Abstract
Mas-related G-protein-coupled receptor X2 (MRGPRX2), expressed on mast cells, is associated with drug-induced pseudo-allergic reactions. Although it is well known that there are differences of sensitivity between species in the pseudo-allergic reactions, no platform for evaluating a human risk of the pseudo-allergic reactions observed in nonclinical studies has been established. Valemetostat tosylate, developed as an anti-cancer drug, induced histamine release in a nonclinical study with dogs. The purpose of the current study was to identify the mechanism and assess the human risk of valemetostat-tosylate-induced histamine release using dog and human MRGPRX2-expressing cells. In an experiment with human or dog MRGPRX2-expressing cells, valemetostat tosylate caused activation of human and dog MRGPRX2. Importantly, the EC50 for dog MRGPRX2 was consistent with the Cmax value at which histamine release was observed in dogs. Furthermore, the EC50 for human MRGPRX2 was ca. 27-fold higher than that for dog MRGPRX2, indicating a species difference in histamine-releasing activity. In a clinical trial, histamine release was not observed in patients receiving valemetostat tosylate. In conclusion, an in vitro assay using human and animal MRGPRX2-expressing cells would be an effective platform to investigate the mechanism and predict the human risk of histamine release observed in nonclinical studies.
INTRODUCTION
In nonclinical studies, drug candidates sometimes induce histamine release in laboratory animals (Mori et al., 2000; Takasuna et al., 1992; Kojima et al., 1984). Because histamine release is potentially associated with serious adverse reactions such as anaphylaxis (Peavy and Metcalfe, 2008; White, 1990), careful evaluation is required. Two pathways, an IgE-dependent pathway and an IgE-independent pathway, are known as the mechanism of drug-induced histamine release (Ferry et al., 2002). In the IgE-dependent pathway, pre-existing anti-drug IgE binds to FcɛRI followed by drug-induced cross-linking of two or more FcɛRI, leading to mast cell degranulation (Kinet, 1999). In contrast, in the IgE-independent pathway, drugs directly bind to a kind of G protein-coupled receptors (GPCR) on mast cells to induce their degranulation. Because this reaction does not depend on IgE, it is distinguished from an allergic reaction and referred to as a “pseudo-allergic” reaction (Wang et al., 2011; Zhang et al., 2018). A characteristic feature of pseudo-allergic reactions is species differences in sensitivity. Therefore, the responses seen in laboratory animals may not be directly extrapolated to humans (Ennis et al., 1985; Hägermark et al., 1978; McNeil et al., 2015). Thus, it is important to identify the mechanism and to clarify species differences for predicting the human risk of histamine release observed in laboratory animals. However, there are no established methods to assess drug-induced pseudo-allergic reactions and to estimate the human risk.
Mas-related G-protein-coupled receptor X2 (MRGPRX2) is a Gi- or Gq-coupled receptor and it was identified as a receptor crucial for drug-induced pseudo-allergic reactions (Subramanian et al., 2016). MRGPRX2 is expressed on mast cells and is activated by drugs via an IgE-independent pathway. Activation of MRGPRX2 induces intracellular calcium mobilization in mast cells, followed by degranulation (Dhyani et al., 2020; Liu et al., 2017). Unlike other GPCRs, MRGPRX2 is known to recognize a wide variety of small molecules and peptides as its ligands, including fluoroquinolones, neuromuscular blocking agents, opioids, and icatibant (Kumar et al., 2021; McGee et al., 2019; Roy et al., 2021). In a recent report, it was described that nearly one-third of 368 structurally diverse small molecules activated MRGPRX2 (Grimes et al., 2019). Therefore, some drug candidates, including small-molecules or peptidergic drugs, have the potential to activate MRGPRX2, followed by degranulation of mast cells.
Dogs, one of the most frequently used non-rodent animals in nonclinical safety studies, are known to exhibit high susceptibility to drug-induced pseudo-allergic reactions compared with rodents (Ennis et al., 1985; Masini et al., 1985; Mori et al., 2000, 2001). Indeed, the dose levels that caused histamine release in dogs were 30 to 100 times lower than in rats (Mori et al., 2000). In addition, intravenous injections of fluoroquinolones, opioids, or neuromuscular blocking agents have been reported to cause severe hypotension in dogs with increasing blood histamine levels (Furuhata et al., 1998; Lee and Johnson, 1971; Robinson et al., 1988; Takasuna et al., 1992). Therefore, in vivo studies using dogs have been conducted generally to detect risks of drug-induced pseudo-allergic reactions. However, little is known about a characteristic of MRGPRX2 ortholog in dogs while many of MRGPRX2-associated studies focused on mice and its Mrgprb2. Although mouse Mrgprb2 is similar to human MRGPRX2 in a distribution or a role in some diseases, the response is markedly weaker than human MRGPRX2 against many ligands such as substance P or fluoroquinolones (McNeil et al., 2015; Kumar et al., 2021; Roy et al., 2021). In a previous report, we identified dog MRGPRX2 as human MRGPRX2 ortholog, with dog MRGPRX2-expressing cells showing higher sensitivity to fluoroquinolones than human MRGPRX2-expressing cells (Hamamura-Yasuno et al., 2020). Regarding its high sensitivity, dogs and dog MRGPRX2 would be a suitable model to screen drugs with pseudo-allergic potential for human use compared with rodents. In the present study, we conducted a more detailed analysis of in vivo dog responses and a construction of an in vitro assay system which can assess different sensitivity against MRGPRX2 ligands between human and dog in order to clarify the usefulness of dog MRGPRX2 in MRGPRX2 research. Because intracellular calcium mobilization in mast cells induced by MRGPRX2 ligands causes degranulation (Dhyani et al., 2020; Liu et al., 2017), evaluation of intracellular calcium levels of MRGPRX2-expressing cells has been widely conducted in many studies as a simple and stable method (Grimes et al., 2019; McNeil et al., 2015; Tatemoto et al., 2006). However, to the best of our knowledge, no studies have focused on evaluating species differences or correlations between responses of an in vivo nonclinical/clinical study and those of an in vitro assay system.
Valemetostat tosylate is an anti-cancer drug developed for treating malignant lymphomas and solid tumors by Daiichi Sankyo, Co., Ltd. This small-molecule drug inhibits enhancer of zeste homolog (EZH)1 and EZH2, which act through histone H3 lysine 27 methylation to regulate gene expression (Margueron et al., 2008). Inhibition of histone H3 lysine 27 methylation by EZH1/2 is considered to prevent the growth of cancer cells by activating tumor suppressor genes (Yamagishi et al., 2019). In nonclinical safety studies, valemetostat tosylate induced histamine release in dogs, but not in rats. Therefore, in this study, we identified the mechanism of valemetostat-tosylate-induced histamine release and assessed the human risk of such release using cell-based assay system with dog and human MRGPRX2.
MATERIALS AND METHODS
Chemicals
Valemetostat tosylate (molecular formula: C26H34ClN3O4·C7H8O3S, structural formula is shown in Figure 1A) was synthesized at Daiichi Sankyo Co., Ltd. (Tokyo, Japan). All doses and concentrations are expressed in terms of valemetostat, a free form of valemetostat tosylate. For the in vivo study, valemetostat tosylate was suspended in vehicle [mixed solution of 80% v/v Polyethylene Glycol 200 (PEG200, Sigma-Aldrich Co. LLC, St. Louis, MO, USA) and 20% v/v sterile water for injection] to make a 5, 10, or 33.3 mg/mL (for the 15, 30, or 100 mg/kg group) suspension of valemetostat. For the in vitro study, valemetostat tosylate was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich Co. LLC) at a valemetostat concentration of 74 mg/mL. Hanks’ balanced salt solution (HBSS; Thermo Fisher Scientific Inc., Waltham, MA, USA), hydroxyethyl piperazine ethanesulfonic acid (HEPES; Sigma-Aldrich Co. LLC), 30% bovine serum albumin (BSA; Sigma-Aldrich Co. LLC), 0.1N NaOH (Fujifilm Wako Pure Chemical Corporation, Osaka, Japan), and compound 48/80 (Sigma-Aldrich Co. LLC) were purchased. Compound 48/80 was dissolved in preparation buffer (HBSS supplemented with 20 mM HEPES and 0.05% BSA, pH 7.4 adjusted with 0.1N NaOH) at a concentration of 3 mg/mL. Valemetostat tosylate and compound 48/80 were diluted with preparation buffer. Preparation buffer and 1% DMSO in preparation buffer were also evaluated as vehicle controls.

For a dog study, male and female beagle dogs (n=17/sex) were purchased from Marshall BioResources (Lyon, France; breeding site: North Rose, NY, USA). At the beginning of the treatment, the dogs were approximately 7–9 months old; males weighed 6.9 to 10.0 kg and females weighed 5.5 to 8.0 kg. The dogs (2 to 5 animals/cage) were housed in stainless steel cages at a controlled temperature of 18 to 22°C and relative humidity of 40% to 70% with a 12-hr light and dark cycle. Each dog was offered approximately 350 g of Teklad 2021 Expanded Diet (Envigo, Bicester, UK) daily. Filtered water from the domestic supply was freely available from an automatic watering system. For a rat study, male and female Crl:CD(SD) rats (n=72/sex) were obtained from Charles River Laboratories Italia S.r.l. (Calco, Italy). At the beginning of the treatment, the rats were approximately 8 weeks old; males weighed 267.4 to 326.8 g and females weighed 168.5 to 237.2 g. The rats (2 to 3 animals/cage) were housed in concrete/solid floored cages with sawdust litter at a controlled temperature of 20 to 22°C and relative humidity of 45% to 65% with a 12-hr light and dark cycle. Gamma irradiated rat and mouse maintenance diet Altromin R (A. Rieper S.p.A., Bolzano, Italy) and filtered water (Eurofins Biolab S.r.l., Vimodrone, Italy) were available ad libitum. All the experimental protocols were approved in advance by the Ethics Review Committee for Animal Experimentation of Daiichi Sankyo Co., Ltd. All animal procedures were performed in accordance with the guidelines of the facility.
Design of dog toxicity study
A study on the toxicity of a single oral dose of valemetostat tosylate was conducted in male and female dogs. Animals were randomly allocated to their treatment group before the start of treatment. Valemetostat was administered to 5 dogs/sex/group at 0 (vehicle), 15, and 30 mg/kg, and 2 dogs/sex/group at 100 mg/kg. Dogs were given a single oral dose of valemetostat tosylate or vehicle at a constant dose volume of 3 mL/kg. The following endpoints/parameters were evaluated: clinical observation, plasma histamine level, and toxicokinetics. After the evaluation, dogs were euthanized by intravenous injection of barbiturate (sodium thiopental 200 mg/mL; 0.75 mL/kg) and sodium heparin (5000 IU/mL; 0.2 mL/kg) followed by exsanguinations.
Plasma histamine level
Plasma samples for histamine analysis were collected in pre-chilled tubes containing 1.6 mg/mL potassium EDTA at the following timepoints: pre-dose, 0.5, 1, 2, 4, 8, 12, and 24 hr after dosing. Histamine concentration in plasma samples was measured by enzyme-linked immunosorbent assay (ELISA).
Toxicokinetics
Plasma samples were collected in tubes containing potassium 1.6 mg/mL EDTA at the following timepoints: 0.5, 1, 2, 4, 8, 12, and 24 hr after dosing. All plasma samples were frozen at approximately –20°C until measurement of drug concentration. The samples were analyzed for quantification of valemetostat using a method based on protein precipitation, followed by liquid chromatography–mass spectrometry (LC-MS/MS) analysis using tandem mass spectrometric detection and a turboionspray interface with positive ion multiple reaction monitoring (chromatography system: Acquity UPLC, Waters Corporation, Milford, MA, USA; mass spectrometer: API-4000, AB Sciex LLC, Framingham, MA, USA). The lower limit of quantification (LLQ) and higher limit of quantification (HLQ) of valemetostat were 10 and 10,000 ng/mL, respectively, using a 10 μL aliquot of dog plasma. Noncompartmental pharmacokinetic analysis was conducted using Phoenix WinNonlin (version 6.3; Certara USA, Inc., Princeton, NJ, USA). Systemic exposure was determined by calculating the area under the plasma concentration time curve (AUC) from the start of dosing to the last quantifiable timepoint (AUC0–24h) using the linear-logarithmic trapezoidal rule. The maximum observed peak plasma concentration (Cmax) and the time at which it was observed (Tmax) were determined by inspection of the observed data.
Rat toxicity study
A study on the toxicity of a single oral dose of valemetostat tosylate was conducted in male and female rats. Animals were randomly allocated to their treatment group before the start of treatment. Valemetostat was administered to 15 rats/sex/group at 0, 60, 200, and 600 mg/kg for clinical observation, and 3 rats/sex/group at 0, 60, 200, and 600 mg/kg for plasma histamine evaluation. Rats were given a single oral dose of valemetostat tosylate or vehicle at a constant dose volume of 5 mL/kg. The following endpoints/parameters were evaluated: clinical observation and plasma histamine levels. Plasma histamine levels were measured at pre-dose, 0.5, 1, 2, 8 hr after dosing using same methods as dogs. After the evaluation, rats were euthanized by exsanguination under deep isoflurane anesthesia.
Transfection of HEK293 cells
HEK293 cells obtained from JCRB Cell Bank (Osaka, Japan). An open reading frame of human MRGPRX2 (accession No. EU883579) and dog MRGPRX2 (accession No. XM_005633812) was inserted into pcDNA3.1(+) in Thermo Fisher Scientific Inc. HEK293 cells were transiently transfected with these vectors including dog or human MRGPRX2. Lipofectamine 2000 Reagent (Thermo Fisher Scientific Inc.) and pcDNA3.1(+) containing each gene were diluted and mixed using Opti-MEM I Reduced Serum Medium (Thermo Fisher Scientific Inc.) to prepare lipid-DNA complexes (final concentrations: lipofectamine 2.5 µL/mL and DNA 2,500 ng/mL). HEK293 cells were detached using TrypLE Express (Thermo Fisher Scientific Inc.) and prepared to 7 × 105 cells/mL with the lipid-DNA complex. Thereafter, 25 µL of cells (1.75 × 104 cells/well) were seeded per well in 384-well flat-bottomed plates (Corning Incorporated, Corning, NY, USA) and incubated overnight at 37°C under 5% CO2 conditions. Cells treated with plasmid-free lipid solution were used as a negative control (untransfected cells).
Calcium mobilization assay
MRGPRX2, which is a Gq-coupled receptor, induces intracellular calcium mobilization after ligand binding. In the present study, intracellular calcium level was measured as endpoint. Intracellular calcium levels were analyzed using Calcium Kit II-iCellux (Dojindo Molecular Technologies, Inc., Kumamoto, Japan), in accordance with the manufacturer’s instructions. HEK293 cells (1.75 × 104 cells/well) were loaded with 1.25 mM probenecid and calcium probe for 45 min at 25°C. Changes in fluorescence intensity between before and after addition of the test articles were measured over time using FLIPR Tetra (Molecular Devices, LLC, Sunnyvale, CA, USA) with excitation at 470 to 495 nm and emission at 515 to 575 nm. The test articles were added 10 sec after beginning the measurements. Compound 48/80, a typical histamine-releasing agent, was evaluated as a positive control. The data were analyzed using ScreenWorks (version 3.2.0.14; Molecular Devices, LLC) to determine the difference between maximal and minimal fluorescence intensity (max – min). The experiments were conducted more than three times independently. The four-parameter sigmoidal model was used for curve-fitting using GraphPad Prism 7.03 (GraphPad Software, La Jolla, CA, USA).
Clinical study
Plasma samples were obtained from patients with acute myeloid leukemia (AML) or acute lymphoblastic leukemia (ALL) enrolled in a Phase I clinical trial of valemetostat tosylate (NCT No. NCT03110354). A total of three subjects were treated with valemetostat at 500 mg QD. The study was carried out in accordance with the Declaration of Helsinki and the International Conference on Harmonization guidelines for Good Clinical Practice. Approval by an independent ethics committee was obtained before study initiation. All patients provided written informed consent before study enrolment. The following endpoints/parameters were evaluated: plasma histamine level and pharmacokinetics.
Statistical analysis
Data are presented as the mean ± S.D. for calcium mobilization and valemetostat tosylate levels in plasma. Half-maximal effective concentration (EC50) of each test article used in the calcium mobilization assay was calculated from individual Emax and E0 for each type of cells using the SAS System Release 9.2 software (SAS Institute Inc., Cary, NC, USA).
RESULTS
Clinical observation in dogs
A study with a single oral dose of valemetostat tosylate was conducted to assess the toxicity in beagle dogs. In dogs given 15, 30, and 100 mg/kg valemetostat, emesis in the cage was observed (Table 1). At 30 mg/kg, subdued behavior, unsteady gait, and lack of coordination were detected. In the 100 mg/kg group, decreased activity, subdued behavior, lateral recumbency, tremors, loose or watery feces, salivating, and labored breathing were observed. In addition, characteristic swelling around the eyes, pinnas, and/or muzzle, and red and/or hot extremities were detected in animals in the 30 and 100 mg/kg groups (Table 1). Many of these clinical signs were detected immediately after dosing and disappeared in most of the animals within 4 hr after dosing. For instance, the typical swelling and redness around the eyes was detected 20 min after dosing, and getting stronger 40 min after doing (Fig. 1B). Thereafter, the swelling and redness was disappeared and recovered to pre-dosing levels at 100 min after dosing. These typical swelling or red extremities were detected in 8 out of 10 animals in 30 mg/kg group and all animals in 100 mg/kg group, but not in 15 mg/kg group. All the animals in the 0 mg/kg group, 5 animals in 15 mg/kg groups, 2 animals in 30 mg/kg group, and no animals in 100 mg/kg groups showed no noteworthy findings. In summary, valemetostat tosylate induced characteristic clinical symptoms to dogs, and the number of types of symptoms and frequency tend to increase in higher dose group.
Table 1. Summary results of clinical observation in dogs treated with valemetostat.
| Dose (mg/kg) |
0 |
15 |
30 |
100 |
| No. of animals |
10 (5/sex) |
10 (5/sex) |
10 (5/sex) |
4 (2/sex) |
| Decreased activity |
0 |
0 |
0 |
2
(M16, F17) |
| Subdued behavior |
0 |
0 |
1
(M15) |
3
(M16, M17, F17) |
| Lateral recumbency |
0 |
0 |
0 |
2
(M16, M17) |
| Tremors |
0 |
0 |
0 |
3
(M16, M17, F17) |
| Unsteady gait |
0 |
0 |
1
(M15) |
0 |
| Lack of coordination |
0 |
0 |
1
(M15) |
0 |
| Emesis in cage |
0 |
5
(M6, M7, M8, M9, M10) |
1
(F13) |
4
(M16, M17, F16, F17) |
| Loose/watery feces |
0 |
0 |
0 |
1
(M16) |
| Salivating |
0 |
0 |
0 |
3
(M16, M17, F17) |
| Labored breathing |
0 |
0 |
0 |
2
(M16, F17) |
| Reddening extremities |
0 |
0 |
8
(M11, M12, M13, M14, M15, F12, F13, F14) |
4
(M16, M17, F16, F17) |
| Hot extremities |
0 |
0 |
0 |
4
(M16, M17, F16, F17) |
| Swollen (around eyes, pinnas, or muzzle) around eyes) |
0 |
0 |
3
(M12, M13, F13) |
4
(M16, M17, F16, F17) |
Valemetostat was orally administered to dogs at 0 (vehicle), 15, 30, or 100 mg/kg. The numbers in the table indicate the number of animals that showed the clinical signs in the respective group. All doses and concentrations are expressed in terms of valemetostat, a free form of valemetostat tosylate. Characters in parentheses indicate animal numbers. M: male, F: female.
Plasma histamine levels in dogs
To assess the correlation between the clinical symptoms observed in the dog toxicity study and histamine release, concentration of plasma histamine was measured. Apparent increased histamine levels were detected in 2 males and 1 female in 30 mg/kg group, and 1 male and 2 females in 100 mg/kg group (Fig. 2). Maximum increases among individual animals when compared to corresponding pre-dose levels were 10-fold (pre-dosing: 3.3 ng/mL and after-dosing: 31.5 ng/mL) in males and 28-fold (pre-dosing: 3.3 ng/mL and after-dosing: 93.8 ng/mL) in females at 30 mg/kg, and 7-fold (pre-dosing: 1.9 ng/mL and after-dosing: 14.2 ng/mL) in males and 80-fold (pre-dosing: 0.6 ng/mL and after-dosing: 47.9 ng/mL) in females at 100 mg/kg. In all the animals with increased histamine levels, red extremities were observed as a clinical symptom. In addition, decreased activity, subdued behavior, lateral recumbency, tremors, emesis, salivating, labored breathing, hot extremities, and swollen were detected as clinical signs in some of the animals with histamine increment. The changes of histamine levels were transient and the plasma histamine levels returned to baseline within 2 hr after dosing. Any increases in histamine levels were not detected in all the animals with no clinical signs. In short, valemetostat tosylate induced increases in plasma histamine levels at 0.5 or 1 hr after dosing to animals at 30 or 100 mg/kg, but not to animals at 15 mg/kg.

The drug concentration in plasma of dogs given a single oral dose of valemetostat tosylate was measured. After single oral administration, systemic exposure to valemetostat increased with increasing dose in both male and female dogs (Table 2). Mean or median Tmax ranged between 0.5 and 1 hr after dosing in both females and males. No notable differences between males and females in terms of systemic exposure of valemetostat were observed following single oral administration. The Cmax in the 15 mg/kg group, which did not show histamine release, was ca. 2 to 2.5 μg/mL, and that in the 30 and 100 mg/kg group, which showed increased plasma histamine levels, were 5.4 to 5.8 μg/mL and 13 to 17 μg/mL, respectively.
Table 2. Toxicokinetic parameters of valemetostat in dogs.
| Sex |
Male |
| Dose (mg/kg) |
15 |
30 |
100 |
| No. of animals |
5 |
5 |
2 |
| AUC0–24h (μg·hr/mL) |
10 ± 4.2 |
30 ± 7.2 |
38 |
| Cmax (μg/mL) |
2.5 ± 0.97 |
5.8 ± 0.55 |
13 |
| Tmax (hr) |
1 |
0.5 |
0.5 |
|
|
|
|
| Sex |
Female |
| Dose (mg/kg) |
15 |
30 |
100 |
| No. of animals |
5 |
5 |
2 |
| AUC0–24h (μg·hr/mL) |
7.9 ± 1.1 |
29 ± 7.6 |
73 |
| Cmax (μg/mL) |
2.0 ± 0.48 |
5.4 ± 1.7 |
17 |
| Tmax (hr) |
1 |
1 |
0.5 |
AUC0–24h indicates area under the plasma concentration-time curve (AUC) from 0 to 24 hr after administration. Plasma samples were collected at the following timepoints: pre-dosing, 0.5, 1, 2, 4, 8, 12, and 24 hr after dosing. For the 15 and 30 mg/kg groups, values represent the mean ± S.D. For the 100 mg/kg group, values represent the mean value. All doses and concentrations are expressed in terms of valemetostat, a free form of valemetostat tosylate.
Toxicity study in rats
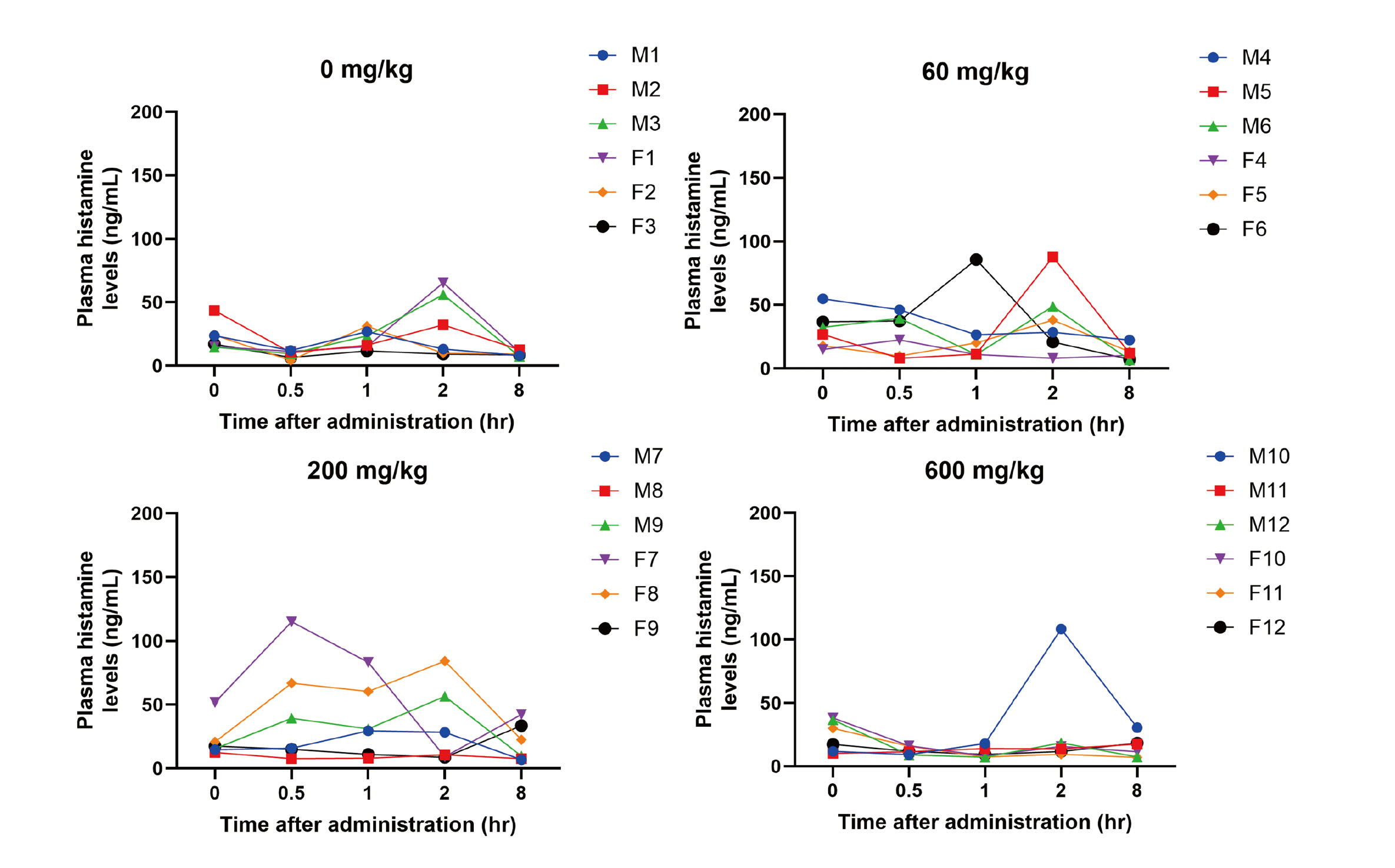
To confirm valemetostat tosylate induces histamine releases and associated clinical symptoms in rats, a study with a single oral dose of valemetostat tosylate using rats was conducted. There were no noteworthy clinical symptoms in all rats administered valemetostat at 60, 200, or 600 mg/kg including the animals for plasma histamine evaluation. Regarding plasma histamine levels, apparent valemetostat tosylate-induced increases were not detected in any rats (Fig. 3). The plasma histamine concentration in rats before dosing in all groups tended to be higher compared with that in dogs before dosing; 10 to 55 ng/mL in rats and 0.1 to 5.6 ng/mL in dogs. The minimal and sporadic variations in histamine plasma concentration compared to pre-dose values were observed in all groups including control. However, these variations were not accompanied by any clinical symptoms and considered related to the stress associated with the sampling procedure (Head et al., 1985), and therefore were not valemetostat tosylate-related. In summary, histamine release or associated clinical symptoms were not detected in rats treated with valemetostat tosylate.
To determine whether MRGPRX2 was associated with valemetostat-tosylate-induced histamine release, intracellular calcium mobilization of MRGPRX2-expressing cells against valemetostat tosylate treatment was evaluated. Because intracellular calcium mobilization in mast cells induced by MRGPRX2 ligands causes degranulation as aforementioned, intracellular calcium levels of MRGPRX2-expressing cells was measured as endpoint in the present study. HEK293 cells were transiently transfected with vectors including dog or human MRGPRX2 and evaluated for reactivity against valemetostat tosylate and positive control compound 48/80. The results showed that valemetostat tosylate concentration-dependently induced intracellular calcium mobilization on HEK293 cells expressing dog or human MRGPRX2 (Fig. 4). In particular, dog MRGPRX2 caused calcium mobilization from lower concentration compared with human MRGPRX2, indicating that the reactivity of dog MRGPRX2 was markedly greater than that of human MRGPRX2. The EC50 value of valemetostat tosylate in human MRGPRX2 was ca. 27-fold higher than that in dog MRGPRX2 (Table 3). The positive control compound 48/80 induced activation of both dog and human MRGPRX2. Untransfected cells, which were used as negative control, did not show any changes as those observed in MRGPRX2-expressing cells. In conclusion, both dog and human MRGPRX2 were activated by valemetostat tosylate, while dog MRGPRX2 showed higher sensitivity than human MRGPRX2.
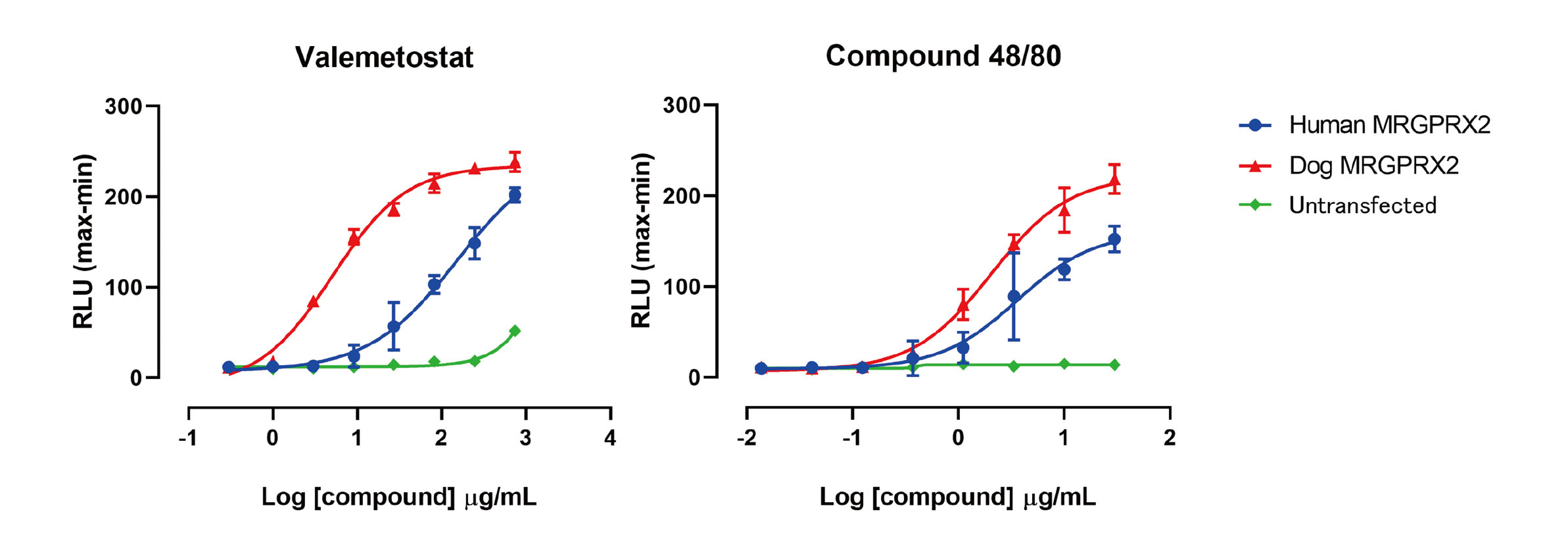
Table 3. Half-maximum effective concentration (EC
50) values of test articles on changes in intracellular calcium levels in dog or human MRGPRX2-expressing cells.
| Test article |
EC50 (µg/mL) ± S.D. |
| Dog MRGPRX2 |
Human MRGPRX2 |
Untransfected |
| Valemetostat |
4.5 ± 2.8 |
121.2 ± 30.4 |
NC |
| Compound 48/80 |
1.9 ± 0.5 |
3.5 ± 0.9 |
NC |
Data represent the mean ± S.D. of more than three independent experiments. All doses and concentrations are expressed in terms of valemetostat, a free form of valemetostat tosylate. Untransfected cells were used as negative control. NC: not calculated.
Clinical study
A Phase I clinical trial with valemetostat tosylate was conducted to determine the highest dose a patient can tolerate or recommended dose of valemetostat tosylate that can be given patients. Valemetostat tosylate at 500 mg QD was a maximum dose level in the trial at the time. The Cmax value of human subjects treated with valemetostat tosylate at 500 mg QD was ca. 4 μg/mL and no allergic-like symptoms associated with histamine release were observed (Table 4).
Table 4. Pharmacokinetic parameters of valemetostat after oral administration in a clinical study.
| Dose |
500 mg QD |
| No. of subjects |
3 |
| AUC0–24h (µg·hr/mL) |
43 ± 40 |
| Cmax (µg/mL) |
3.9 ± 3.6 |
| Tmax (hr) |
4 |
AUC0–24h indicates area under the plasma concentration-time curve (AUC) from 0 to 24 hr after dose administration over a dosing interval. For AUC0–24h and Cmax, values represent the arithmetic mean ± S.D. For Tmax, the median is reported. The results of Day 1 at cycle 1 in the clinical trial were extracted. All doses and concentrations are expressed in terms of valemetostat, a free form of valemetostat tosylat
DISCUSSION
Symptoms of anaphylaxis, which are associated with histamine release, are observed mainly in the skin, respiratory system, gastrointestinal (GI) tract, cardiovascular system, and central nervous system (CNS) (Ring et al., 2021). Single administration of valemetostat tosylate in dogs resulted in flush responses in the skin observed as red/hot extremities as well as flushing/swelling around the eyes, pinnas, and muzzle (Table 1). In addition, dogs showed salivation and labored breathing as respiratory symptoms, vomiting and loose/watery feces as GI tract symptoms, and subdued behavior, tremors, unsteady gait, and lack of coordination as CNS symptoms (Table 1), which were consistent with clinical signs associated with histamine release in anaphylaxis (Ring et al., 2021). Many of these symptoms were transient and disappeared within 4 hr after dosing (Fig. 1B). Moreover, plasma histamine release was observed in dogs within 0.5 to 1 hr after the administration of valemetostat tosylate (Fig. 2). Based on the above information, these clinical signs were related to histamine release. Some of the dogs with marked clinical signs showed no detectable histamine release in plasma (Table 1, Fig. 2). It is considered that the peak of histamine release may not have been captured at the timepoints set in the present study, since histamine release is a very rapid reaction with possible peaks at 5 to 10 min after administration in certain cases (Schwartz et al., 1989). In the present study, the maximum histamine increases associated with valemetostat tosylate in individual animals when compared to pre-dose levels were tended to be stronger in female dogs compared with male dogs as described above. In an IgE-dependent anaphylaxis model, female mice have reported to exhibit greater histamine levels compared with male mice, which is considered to be involved with the difference of mediator concentration, storage, and release between male and female (Mackey et al., 2020). Furthermore, in a retrospective study using data from patients treated with MRGPRX2-activating drugs, female sex was associated with increased odds of adverse reactions (Foer et al., 2023). These results suggest that valemetostat tosylate may have a potential to induce a stronger histamine release in females compared with males.
As a possible mechanism of the histamine release in valemetostat-tosylate-treated dogs, we speculated that it involves the IgE-independent pathway associated with MRGPRX2, which is a key receptor of drug-induced pseudo-allergic reactions expressed in mast cells, for the following reasons. First, single administration of valemetostat induced histamine release and associated clinical symptoms at a concentration of 30 mg/kg or more (Fig. 2). The observation suggested that the histamine release was dose-dependent response and did not require sensitization. Second, valemetostat tosylate caused marked histamine release in dogs, although this was not detected in rats (Fig. 3). As described above, dog is known as the species that is most sensitive to drug-induced pseudo-allergic reactions involving a variety of drugs (Ennis et al., 1985; Masini et al., 1985; Mori et al., 2000, 2001). Lastly, valemetostat is a zwitterionic compound carrying both positive and negative charges, and has two aromatic rings (Fig. 1A), consistent with the characteristics of MRGPRX2 ligands (McNeil, 2021; Roy et al., 2021). To verify this hypothesis, we conducted in vitro evaluation using MRGPRX2-expressing cells and found that valemetostat tosylate induced intracellular calcium mobilization of the cells (Fig. 4). This indicated that the valemetostat-tosylate-induced histamine release occurred via MRGPRX2 activation depending on a chemical-characteristic of valemetostat tosylate. These findings suggest that this in vitro MRGPRX2 assay system would be helpful to identify the mechanism of histamine releases observed in nonclinical studies.
Although the factors involved in species differences in MRGPRX2 activation are not fully understood, structural differences in MRGPRX2 between species might cause differences in sensitivity (Kumar et al., 2021). Orthologs of experimental animals have low sequence identity to human MRGPRX2, with whole-receptor amino acid sequence identities of 53% for mouse Mrgprb2, 56% for rat Mrgprb3, and 62% for dog MRGPRX2 (Hamamura-Yasuno et al., 2020). Further analysis is required to elucidate the mechanism underlying species differences in MRGPRX2 activation.
In the present study, valemetostat tosylate induced histamine release along with associated symptoms in dogs (Table 1), but not in humans or rats. Our results support the high sensitivity of dogs to pseudo-allergy. One major issue associated with MRGPRX2 research is the difficulty in extrapolating the nonclinical findings to clinical ones or vice versa (Kumar et al., 2021) because there are significant differences in the binding affinity and selectivity of ligands between human MRGPRX2 and MRGPRX2 ortholog of mouse, which is the primary species used in nonclinical MRGPRX2 research (Kumar et al., 2021; Mencarelli et al., 2020; Ogasawara et al., 2019; Zhang et al., 2017). The low sensitivity or specific ligand selectivity in mice is a significant limitation to mechanistic research or drug toxicity/efficacy studies. Therefore, it would be more useful to employ other species that are more sensitive than mice or have ligand selectivity similar to that of humans. We previously identified dog MRGPRX2 as a human MRGPRX2 ortholog (Hamamura-Yasuno et al., 2020). In addition, valemetostat-tosylate-induced pseudo-allergy was captured in an in vivo dog study and an in vitro assay using dog MRGPRX2-expressing cells, suggesting the usefulness of dogs as an animal model in MRGPRX2 research. However, dogs might be even more sensitive to certain drugs than humans, as also shown in the present study. Therefore, an in vitro assay platform that can evaluate species differences in MRGPRX2 reactivity is required for the extrapolation of nonclinical data to humans.
In the present study, we acquired comprehensive data from a nonclinical safety study, clinical study, and in vitro assessment using MRGPRX2-expressing cells. In dogs, valemetostat at 30 mg/kg or more caused histamine release and associated symptoms, with a Cmax of ca. 5 to 6 μg/mL in the 30 mg/kg group (Table 2). Both the timing of the histamine release and Tmax in the dogs ranged from 0.5 to 1 hr after dosing (Fig. 2), suggesting that histamine release occurred when drug levels exceeded a certain threshold. Considering that the Cmax in the 15 mg/kg group, which did not show histamine release, was ca. 2 to 2.5 μg/mL, thresholds for valemetostat-induced histamine release in dogs would be between ca. 2.5 and 5 μg/mL. Remarkably, the EC50 of valemetostat in dog MRGPRX2-expressing cells was 4.5 μg/mL (Table 3), consistent with the response in the dog in vivo study. Although valemetostat also caused activation of human MRGPRX2-expressing cells, the EC50 (121.2 μg/mL) was 27-fold higher than that for dog MRGPRX2, reflecting the marked species differences in valemetostat-tosylate-induced histamine release. Actually, Cmax in human subjects receiving 500 mg of valemetostat once daily was ca. 4 μg/mL (Table 4), and no histamine release or pseudo-allergic symptoms were observed. In human MRGPRX2-expressing cells, no activation was observed at 4 μg/mL (Fig. 4), which correlates with results in human studies. These findings strongly suggest that IC50 values of MRGPRX2-expressing cells correlate with plasma drug concentrations causing histamine release in vivo. Then, the assay system would be an effective tool to predict human risk of drug-induced pseudo-allergy. The approved dosage of valemetostat tosylate in patients with adult T-cell leukemia/lymphoma is 200 mg once daily, and 500 mg QD was maximum dose in the clinical trials conducted at that time. Because any histamine releases or associated clinical symptoms were not detected in humans even in the 500 mg QD group, the actual concentration causing histamine release in humans is still unclear. However, the Cmax value of patients treated with 500 mg was ca. 4 μg/mL, and the value was almost comparable to the Cmax value of dogs treated with 30 mg/kg (ca. 5 to 6 μg/mL), in which 8 out of 10 dogs showed apparent clinical symptoms associated with histamine release. Regarding this point, species difference between human and dog in sensitivity to valemetostat-induced histamine release in vivo would exist as shown in our in vitro assay system. Further investigation with other drugs is needed to validate the correlation between in vivo/in vitro response.
In conclusion, we demonstrated that valemetostat-tosylate-induced histamine release observed in dogs was associated with MRGPRX2 activation. In addition, the lower sensitivity of human MRGPRX2 to valemetostat tosylate in an in vitro assay system was consistent with the result of a phase I clinical trial showing that the risk for pseudo-allergic reaction in humans related to valemetostat tosylate was low. Therefore, this assay system using MRGPRX2-expressing cells would be an effective tool for mechanistic analysis of histamine release observed in nonclinical studies and its human risk assessment.
ACKNOWLEDGMENTS
The authors would like to thank Mr. Noriaki Iwata (Daiichi Sankyo Co., Ltd.) for help with the animal experiments and Ms. Chikako Maru (Daiichi Sankyo Co., Ltd.) for help with the histamine measurement in the preliminary studies.
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Dhyani, V., Gare, S., Gupta, R.K., Swain, S., Venkatesh, K.V. and Giri, L. (2020): GPCR mediated control of calcium dynamics: A systems perspective. Cell. Signal., 74, 109717.
- Ennis, M., Lorenz, W., Kapp, B., Lüben, L. and Schmal, A. (1985): Comparison of the histamine-releasing activity of cremophor E1 and some of its derivatives in two experimental models: the in vivo anaesthetized dog and in vitro rat peritoneal mast cells. Agents Actions, 16, 265-268.
- Ferry, X., Brehin, S., Kamel, R. and Landry, Y. (2002): G protein-dependent activation of mast cell by peptides and basic secretagogues. Peptides, 23, 1507-1515.
- Foer, D., Wien, M., Karlson, E.W., Song, W., Boyce, J.A. and Brennan, P.J. (2023): Patient characteristics associated with reactions to Mrgprx2-activating drugs in an electronic health record-linked biobank. J. Allergy Clin. Immunol. Pract., 11, 492499.e2.
- Furuhata, K., Hayakawa, H., Soumi, K., Arai, H., Watanabe, Y. and Narita, H. (1998): Histamine-releasing properties of T-3762, a novel fluoroquinolone antimicrobial agent in intravenous use. I. Effects of doses and infusion rate on blood pressure, heart rate and plasma histamine concentration. Biol. Pharm. Bull., 21, 456-460.
- Grimes, J., Desai, S., Charter, N.W., Lodge, J., Moita Santos, R., Isidro-Llobet, A., Mason, A.M., Wu, Z., Wolfe, L.A. 3rd, Anantharaman, L., Green, A., Bridges, A.M., Dalmas Wilk, D.A. and Brown, A.J. (2019): MrgX2 is a promiscuous receptor for basic peptides causing mast cell pseudo-allergic and anaphylactoid reactions. Pharmacol. Res. Perspect., 7, e00547.
- Hägermark, O., Hökfelt, T. and Pernow, B. (1978): Flare and itch induced by substance P in human skin. J. Invest. Dermatol., 71, 233-235.
- Hamamura-Yasuno, E., Iguchi, T., Kumagai, K., Tsuchiya, Y. and Mori, K. (2020): Identification of the dog orthologue of human MAS-related G protein coupled receptor X2 (MRGPRX2) essential for drug-induced pseudo-allergic reactions. Sci. Rep., 10, 16146.
- Head, R.J., Jarrott, B., Libys, J., Robinson, R.L., Stitzel, R.E. and Zavisca, F. (1985): Influence of blood sampling conditions upon histamine concentrations in rat plasma: a study of a complex relationship with plasma epinephrine. Neurochem. Int., 7, 473-479.
- Kinet, J.P. (1999): The high-affinity IgE receptor (Fc epsilon RI): from physiology to pathology. Annu. Rev. Immunol., 17, 931-972.
- Kojima, H., Hirohashi, M., Sakurai, T., Kasai, Y. and Akashi, A. (1984): General pharmacology of DL-8280. Chemotherapy, 32, 1148-1161.
- Kumar, M., Duraisamy, K. and Chow, B.K. (2021): Unlocking the Non-IgE-Mediated Pseudo-Allergic Reaction Puzzle with Mas-Related G-Protein Coupled Receptor Member X2 (MRGPRX2). Cells, 10.
- Lee, D. and Johnson, D.L. (1971): Effect of D-tubocurarine and anaesthesia upon cardiac output in normal and histamine-depleted dogs. Can. Anaesth. Soc. J., 18, 157-165.
- Liu, R., Che, D., Zhao, T., Pundir, P., Cao, J., Lv, Y., Wang, J., Ma, P., Fu, J., Wang, N., Wang, X., Zhang, T., Dong, X. and He, L. (2017): MRGPRX2 is essential for sinomenine hydrochloride induced anaphylactoid reactions. Biochem. Pharmacol., 146, 214-223.
- Mackey, E., Thelen, K.M., Bali, V., Fardisi, M., Trowbridge, M., Jordan, C.L. and Moeser, A.J. (2020): Perinatal androgens organize sex differences in mast cells and attenuate anaphylaxis severity into adulthood. Proc. Natl. Acad. Sci. USA, 117, 23751-23761.
- Margueron, R., Li, G., Sarma, K., Blais, A., Zavadil, J., Woodcock, C.L., Dynlacht, B.D. and Reinberg, D. (2008): Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell, 32, 503-518.
- Masini, E., Planchenault, J., Pezziardi, F., Gautier, P. and Gagnol, J.P. (1985): Histamine-releasing properties of Polysorbate 80 in vitro and in vivo: correlation with its hypotensive action in the dog. Agents Actions, 16, 470-477.
- McGee, E.U., Samuel, E., Boronea, B., Dillard, N., Milby, M.N. and Lewis, S.J. (2019): Quinolone Allergy. Pharmacy (Basel), 7.
- McNeil, B.D. (2021): Minireview: mas-related G protein-coupled receptor X2 activation by therapeutic drugs. Neurosci. Lett., 751, 135746.
- McNeil, B.D., Pundir, P., Meeker, S., Han, L., Undem, B.J., Kulka, M. and Dong, X. (2015): Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature, 519, 237-241.
- Mencarelli, A., Gunawan, M., Yong, K.S., Bist, P., Tan, W.W., Tan, S.Y., Liu, M., Huang, E.K., Fan, Y., Chan, J.K., Choi, H.W., Abraham, S.N. and Chen, Q. (2020): A humanized mouse model to study mast cells mediated cutaneous adverse drug reactions. J. Leukoc. Biol., 107, 797-807.
- Mori, K., Maru, C. and Takasuna, K. (2000): Characterization of histamine release induced by fluoroquinolone antibacterial agents in-vivo and in-vitro. J. Pharm. Pharmacol., 52, 577-584.
- Mori, K., Shibano, M., Satoh, H., Takasuna, K. and Furuhama, K. (2001): Differential response of mast cells separated from various organs and basophils of dogs to the fluoroquinolone antimicrobial levofloxacin. Arch. Toxicol., 75, 227-233.
- Ogasawara, H., Furuno, M., Edamura, K. and Noguchi, M. (2019): Novel MRGPRX2 antagonists inhibit IgE-independent activation of human umbilical cord blood-derived mast cells. J. Leukoc. Biol., 106, 1069-1077.
- Peavy, R.D. and Metcalfe, D.D. (2008): Understanding the mechanisms of anaphylaxis. Curr. Opin. Allergy Clin. Immunol., 8, 310-315.
- Ring, J., Beyer, K., Biedermann, T., Bircher, A., Fischer, M., Fuchs, T., Heller, A., Hoffmann, F., Huttegger, I., Jakob, T., Klimek, L., Kopp, M.V., Kugler, C., Lange, L., Pfaar, O., Rietschel, E., Rueff, F., Schnadt, S., Seifert, R., Stöcker, B., Treudler, R., Vogelberg, C., Werfel, T., Worm, M., Sitter, H. and Brockow, K. (2021): Guideline (S2k) on acute therapy and management of anaphylaxis: 2021 update: S2k-Guideline of the German Society for Allergology and Clinical Immunology (DGAKI), the Medical Association of German Allergologists (AeDA), the Society of Pediatric Allergology and Environmental Medicine (GPA), the German Academy of Allergology and Environmental Medicine (DAAU), the German Professional Association of Pediatricians (BVKJ), the Society for Neonatology and Pediatric Intensive Care (GNPI), the German Society of Dermatology (DDG), the Austrian Society for Allergology and Immunology (ÖGAI), the Swiss Society for Allergy and Immunology (SGAI), the German Society of Anaesthesiology and Intensive Care Medicine (DGAI), the German Society of Pharmacology (DGP), the German Respiratory Society (DGP), the patient organization German Allergy and Asthma Association (DAAB), the German Working Group of Anaphylaxis Training and Education (AGATE). Allergo J. Int., 30, 1-25.
- Robinson, E.P., Faggella, A.M., Henry, D.P. and Russell, W.L. (1988): Comparison of histamine release induced by morphine and oxymorphone administration in dogs. Am. J. Vet. Res., 49, 1699-1701.
- Roy, S., Chompunud Na Ayudhya, C., Thapaliya, M., Deepak, V. and Ali, H. (2021): Multifaceted MRGPRX2: new insight into the role of mast cells in health and disease. J. Allergy Clin. Immunol., 148, 293-308.
- Schwartz, L.B., Yunginger, J.W., Miller, J., Bokhari, R. and Dull, D. (1989): Time course of appearance and disappearance of human mast cell tryptase in the circulation after anaphylaxis. J. Clin. Invest., 83, 1551-1555.
- Subramanian, H., Gupta, K. and Ali, H. (2016): Roles of Mas-related G protein-coupled receptor X2 on mast cell-mediated host defense, pseudoallergic drug reactions, and chronic inflammatory diseases. J. Allergy Clin. Immunol., 138, 700-710.
- Takasuna, K., Kasai, Y., Usui, C., Takahashi, M., Hirohashi, M., Tamura, K. and Takayama, S. (1992): General pharmacology of the new quinolone antibacterial agent levofloxacin. Arzneimittelforschung, 43 (3A), 408-418.
- Tatemoto, K., Nozaki, Y., Tsuda, R., Konno, S., Tomura, K., Furuno, M., Ogasawara, H., Edamura, K., Takagi, H., Iwamura, H., Noguchi, M. and Naito, T. (2006): Immunoglobulin E-independent activation of mast cell is mediated by Mrg receptors. Biochem. Biophys. Res. Commun., 349, 1322-1328.
- Wang, H., Wang, H.S. and Liu, Z.P. (2011): Agents that induce pseudo-allergic reaction. Drug Discov. Ther., 5, 211-219.
- White, M.V. (1990): The role of histamine in allergic diseases. J. Allergy Clin. Immunol., 86, 599-605.
- Yamagishi, M., Hori, M., Fujikawa, D., Ohsugi, T., Honma, D., Adachi, N., Katano, H., Hishima, T., Kobayashi, S., Nakano, K., Nakashima, M., Iwanaga, M., Utsunomiya, A., Tanaka, Y., Okada, S., Tsukasaki, K., Tobinai, K., Araki, K., Watanabe, T. and Uchimaru, K. (2019): Targeting Excessive EZH1 and EZH2 Activities for Abnormal Histone Methylation and Transcription Network in Malignant Lymphomas. Cell Rep., 29, 2321-2337.e7.
- Zhang, B., Li, Q., Shi, C. and Zhang, X. (2018): Drug-Induced Pseudoallergy: A Review of the Causes and Mechanisms. Pharmacology, 101, 104-110.
- Zhang, T., Che, D., Liu, R., Han, S., Wang, N., Zhan, Y., Pundir, P., Cao, J., Lv, Y., Yang, L., Wang, J., Ding, M., Dong, X. and He, L. (2017): Typical antimicrobials induce mast cell degranulation and anaphylactoid reactions via MRGPRX2 and its murine homologue MRGPRB2. Eur. J. Immunol., 47, 1949-1958.