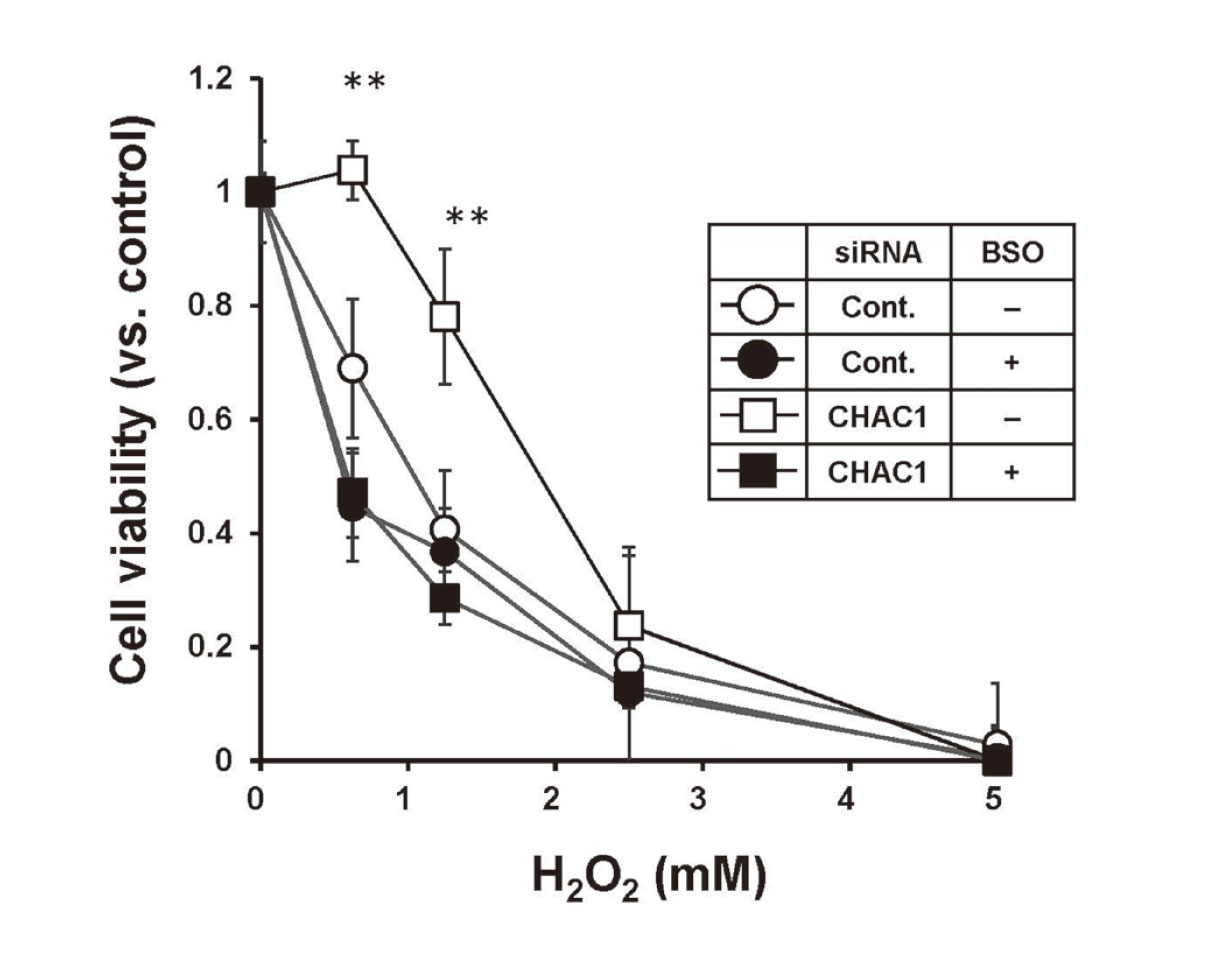
Effects of CHAC1 siRNA on H2O2-induced cytotoxicity. HaCaT cells transfected with Control (Cont.) or CHAC1 siRNA for 36 hr were treated with BSO for 12 hr followed by exposure to the indicated concentrations of H2O2 for 24 hr. The cell viability was assessed by alamarBlue assay. Data are presented as the percentages of the viable cells relative to the control. The values are mean ± SD. **p < 0.01 vs. control siRNA. Data were obtained from three separate experiments.

Effects of As(III) exposure on CHAC1 expression. HaCaT cells were exposed to the indicated concentrations of As(III) for 24 hr. A: The mRNA (A) and protein (B) levels of CHAC1 in HaCaT cells were analyzed by RT-qPCR and western blotting, respectively. The values are mean ± SD. **p < 0.01 vs.control. Data were obtained from three separate experiments. B: The protein levels of CHAC1 in HaCaT cells were analyzed by western blotting. Representative data from three individual determinations are shown.
In order to examine whether CHAC1 is implicated in the sensitivity to As(III), we first verified the effect of CHAC1 siRNA at two different target sites on HaCaT cells. As shown in Fig. 1A, both the No. 1 and No. 2 siRNAs targeting CHAC1 mRNA suppressed the levels of endogenous CHAC1 expression in HaCaT cells. The No. 2 siRNA showed a greater knockdown efficiency at the level of mRNA (Fig. 1A). Therefore, the No. 2 siRNA was used in subsequent experiments. When the intracellular GSH levels in HaCaT cells transfected with CHAC1 siRNA were measured, the intracellular GSH levels were found to be significantly increased compared to those in HaCaT cells transfected with the control siRNA (Fig. 1B). These results suggest that CHAC1 siRNA decreased the levels of CHAC1 expression and inhibited the degradation of intracellular GSH in HaCaT cells, resulting in an increase in the intracellular GSH levels.
We next examined whether the decreased levels of CHAC1 expression affected the sensitivity to As(III) in HaCaT cells. The results showed that As(III)-induced cytotoxicity was reduced in HaCaT cells transfected with CHAC1 siRNA (Fig. 2). To investigate whether intracellular GSH levels were involved in the reduction in sensitivity to As(III) induced by decreased CHAC1 expression, we examined HaCaT cells in which intracellular GSH levels were reduced by BSO (an inhibitor of GSH biosynthesis) (Fig. 3A). The results showed that As(III)-induced cytotoxicity was reduced in HaCaT cells transfected with CHAC1 siRNA in the absence of BSO (Fig. 3B: open circles vs. open squares), but such a reduction was not observed in the presence of BSO (Fig. 3B: closed circles vs. closed squares). These results suggest that, in HaCaT cells, the reduction in sensitivity to As(III) induced by a decrease in CHAC1 expression was attributable to an increase in intracellular GSH levels.
ROS are involved in As(III)-induced cytotoxicity (Liu et al., 2003; Partridge et al., 2007). Therefore, we hypothesized that the reduction in sensitivity to As(III) in HaCaT cells transfected with CHAC1 siRNA might be due to enhanced scavenging of ROS by GSH, and examined whether the decreased levels of CHAC1 expression affected the sensitivity to hydrogen peroxide (H2O2) in HaCaT cells. Similar to the sensitivity to As(III), H2O2-induced cytotoxicity was reduced in HaCaT cells transfected with CHAC1 siRNA, and this reduction was not detected in the presence of BSO (Fig. 4).
Because apoptosis is known to be associated with As(III)-induced cell death (Liu et al., 2003; Partridge et al., 2007) and CHAC1 has been reported to play a proapoptotic role (Mungrue et al., 2009), we next examined whether As(III) exposure affects the levels of CHAC1 expression in HaCaT cells. When HaCaT cells were exposed to As(III) for 24 hr, 5 μM As(III) significantly enhanced the levels of CHAC1 mRNA (Fig. 5A), which also increased the protein levels of CHAC1 (Fig. 5B). To further clarify whether CHAC1 is involved in As(III)-induced apoptosis, we exposed CHAC1 siRNA-transfected HaCaT cells to As(III) and observed apoptotic cells and detected cleaved caspase-3 as an indicator of apoptosis. While early stage of apoptotic cells (green) were observed in an As(III) concentration-dependent manner in control siRNA-transfected cells, the appearance of apoptotic cells was remarkably reduced in the cells transfected with CHAC1 siRNA (Fig. 6). As shown in Fig. 7, cleaved caspase-3 was detected following As(III) exposure in HaCaT cells transfected with the control siRNA, but not in HaCaT cells transfected with CHAC1 siRNA (Fig. 7). These results suggest that CHAC1 is involved in the induction of apoptosis by As(III) exposure in HaCaT cells.
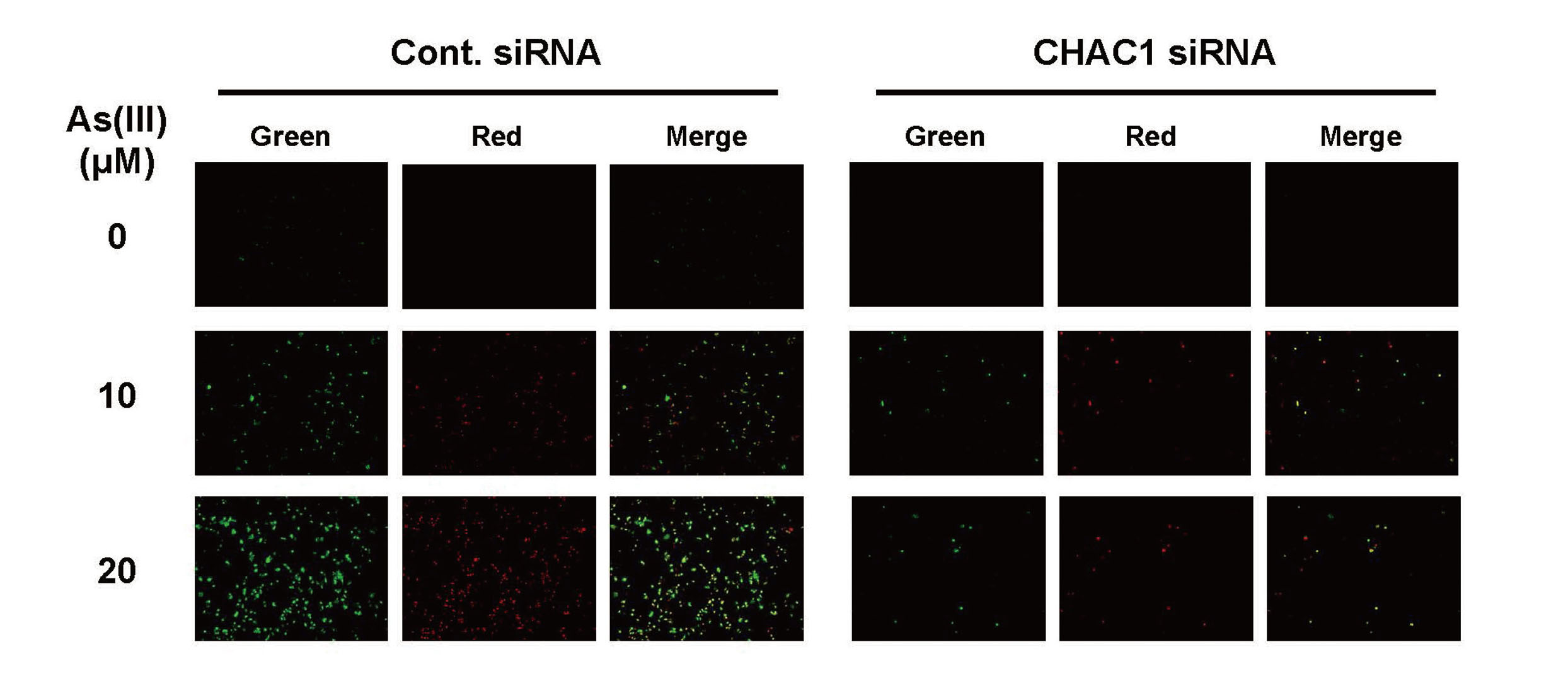
Effects of CHAC1 siRNA on As(III)-induced apoptosis. HaCaT cells transfected with Control (Cont.) or CHAC1 siRNA for 48 hr were exposed to 10 and 20 μM As(III) for 24 hr. Cells were stained with Annexin V-FITC Apoptosis Detection Kit according to the manufacturer’s protocol. Cells stained with Annexin V (Green) show early stages of apoptosis. Representative data from three individual determinations are shown.
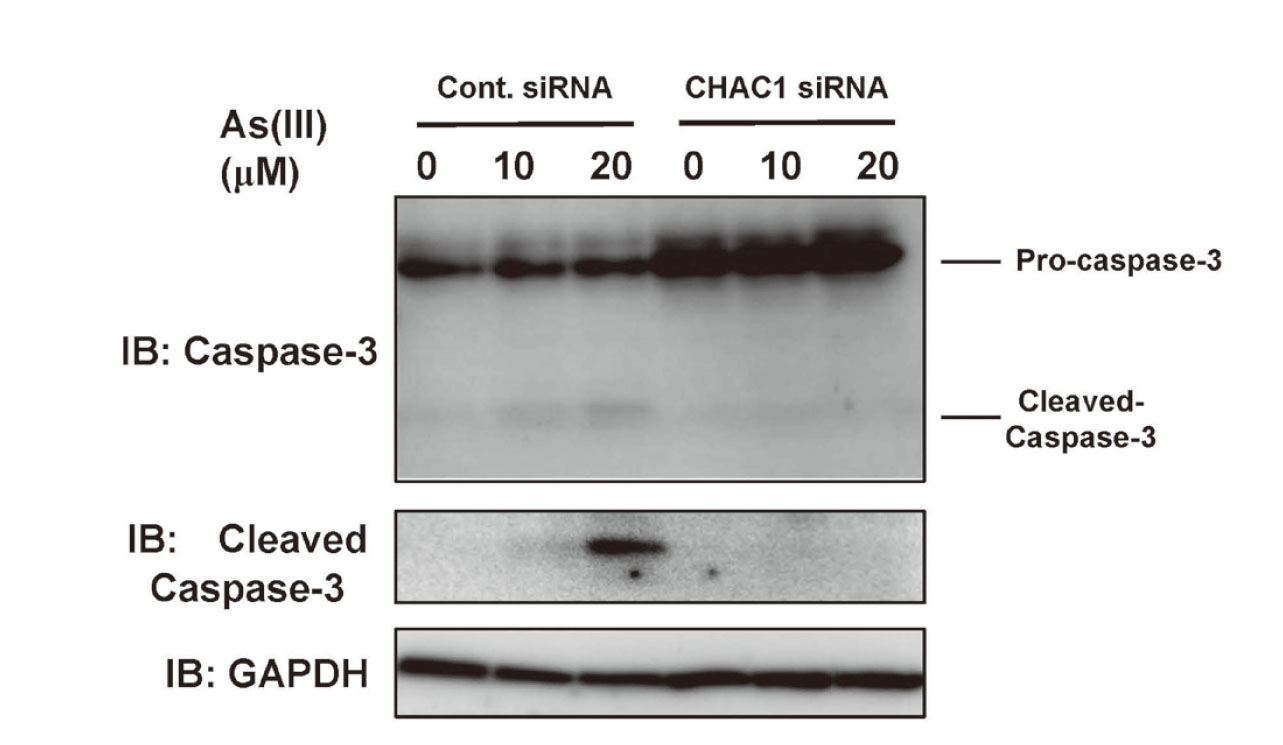
Effects of CHAC1 siRNA on As(III)-induced caspase-3 activation. HaCaT cells transfected with Control (Cont.) or CHAC1 siRNA for 48 hr were exposed to 10 and 20 μM As(III) for 24 hr. Cells were harvested and caspase-3 and cleaved caspase-3 were detected by western blotting. Representative data from three individual determinations are shown.
CHAC1 is a molecule whose expression is upregulated in unfolded protein response (UPR)/endoplasmic reticulum (ER) stress (Mungrue et al., 2009), amino acid restriction (Tattoli et al., 2012), cancer (Mehta et al., 2022; He et al., 2021), and diabetes (Li et al., 2020). Kumar et al. (2012) showed that CHAC1 specifically degrades GSH via the actions of recombinant proteins of CHAC1 and YER163c (a yeast homologue of CHAC1). The intracellular expression of CHAC1 led to the introduction of a new concept-namely, that a cycle of biosynthesis and degradation functions to maintain intracellular GSH homeostasis. On the other hand, GSH is a critical factor in the methylation metabolism of arsenic compounds (Aposhian and Aposhian, 2006; Hayakawa et al., 2005; Kumagai and Sumi, 2007) and in the response to arsenic-induced oxidative stress (Jing et al., 1999; Lu et al., 2007; Liu et al., 2003; Smith et al., 2001), and the toxicity of arsenic compounds is remarkably enhanced by the regulation of gene expression or inhibitors of GSH biosynthesis (Chen et al., 2006; Shen et al., 2012; Sumi et al., 2011). However, there has been no report on whether intracellular degradation of GSH is involved in arsenic toxicity. Accordingly, we here investigated the role of CHAC1 in arsenic toxicity.
The results of Figs. 2 and 3 strongly suggested that the silencing of CHAC1 expression by siRNA reduces the sensitivity to As(III), and that this reduction in sensitivity was related to the intracellular GSH levels. As factors that determine the As(III) toxicity in cells, these two factors are related: whether As(III) undergoes methylation metabolism and/or whether cells are able to respond to oxidative stress. This is also evident from the fact that the activation of the transcription factor Nrf2 by As(III) suppresses the As(III) toxicity by increasing the expression of metabolic and antioxidant enzymes (Kumagai and Sumi, 2007; Pi et al., 2003). As shown in Fig. 4, the suppression of CHAC1 in HaCaT cells also reduced the sensitivity to H2O2, suggesting that, at least in part, HaCaT cells transfected with CHAC1 siRNA have an increased resistance to oxidative stress by As(III) exposure.
Based on the results of our experiments, we initially thought that the HaCaT cells responded to As(III)-induced cytotoxicity by increasing GSH levels through a decrease in the levels of CHAC1 expression, thereby avoiding the cytotoxicity. However, we found that CHAC1 levels were elevated by As(III) exposure, appearance of apoptotic cells by As(III) exposure were reduced and cleavage of caspase-3 by As(III) was not detected in cells transfected with CHAC1 siRNA (Figs. 5, 6 and 7). This suggests that CHAC1 induces apoptosis in response to As(III). Örd et al. (2016) showed that CHAC1 expression is elevated by As(III) exposure through suppression of TRIB3 expression, which negatively regulates CHAC1 expression in mouse embryonic fibroblasts, but there is no result on whether GSH degradation mediated by increased CHAC1 expression by As(III) exposure is related to apoptosis. The novelty of our study is that we have clarified that GSH degradation by CHAC1 is involved in As(III)-induced apoptosis. Arsenicals have long been used therapeutically as Chinese medicines. Over the last decade, arsenic trioxide (ATO) was adopted for the treatment of relapsed or intractable acute promyelocytic leukemia (APL) (Chen et al., 1997). Several mechanisms have been found to underlie the efficacy of ATO in the treatment of APL, including induction of apoptosis (Chen et al., 1996). ATO induces apoptosis not only in hematological disorders such as APL but also in solid cancer cells (Ling et al., 2002). If CHAC1 is elevated in the induction of cancer cell apoptosis by ATO, combination treatment with ATO and a CHAC1 inducer might be used as a highly effective anticancer therapy.
5-oxoproline, a metabolite, is produced when the degradation of GSH is promoted by upregulation of CHAC1. Accumulation of 5-oxoproline is known to occur in GSH synthase deficiency, an inborn metabolic defect of the γ-glutamyl cycle that is clinically characterized by hemolytic anemia, metabolic acidosis and neurological disorders (Njålsson, 2005). Pederzolli et al. (2007) reported that exposure of the isolated cerebral cortex of rats to 5-oxoproline increased indicators of oxidative stress. On the other hand, in an investigation of the relationship between metabolites in blood and urine and the onset of diabetes in residents living in an arsenic-contaminated area in Chihuahua, Mexico, arsenic exposure was found to increase the 5-oxoproline levels in urine (Martin et al., 2015). Based on these findings, the increased expression of CHAC1 in response to As(III) exposure may lead to an accumulation of 5-oxoproline levels, and thus an increase in oxidative stress might be a factor in the induction of apoptosis. Future studies using an inhibitor and siRNA for 5-oxoprolinase will be needed to fully elucidate the implications of 5-oxoproline in As(III)-induced cytotoxicity.
This work was partly supported by a Grant-in-Aid for Scientific Research on Innovative Areas (No. 19H05770).
Conflict of interestThe authors declare that there is no conflict of interest.