Abstract
Selenoproteins, the functional form of the essential trace element selenium, play a vital role in maintaining redox homeostasis in humans. Selenocysteine (Sec), which constitutes the active center of many selenoproteins, is introduced to the polypeptide chain by a unique biosynthetic insertion mechanism, making the expression of selenoproteins through biological means challenging. Compared to its analogue cysteine (Cys), Sec exhibits a lower redox potential, facilitating the oxidation of selenol groups to form diselenide bonds. These diselenide bonds are more resistant to reduction than disulfide bonds, providing an enhanced stability to peptides under reducing conditions. On the other hand, due to the larger atomic radius of selenium, the dissociation energy of the diselenide bond is lower than that of the disulfide bond, rendering them more prone to diselenide metathesis. This mini-review summarizes the use of Sec for the chemical synthesis of proteins, Sec-containing peptides and the selenoproteins. The diselenide metathesis reaction of Sec-containing peptides is also reviewed.
1. Introduction
-
1.1 Selenium and selenoproteins
Selenoproteins are the primary form of selenium, an essential trace element, that functions in our body. The selenium atom in selenoproteins exists as Sec, the 21st genetically encoded amino acid, which usually constitutes the active center of selenoproteins and has garnered significant attention in biochemistry and biology.
The Swedish chemist Jöns Jacob Berzelius first discovered selenium while re-examining a red sediment found in a sulfuric acid plant in 1818 [1]. Initially, selenium was considered as a toxic element, largely due to reports by Kurt Franke and others linking it to a series of livestock diseases [2]. However, in 1973, Rotruck made a pivotal discovery by identifying selenium as a crucial component of erythrocyte glutathione peroxidase (RBC-GPx) [3]. Subsequently, in 1976, Stadtman’s group demonstrated the presence of selenium, in the form of Sec, as the active center of clostridial glycine reductase selenoprotein in Clostridium sticklandii [4]. In 1978, Tappel’s group further identified Sec as an essential component of the catalytic site of glutathione peroxidase (GPx) in rat liver [5]. These findings demonstrated the biological significance of selenium. With the advancement of molecular biology, 25 selenoproteins have been identified in humans [6], in which Sec constitutes the active center, playing a critical role in maintaining redox homeostasis. These proteins are related to human diseases such as diabetes, cancer and immune diseases [7].
-
1.2 The comparison of selenocysteine and cysteine
Sec is an analogue of Cys, in which the sulfur atom is replaced by selenium (Fig. 1). Both selenium and sulfur belong to the chalcogen family and share similar chemical properties. However, compared to sulfur, selenium has a higher polarizability due to its larger atomic radius, which makes it more nucleophilic.
On the other hand, compared to Cys, Sec has a lower pKa (pKa of Sec selenol = 5.2, pKa of Cys thiol = 8.3), which means that the selenol group in Sec more readily liberates a proton to form selenolate, whereas the thiol group in Cys remains as the protonated thiol form at physiological pH [8]. As a result, under physiological conditions, Sec exhibits a higher nucleophilicity. In addition, the diselenide bond (E0 = −381 mV) in Sec has a lower reduction potential than the disulfide bond (E0 = −180 mV) in Cys, meaning Sec is more easily oxidized to form diselenide dimers, which are less prone to be reduced compared to disulfides [9].
Given these chemical properties, Sec has broad applications in biochemistry. Its higher nucleophilicity can enhance reaction rates and improve enzyme catalytic activity. The lower redox potential of the diselenide bond realizes the preferential formation of the diselenide bond to the disulfide bond, which makes it useful in studying the oxidative protein folding pathway of the disulfide-rich peptide by substiuting a part of the disulfide to diselenide bond. The difference in the pKa between selenol and thiol also provides a detection method for Sec. For example, as shown in Fig. 2, BESThio (3'-(2,4-dinitrobenzenesulfonyl)-2',7'-dimethylfluorescein) is a fluorescent probe for selenols, which only reacts with Sec at pH 5.8, while keeping thiols intact [10].
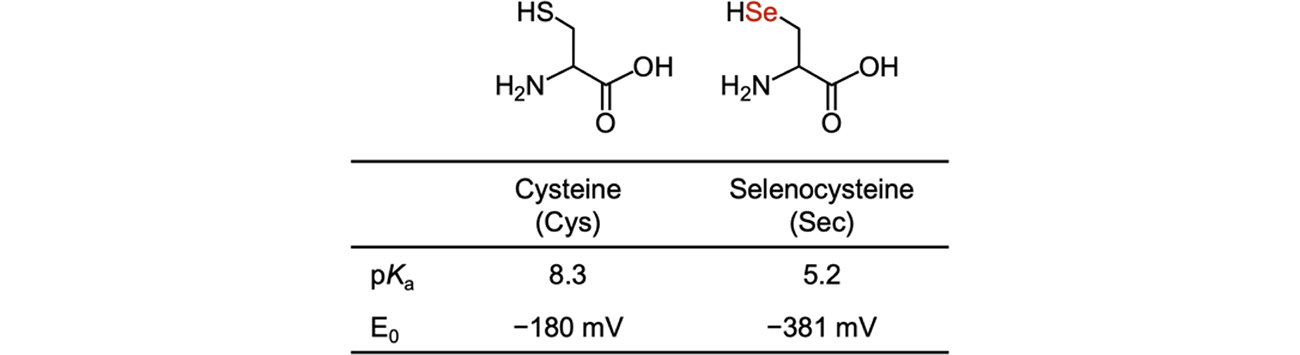
Fig. 1.
Structure, pKa, and redox potential of Cys and Sec.

Fig. 2.
Detection of Sec based on the different pKa of Sec and Cys.
-
1.3 Biosynthesis of selenoproteins
Sec is a genetically-encoded amino acid. As the stop codon, UGA is used for its incorporation in the growing polypeptide chain, a noncanonical insertion mechanism has been developed to direct the selenoprotein synthesis. The Sec insertion sequence (SECIS) element guides the translation of the stop codon UGA into Sec [11]. In prokaryotes, the SECIS element is located downstream of the UGA codon encoding Sec, whereas in eukaryotes, the SECIS element resides in the 3′ untranslated region (3′UTR) of the mRNA.
As shown in Fig. 3, the first step in Sec biosynthesis involves the aminoacylation of tRNASec with serine (Ser), catalyzed by seryl-tRNA synthetase (SerRS), to form Ser-tRNASec. In prokaryotes, Ser in Ser-tRNASec is converted to Sec by selenocysteine synthase (SelA). The structure of SelA contains pyridoxal 5'-phosphate (PLP). PLP forms a Schiff base with the amino group of Ser-tRNA, leading to the 2,3-elimination of H2O, resulting in the formation of an aminoacrylyl-tRNA intermediate. This intermediate then reacts with a selenium donor (selenophosphate), yielding Sec, which is subsequently released from SelA [12]. In eukaryotes, after the formation of Ser-tRNASec, O-phosphoseryl-tRNASec kinase (PSTK) phosphorylates Ser-tRNASec to generate O-phosphoseryl-tRNASec (Sep-tRNASec). Subsequently, Sec-tRNASec is formed from Sep-tRNASec through selenocysteinyl-tRNA synthase (SepSecS) [13].
In prokaryotes, the insertion of Sec requires the Sec-specific elongation factor SelB, which recognizes the SECIS element and facilitates the incorporation of Sec into the polypeptide chain [14]. The insertion of Sec in eukaryotes involves the SECIS binding protein 2 (SBP2) and Sec-specific elongation factor (eEFSec) to recognize the SECIS element and to recruit Sec-tRNASec to the ribosome [15]. However, other components involved in this process, as well as their specific roles, remain poorly understood.
Since the biosynthesis of the selenoprotein is regulated by both cis-acting elements and trans-acting factors, the recombinant expression of selenoproteins is highly complex and challenging.

Fig. 3.
Biosynthesis of Sec in prokaryotes and in eukaryotes.
-
1.4 Glutathione Peroxidases (GPxs)
Glutathione peroxidases (GPxs) are a well-characterized family of antioxidant enzymes in the human body that play a critical role in maintaining the cellular redox balance by reducing reactive oxygen species (ROS), by-products of cellular oxidative respiration. GPxs reduce hydrogen peroxide (H2O2) to water and organic hydroperoxides (ROOH) to alcohol (ROH), thereby preventing oxidative damage of biomolecules like DNA, proteins and lipids. In humans, there are eight types of GPx, five of which (GPx-1, GPx-2, GPx-3, GPx-4, and GPx-6) contain Sec at their active centers, while the other three (GPx-5, GPx-7, GPx-8) use Cys as their active site residue [16]. The active center of GPx consists of four amino acids: Gln, Asn, Trp, and either Sec or Cys [17]. Studies have shown that mutating Sec to Cys in murine GPx results in a 1000-fold decrease in enzymatic activity [18].
GPx-1 is one of the most abundantly expressed selenoproteins in humans. It presents in all cells and is known as cytosolic GPx or classic GPx [16, 19]. As shown in Fig. 4, the catalytic mechanism of GPx-1 is as follows: upon interaction with peroxides, the selenol group in the active center of GPx-1 is oxidized to the selenenic acid (GPx-SeOH) intermediate, while the peroxide is reduced. GPx-SeOH then reacts with one reduced glutathione (GSH) molecule to form the selenosulfide intermediate (GPx-SeSG). A second molecule of GSH subsequently reduces GPx-SeSG back to the original selenol group (GPx-SeH) restoring its activity, and generates one molecule of oxidized glutathione (GSSG) at the same time [20]. GPx-1 is closely linked to human health and is associated with several cancers, including breast, lung, and prostate cancers, as well as diabetes and cardiovascular diseases [19].
In conclusion, although selenium is the only trace element whose incorporation as Sec is genetically regulated and is closely linked to human health, the functions of all the selenoproteins have not yet been fully elucidated. However, the biosynthesis of selenoproteins is complicated due to the unique mechanism of Sec incorporation regulated by both the cis- and trans-acting factors.
In this mini-review, we focus on the chemical synthesis of selenopeptides (Sec-peptides) and selenoproteins by solid-phase peptide synthesis (SPPS), Sec-mediated native chemical ligation (NCL), and the recently discovered but still enigmatic diselenide metathesis found in diselenide bond-containing peptides.

Fig. 4.
Catalytic cycle of GPx.
2. Chemical synthesis of Sec-containing peptides
-
2.1 Solid-phase peptide synthesis (SPPS)
Solid-phase peptide synthesis, which is routinely used for the synthesis of peptides, was invented by Merrifield in 1963 [21]. The significant contribution of this method to this field also earned him the Nobel Prize in Chemistry in 1984. The main concept of the method is to couple the N-protected amino acid to a solid support, followed by deprotection of the amino group and subsequent amide bond formation with the next N-protected amino acid, allowing stepwise peptide elongation. Compared to solution-phase synthesis, SPPS is simpler to operate, because the synthesis can be simply realized by the addition of an activated amino acid and removal of the deprotection reagent. However, a major limitation is that the polypeptides exceeding 50 amino acids are usually obtained with a significantly lower purity and yield by this method.
In SPPS, the N-terminal protection of amino acids is typically achieved using either the 9-fluorenylmethoxycarbonyl (Fmoc) or tert-butoxycarbonyl (Boc) group, which are removed under basic or acidic conditions, respectively. Due to the high reactivity, protection of the -SeH group of Sec is required during the chain elongation. The most common and commercially-available protecting group is the 4-methoxyphenylmethyl (PMB) group. PMB deprotection is achieved under various conditions, such as HF, Lewis acids, or oxidative methods like iodine or dimethyl sulfoxide (DMSO) in TFA [22]. Inspired by the work of Tung about sulfur-based linkages on resin [23], Hondal developed a milder deprotection method using 2,2′-dithiobis(5-nitropyridine) (DTNP), an electrophilic disulfide, in TFA [24]. The method was further improved into the two-step procedure combining the ascorbolysis [25].
Another recently developed commercially-available Fmoc building block has the xanthenyl (Xan) protecting group [26], which can be removed using the TFA cocktail (TFA/TIS/H2O). Flemer successfully used Fmoc-Sec(Xan)-OH to synthesize the Sec-substituted analogue of the Stromelysin 1 matrix metalloproteinase inhibitor (MMP3-I) and the Sec-containing glutaredoxin analogue fragment, Lys16-Grx(10–17) designed by Moroder [27], observing minor by-products containing such as dehydroalanine (dHA) and selenenic acid derivative [26].
-
2.2 Sec-substituted peptide analogue
Due to the chemical similarity between selenium and sulfur, as well as the lower redox potential of Sec compared to Cys, the diselenide bond is more stable than the disulfide bond in the presence of reducing agents. The approach of replacing Cys with Sec in peptides was first used by Moroder to study the oxidative folding of the reduced Sec-analogue of human endothelin-1 (ET-1) which contains two disulfide bridges [28]. By replacing Cys3 and Cys11 with Sec, Moroder and colleagues synthesized [Sec3, Sec11, Nle7]-ET-1. Air oxidation achieved the target peptide with the diselenide bond between Sec3 and Sec11 and the disulfide bond between Cys1 and Cys15. No mixed Se-S bond isomer by the thiol/diselenide exchange reaction was observed. The results also confirmed that the Sec-analogue retained the same structure and receptor-binding properties as the native ET-1. The results showed that by replacing one of the Cys pairs in the Cys-rich peptides by Sec residues, the oxidative folding can be steered so that the diselenide bond is preferentially formed. This method has been successfully used for analysis of the folding pathway and accelerating folding reaction [29, 30]. Similar Sec-analogues have also been investigated, such as the bee venom apamin, a peptide highly resistant to isomer formation due to its stable disulfide bonds [31, 32], conotoxins, neurotoxic peptides from marine cone snail venom [33-37], interleukin-8 [38], and the Ecballium elaterium trypsin inhibitor II (EETI-II) [39].
Notably, Prof. Iwaoka, Inaba and our group also replaced the A-chain Cys7 and B-chain Cys7 residues in bovine pancreatic insulin (BPIns) to synthesize the [C7UA, C7UB]-BPIns, and through biological assays, we found that this analogue exhibited an increased resistance to degradation by the insulin-degrading enzyme (IDE) and had an extended half-life [40]. This enhanced activity was attributed to the higher propensity of oligomer formation of the diselenide-substituted BPIns compared to BPIns, due to the stronger interaction between the diselenide bonds than the disulfide bonds [41]. Similarly, Metanis’ group designed a Sec-analogue of human insulin by replacing Cys6 and Cys11 in the A-chain with Sec while retaining the two interchain disulfide bonds, demonstrating that [C6UA, C11UA]-insulin also retained biological activity with an enhanced thermodynamic stability and increased resistance to reduction and enzymatic digestion [42]. The similar internal diselenide bond replacement applied to the insulin prescription drug, glargine, also enhanced its thermodynamic stability [43].
In 2022, our laboratory replaced all six Cys in the human epidermal growth factor (EGF) with Sec, successfully synthesizing Se-EGF with a comparable biological activity, structure and enhanced stability under reducing conditions in the presence of reduced glutathione [44]. Despite the presence of three diselenide bonds, the folding of Se-EGF was efficient and obtained isomorphous products.
These studies highlight the possibility and potential of Sec-substitution to achieve directional folding and increased stability of Cys-rich peptides in therapeutic use.
3. Synthesis of Sec-containing proteins by the ligation method
-
3.1 Native chemical ligation (NCL) and thioester method
The SPPS is a well-established and widely used method for chemical peptide synthesis. However, it is not suitable for the synthesis of peptides containing more than 50 amino acid residues due to several challenges that arise as the peptide chain lengthens. These challenges include peptide aggregation and accumulation of missing amino acid by-products, all of which lead to a reduced purity and yield. To overcome the limitation of SPPS, various chemical ligation methods have been developed to synthesize longer polypeptides by assembling shorter peptide segments.
The most commonly used method is the native chemical ligation (NCL, Fig. 5. (A)) method, developed by Kent and colleagues in 1994 [45]. NCL is an efficient and chemoselective ligation method performed in an aqueous environment at neutral pH without protection of the side-chain functional groups. NCL enables the ligation of the peptide segments with a C-terminal thioester and an N-terminal Cys. The thiol group of the N-terminal Cys attacks the carbonyl carbon of the C-terminal thioester of the other segment, forming a thioester bond, which then undergoes an intramolecular S-N acyl shift to form the native amide bond. They successfully synthesized human interleukin-8 (IL-8), a 72-residue protein by NCL (Fig. 6). The basics of this reaction was already developed by Wieland and coworkers in 1953 [46] by achieving ligation between the valine thioester and Cys.
NCL represents a milestone in protein chemical synthesis. However, its application is limited by the low abundance of Cys in proteins (1.9%). To expand the applicability of this method, Dawson introduced a metal-based desulfurization reaction in 2001 [47].
This approach realizes the ligation at the abundant (7.8%) alanine (Ala) site by ligation at the Cys site, followed by the Raney nickel or Pd/Al2O3 mediated desulfurization reaction to convert Cys to Ala. In 2007, Danishefsky, inspired by Hoffmann’s report on the desulfurization reaction between mercaptans and trialkylphosphine in 1956 [48], introduced a mild, radical-mediated desulfurization reaction [49]. The desulfurization conditions involved the use of a water-soluble radical initiator, 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044), a thiol to provide a hydrogen atom, and TCEP for the phosphoranyl radical. These mild conditions are compatible with the synthesis of various peptides and glycopeptides. Subsequent studies combined desulfurization with the design of thiolated amino acids to expand the NCL-desulfurization chemistry’s scope [50-57]. However, the lack of selectivity in desulfurization has limited its use in biologically-expressed proteins.
Before the development of NCL, our laboratory developed the thioester method shown in Fig. 5. (D) for chemical protein synthesis in 1991 [58] inspired by Blake’s work about the Ag+-activated condensation of a peptide bearing a thiocarboxy group at its C-terminus with another peptide segment [59]. In the thioester method, the C-terminal thioester group is activated by a silver ion, converting the segment into the corresponding active ester, which is then attacked by the amino group of the N-terminal segment, forming an amide bond. To realize the chemoselective reaction, the side chain amino and thiol groups requires protection. Instead, in principle, there is no limitation in the selection of the ligation site. We also discovered that aryl thioesters can undergo aminolysis without Ag+ activation [60]. By exploiting the difference in the reactivity of the aryl and alkyl thioesters, it is possible to assemble three peptide fragments in a one-pot reaction [61-65]. Additionally, our laboratory utilized selenoester chemistry to develop a one-pot four-segment coupling strategy by the thioester method, which was successfully applied to the synthesis of superoxide dismutase (SOD) [66].
In 2020, we successfully synthesized Se-ferredoxin (Se-Fd), in which the four Cys residues coordinating the [2Fe-2S] cluster were replaced with Sec using a one-pot three-segment coupling by the thioester method as shown in Fig. 7 [67]. The 97-residue ferredoxin (Fd) was divided into three peptide segments. These included the side chain amino and selenol group protected Fd (1–33) with a C-terminal aryl thioester, Fd (34–73) with a C-terminal alkyl thioester, and Fd (74–97). To prepare the thioesters, we introduced N-alkylcysteine at the C-terminus of the peptide chains. This allowed for an intramolecular N-to-S acyl shift, followed by an intermolecular thioester exchange reaction to obtain the Fd (1–33) and Fd (34–73) thioesters. This approach, developed by our laboratory, has proven effective for generating peptide thioester by the Fmoc method [68]. The first ligation step, achieved using a Ag+-free thioester method, involved coupling of the Fd (1–33) aryl thioester with the Fd (34–73) alkyl thioester to produce Fd (1–73) alkyl thioester. Subsequently, Fd (74–97) was added together with Ag+ to promote the second ligation via a Ag+-activated thioester method, producing the full-length Fd (1–97) in one-pot. After deprotection and [2Fe-2S] cluster incorporation, Se-Fd was successfully synthesized. Circular dichroism (CD) and biochemical assays indicated that Se-Fd retained a structure similar to the wild-type Fd, though its catalytic activity was slightly decreased. This decrease in activity is likely due to the lower redox potential of selenium, which may impact the efficiency of electron transfer in the Fe cluster.

Fig. 5.
(A) Native chemical ligation (NCL) and desulfurization of Cys. (B) Sec-mediated NCL and deselenization of Sec. (C) Diselenide-selenoester ligation (DSL) and deselenization of Sec. (D) Thioester method and deprotection of amino and thiol protecting groups (PG).

Fig. 6.
Synthesis of [A33]IL-8 by the NCL.

Fig. 7.
One-pot ligation of Se-ferredoxin (Se-Fd) by thioester method.
-
3.2 Sec-mediated NCL
To overcome the inherent limitations of the NCL-desulfurization strategy, a method using the N-terminal Sec instead of Cys for site-specific ligation was developed (Fig. 5. (B)). The mechanism of the Sec-mediated NCL is similar to traditional NCL. The selenol group attacks the acyl carbon, forming a selenoester, followed by a Se-N acyl shift to form a native peptide bond. This approach was independently proposed by three different groups in 2001 [69-71].
Due to the higher nucleophilicity of selenol compared to thiol, Sec-mediated NCL proceeds faster than the conventional NCL. Because of Sec's lower pKa, selenol is more readily deprotonated, allowing the reaction to occur at a lower pH, which helps to avoid potential side reactions such as thioester hydrolysis. Another advantage of Sec is its selective deselenization even in the presence of Cys. In 2005, Metanis and Payne reported methods to convert Sec to Ala or Ser via deselenization [72, 73]. Compared to desulfurization, the deselenization conditions are milder and do not require the use of radical initiators. In 2010, Dawson demonstrated the deselenization of a 10-residue peptide containing a Se-S bond using excess TCEP and DTT, achieving Sec-to-Ala conversion without affecting Cys [74]. They also applied this method to 38 residues of glutaredoxin 3 (Grx3(1-38)), where Cys11 and Cys14 were mutated to Sec, and Ala38 was mutated to Cys. Sec11 and Sec14 were successfully converted to Ala with only minimal desulfurization observed at Cys38 by their method [74].
Due to the lower redox potential of Sec, the segment with the N-terminal Sec often forms the diselenide dimer or Se-S bond with other compounds [75]. Therefore, the use of a reducing agent is essential in Sec-NCL. Thiol-based reducing agents are preferable in Sec-NCL since commonly used reducing agents like TCEP can cause deselenization [76].
In recent years, there have been several examples of proteins synthesized using Sec-NCL. The bovine pancreatic trypsin inhibitor (C38U-BPTI), whose Cys38 was substituted with Sec, was synthesized, followed by oxidative folding of two disulfide bonds and one Se-S bond (Fig. 8). Cyclization of a peptide containing an N-terminal Sec and a C-terminal thioester can also be achieved by Sec-NCL [69, 77]. Additionally, Sec-NCL has been applied in semi-synthetic strategies, such as the synthesis of C110U-RNase A and C112U-azurin [70, 78].

Fig. 8.
Synthesis of C38U-BPTI by the Sec-mediated NCL.
-
3.3 Diselenide-selenoester ligation (DSL) and reductive DSL (rDSL)
Due to the low redox potential of Sec, selenopeptides tend to exist as diselenide dimers. Therefore, as already described, a reducing agent is needed to reduce diselenide or Se-S bonds into reactive selenolates during the Sec-NCL process. The choice of the reducing agent is critical as it directly affects the rate and the yield of Sec-NCL. Overly strong reducing agents may induce deselenization, while weak reducing agents may not fully reduce the diselenide or Se-S bonds, leading to a decreased ligation efficiency.
To address this issue, Payne’s group introduced a method called diselenide-selenoester ligation (DSL), shown in Fig. 5 (C), in 2015, which does not require reducing agents [79]. This reaction occurs between a C-terminal aryl selenoester and an N-terminal diselenide dimer. Compared to NCL, DSL proceeds extremely fast, completing in just a few minutes, and it resolves the challenge of the ligation at sterically-hindered sites. The proposed mechanism involves the release of phenylselenoate from the C-terminal aryl selenoester through hydrolysis, aminolysis or acyl substitution. This phenylselenoate then attacks the diselenide bond, generating selenolate, which forms a native peptide bond through a mechanism similar to NCL.
In 2020, the same group developed a refined approach, known as reductive DSL (rDSL), by employing TCEP as a reducing agent to generate selenolate and diphenyl diselenide (DPDS) as a radical scavenger to avoid deselenization while accelerating the formation of selenolate [80]. This method realized the ligation at a concentration as low as 50 nM. Additionally, after extracting DPDS by ether, they succeeded in a viable and efficient photodeselenization reaction in the presence of TCEP and GSH under light irradiation (254 nm) for 30 seconds.
The possibility of ligation at a low concentration by rDSL allows it to be used for synthesizing hydrophobic proteins without the need for hydrophilic tags, and subsequent one-pot photodeselenization further improved both the yield and efficiency of the synthesis. For example, the synthesis of various therapeutic tesamorelins, the palmitoylated variants of the transmembrane lipoprotein phospholemman (FXYD1, Fig.9), adiponectin (19-107), haemathrin 1, haemathrin 2, and peptide-oligonucleotide conjugates [80-82].

Fig. 9.
The synthesis of palmitoylated FXYD1 by one-pot rDSL-alkylation and rDSL-photodeselenization.
-
3.4 Chemical synthesis of selenoproteins
The functions of many selenoproteins remain unclear, due to the complexity involved in their biosynthetic pathway as already described. However, the application of the aforementioned chemical synthetic methods has enabled the synthesis of several human selenoproteins.
In 2016, Metanis and colleagues successfully synthesized human selenoprotein M (SelM) and W (SelW) [83]. Mature human SelM (24-145) consists of 122 amino acids and is widely expressed in mammalian tissues, particularly in the brain, implying that it is closely associated with brain function [84]. SelM contains a CXXU redox motif which is similar to the CXXC active site in thioredoxins (Trx), conferring antioxidant properties [83, 85]. The protein was divided into four segments with ligation sites at Gly47-Sec48, Gly77-Ala78, and Asn106-Ala107. Ala78 and Ala107 were mutated to Sec to facilitate Sec-NCL, followed by deselenization to convert them to Ala. The introduction of the C-terminal thioester employed the Dawson linker [86, 87]. The four peptide segments were: SelM (24-47) with a C-terminal thioester, SelM (48-77) and SelM (78-106) with N-terminal selenazolidine (Sez) and C-terminal thioesters, and SelM (107-145). Sez48,78 were used to protect the N-terminal Sec and prevent deselenization. Methionine (Met) was replaced with the norleucine (Nle) to prevent oxidation. The first Sec-NCL ligated SelM (78-106) and SelM (107-145), and subsequent MeONH2 treatment converted Sez78 to Sec, forming SelM (78-145) with unprotected Sec78 and Sec107. The second Sec-NCL ligated SelM (48-77) to SelM (78-145) followed by deselenization of Sec78 and Sec107 under anaerobic conditions using DTT and TCEP to form Ala78 and Ala107, then followed by the conversion of Sez48 to Sec. During deselenization, partial Sez deprotection led to the observation of minor three deselenized products. The final Sec-NCL yielded full-length SelM with a Se-S bond. The folding of the final protein was confirmed by CD comparison with structurally similar Trx proteins.
Selenoprotein W (SelW), which contains 86 residues, is expressed in muscle, brain, and spleen tissues [88]. It also features a CXXU motif and is associated with intracellular redox processes, bone remodeling, and diseases such as non-alcoholic fatty liver disease (NAFLD), although its functions are not fully understood [89, 90]. SelW was divided into two segments with a ligation site between Ile36 and Sec37. Met was similarly replaced with Nle. The C-terminal thioester of SelW (2-36) was prepared using the Dawson linker. The two segments SelW (2-36) and SelW (37-87) were successfully ligated via Sec-NCL, forming a SelW with a Se-S bond and a free Cys. The folding of the final protein was verified by CD analysis [83].
In 2017, Payne et al. synthesized a homodimeric Selenoprotein K (SelK) via one-pot DSL-deselenization [91]. SelK consists of 93 amino acids. SelK was divided into two segments: SelK (2-60) with a selenoester and SelK (61-94) with an N-terminal (β-Se)-Asp. Met was replaced with Nle. DSL gave the full-length SelK (2-94) forming a diselenide bond between Sec61 and Sec92 with part of the product retaining the extra SelK (2-60) linked to the side chain selenol group. Hydrazinolytic removal of the extra SelK (2-60), followed by selective deselenization of (β-Se)-Asp using TCEP gave the desired SelK dimer.
In 2022, He and colleagues achieved the semi-synthesis of selenoprotein F (SelF) (Fig. 10), [92]. Mature SelF consists of 134 amino acids, including one Sec and seven Cys, and its function remains unclear. SelF was synthesized in three segments, ligated by two NCL at Gly41-Cys42 and Gln74-Ala75. To stabilize the peptide hydrazide precursor, Gln74 was mutated to Ala, and Ala75 was mutated to Cys to afford NCL. The three segments were SelF (1-41) with a C-terminal thioester, Cys- and Sec-protected SelF (42-74) with a C-terminal thioester, and SelF (75-134). SelF (75-134) was biologically expressed due to aspartimide formation. SelF (42-134) was synthesized by the first NCL followed by desulfurization under mild condition by reducing the amount of radical initiator VA044 and reaction time. The deprotection of acetamidomethyl (Acm) and also PMB group was achieved by Pd2+ followed by DTT treatment. The reduction of diselenide dimer was then performed with TCEP and ascorbate. Finally, the second NCL yielded the full-length SelF (1-134). One Se-S bond and three disulfide bonds were formed by oxidative folding. Other selenoproteins, such as selenoprotein H and S, were also synthesized via semi-synthetic methods [93, 94].

Fig. 10.
Semi-synthesis of selenoprotein F.
4. Diselenide metathesis of Sec-containing peptides
Although the isomorphic substitution of disulfide bonds with diselenide bonds is commonly used to enhance protein stability under reducing conditions, the larger atomic radius of selenium leads to a lower bond dissociation energy for diselenide bonds compared to disulfide bonds (Se-Se bond: 172 kJ/mol, S-S bond: 240 kJ/mol), making diselenide bonds more prone to be broken under external influences [95]. Diselenide bonds function as dynamic covalent bonds (DCBs) and can undergo metathesis reactions under defined conditions, such as light irradiation or heat. This property has garnered significant attention in recent years for application in self-healing materials, fluorescent probes, organic synthesis, selective surface modification, and drug delivery systems [96-101].
In 2019, Stefanowicz discovered that the diselenide bond metathesis also occurs in short peptides containing Sec (Fig. 11). They dissolved two selenopeptide dimers in methanol and observed the formation of heterodimers after 30 minutes of light irradiation (400-700 nm wavelength) [102].

Fig. 11.
Diselenide metathesis of model peptide in methanol by light irradiation.
Following this, our group found that the metathesis reaction can even occur in neutral aqueous buffer solutions without reducing agents in the dark and exhibits a pH dependency. This suggests that diselenide bonds may be inherently unstable [103]. To find the conditions that can keep the diselenide bonds stable and to further investigate the reaction mechanism for broader applications, we studied the inter- and intramolecular metathesis reactions of three bioactive peptides (both the wild type and Sec-substituted analogues) under physiological conditions in the dark; i.e., oxytocin, α-conotoxin ImI, and apamin. Oxytocin has one intramolecular disulfide bond, while conotoxin and apamin have two intramolecular disulfide bonds. We found that Se-oxytocin was stable under a low-concentration (10 μM) in the dark. However, the intermolecular diselenide metathesis happened at higher concentrations, leading to polymerization. Se-α-conotoxin ImI showed similar results to Se-oxytocin. Additionally, it formed isomers via intramolecular diselenide metathesis, regardless of the concentration (Fig. 12). Interestingly, the ratio of the diselenide isomer to Se-conotoxin matched that observed during oxidative folding of Se-conotoxin. In contrast, Se-apamin exhibited no metathesis which might be due to the inherent stability of its tertiary structure, as the wild-type apamin explicitly forms the product with native disulfide bond pairing in the oxidative folding reaction [31]. This indicates that selenopeptides with stable tertiary structures are resistant to diselenide metathesis reactions. We observed a similar behavior when synthesizing the Sec-substituted epidermal growth factor (Se-EGF) [44].

The mechanism behind diselenide metathesis remains debated. In 2014, Xu proposed that the light-driven diselenide metathesis is facilitated by light-induced selenium radicals, a hypothesis supported by the inhibition of the reaction upon adding the radical scavenger (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) [104]. However, the diselenide isomerization observed by us occurs under dark conditions, which excludes the proposed reaction mechanism by Xu in our case. We found that the production of diselenide isomers was accelerated under reducing conditions in the presence of GSH, suggesting that the selenol group generated by reducing agents can promote isomer formation. Therefore, our isomerization reaction is not selenol mediated. In 2022, Zhu suggested a mechanism of metathesis in the absence of light driven by the ease of polarization of the diselenide bond, which is accelerated in polar solvents [105]. This mechanism agrees with our group’s observations.
5. Conclusion and outlook
Selenopeptides and selenoproteins remain an important topic of research in molecular biology, biochemistry, and medicinal chemistry. However, the unique biosynthetic insertion mechanism of Sec into proteins makes the biological expression of selenoproteins challenging. To address this, chemical synthesis methods for selenopeptide and selenoprotein have been developed, such as NCL, Sec-NCL, DSL, and rDSL. Moreover, deselenization reactions allow for selective conversion of Sec to Ala or Ser in the presence of the unprotected Cys, broadening the scope of NCL. Due to the higher nucleophilicity of the selenol group compared to the thiol group, these methods offer faster reaction rates and extend the reaction concentration down to 50 nM, making the synthesis of highly-hydrophobic proteins feasible. Selenoproteins can also be synthesized by the thioester method.
Sec also has a significant potential for various applications. With a lower redox potential than thiol, selenol can form diselenide bonds more rapidly in the presence of Cys and is more resistant to reduction than disulfide bonds, which can be exploited in protein folding studies. Additionally, given the similar chemical properties of Se and S, Sec-substituted peptide analogues generally retain a very similar biological activity with the native Cys peptides while being more stable under reducing conditions.
On the other hand, diselenide bonds, as dynamic covalent bonds, can undergo diselenide metathesis under certain conditions, making them suitable for applications in self-healing materials, drug delivery systems, and fluorescent probes. This diselenide metathesis reaction has also been observed in proteins, and although the exact mechanism remains unclear, it is believed to hold a significant potential for further applications.
Acknowledgments
This article has no funding.
Statements about COI
The authors declare no conflict of interest associated with this manuscript.
Author contributions
Y. He wrote the draft of this paper. H. Hojo revised this manuscript.
References
- [1]
Weeks
ME
.
The discovery of the elements. VI. Tellurium and selenium.
J. Chem. Educ.
1932;
9(3):
474-485.
- [2]
Franke
KW
.
A new toxicant occurring naturally in certain samples of plant foodstuffs I: Results obtained in preliminary feeding trials.
J. Nutr.
1934;
8:
597−608.
- [3]
Rotruck
JT
,
Pope
AL
,
Ganther
HE
,
Swanson
AB
,
Hafeman
DG
,
Hoekstra
W
.
Selenium: biochemical role as a component of glutathione peroxidase.
Science.
1973;
179(4073):
588-590.
- [4]
Cone
JE
,
Del Rio
RM
,
Davis
JN
,
Stadtman
TC
.
Chemical characterization of the selenoprotein component of clostridial glycine reductase: identification of selenocysteine as the organoselenium moiety.
Proc. Natl. Sci. U. S. A.
1976;
73(8):
2659-2663.
- [5]
Forstrom
JW
,
Zakowski
JJ
,
Tappel
AL
.
Identification of the catalytic site of rat liver glutathione peroxidase as selenocysteine.
Biochemistry
1978;
17(13):
2639-2644.
- [6]
Kryukov
GV
,
Castellano
S
,
Novoselov
SV
,
Lobanov
AV
,
Zehtab
O
,
Guigó
R
,
Gladyshev
VN
.
Characterization of mammalian selenoproteomes.
Science.
2003;
300(5624):
1439-1443.
- [7]
Rayman
MP
.
Selenium and human health.
Lancet.
2012;
379(9822):
1256–1268.
- [8]
Poole
LB
.
The basics of thiols and cysteines in redox biology and chemistry.
Free Radic. Biol. Med.
2015;
80:
148–157.
- [9]
Besse
D
,
Siedler
F
,
Diercks
T
,
Kessler
H
,
Moroder
L
.
The redox potential of selenocystine in unconstrained cyclic peptides.
Angew. Chem. Int. Ed.
1997;
36(8):
883–885.
- [10]
Maeda
H
,
Katayama
K
,
Matsuno
H
,
Uno
T
.
3′-(2, 4-Dinitrobenzenesulfonyl)-2′, 7′-dimethylfluorescein as a fluorescent probe for selenols.
Angew. Chem. Int. Ed.
2006;
45(11):
1810-1813.
- [11]
Walczak
R
,
Westhof
E
,
Carbon
P
,
Krol
A
.
A novel RNA structural motif in the selenocysteine insertion element of eukaryotic selenoprotein mRNAs.
RNA.
1996;
2(4):
367–379.
- [12]
Forchhammer
K
,
Böck
A
.
Selenocysteine synthase from Escherichia coli. Analysis of the reaction sequence.
J. Biol. Chem.
1991;
266(10):
6324–6328.
- [13]
Xu
XM
,
Carlson
BA
,
Mix
H
,
Zhang
Y
,
Saira
K
,
Glass
RS
,
Berry
MJ
,
Gladyshev
VN
,
Hatfield
DL
.
Biosynthesis of selenocysteine on its tRNA in eukaryotes.
PLoS Biol.
2006;
5(1):
e4. DOI:
10.1371/journal.pbio.0050004
- [14]
Baron
C
,
Heider
J
,
Böck
A
.
Interaction of translation factor SELB with the formate dehydrogenase H selenopolypeptide mRNA.
Proc. Natl. Acad. Sci.
1993;
90(9):
4181-4185.
- [15]
Cain
A
,
Krahn
N
.
Overcoming challenges with biochemical studies of selenocysteine and selenoproteins.
Int. J. Mol. Sci.
2024;
25(18):
10101-10118.
- [16]
Stolwijk
JM
,
Garje
R
,
Sieren
JC
,
Buettner
GR
,
Zakharia
Y
.
Understanding the redox biology of selenium in the search of targeted cancer therapies.
Antioxidants (Basel).
2020;
9(5):
420-444.
- [17]
Tosatto
SCE
,
Bosello
V
,
Fogolari
F
,
Mauri
P
,
Roveri
A
,
Toppo
S
,
Flohé
L
,
Ursini
F
,
Maiorino
M
.
The catalytic site of glutathione peroxidases.
Antioxid. Redox Signal.
2008;
10(9):
1515-1526.
- [18]
Rocher
C
,
Lalanne
JL
,
Chaudiere
J
.
Purification and properties of a recombinant sulfur analog of murine selenium-glutathione peroxidase.
Eur. J. Biochem.
1992;
205(3):
955-960.
- [19]
Lubos
E
,
Loscalzo
J
,
Handy
DE
.
Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities.
Antioxid. Redox Signal.
2011;
15(7):
1957-1997.
- [20]
Maia
LB
,
Maiti
BK
,
Moura
I
,
Moura
JJG
.
Selenium—more than just a fortuitous sulfur substitute in redox biology.
Molecules.
2023;
29(1):
120-155.
- [21]
Merrifield
RB
.
Solid phase peptide synthesis. I. The synthesis of a tetrapeptide.
J. Am. Chem. Soc.
1963;
85(14):
2149-2154.
- [22]
Flemer
SJr
.
Selenol protecting groups in organic chemistry: Special emphasis on selenocysteine Se-protection in solid phase peptide synthesis.
Molecules.
2011;
16(4):
3232-3251.
- [23]
Galande
AK
,
Weissleder
R
,
Tung
CH
.
An effective method of on-resin disulfide bond formation in peptides.
J. Comb. Chem.
2005;
7(2):
174–177.
- [24]
Harris
KM
,
Flemer
SJr
,
Hondal
RJ
.
Studies on deprotection of cysteine and selenocysteine side-chain protecting groups.
J. Pep. Sci.
2007;
13(2):
81-93.
- [25]
Marie
SEJ
,
Ruggles
EL
,
Hondal
RJ
.
Removal of the 5-nitro-2-pyridine-sulfenyl protecting group from selenocysteine and cysteine by ascorbolysis.
J. Pep. Sci.
2016;
22(9):
571-576.
- [26]
Flemer
SJr
.
Fmoc-Sec(Xan)-OH: synthesis and utility of Fmoc selenocysteine SPPS derivatives with acid-labile sidechain protection.
J. Pep. Sci.
2015;
21(1):
53-59.
- [27]
Besse
D
,
Moroder
L
.
Synthesis of selenocysteine peptides and their oxidation to diselenide-bridged compounds.
J. Pep. Sci.
1997;
3(6):
442-453.
- [28]
Pegoraro
S
,
Fiori
S
,
Rudolph
BS
,
Watanabe
TX
,
Moroder
L
.
Isomorphous replacement of cystine with selenocystine in endothelin: Oxidative refolding, biological and conformational properties of [Sec3, Sec11,Nle7]-endothelin-1.
J. Mol. Biol.
1998;
284(3):
779-792.
- [29]
Metanis
N
,
Hilvert
D
.
Strategic use of non-native diselenide bridges to steer oxidative protein folding.
Angew. Chem. Int. Ed.
2012;
51(23):
5585-5588.
- [30]
Metanis
N
,
Hilvert
D
.
Harnessing selenocysteine reactivity for oxidative protein folding.
Chem. Sci.
2015;
6(1):
322-325.
- [31]
Pegoraro
S
,
Fiori
S
,
Cramer
J
,
Rudolph
BS
,
Moroder
L
.
The disulfide-coupled folding pathway of apamin as derived from diselenide-quenched analogs and intermediates.
Protein Sci.
1999;
8(8):
1605–1613.
- [32]
Fiori
S
,
Pegoraro
S
,
Rudolph
BS
,
Cramer
J
,
Moroder
L
.
Synthesis and conformational analysis of apamin analogues with natural and non-natural cystine/selenocystine connectivities.
Biopolymers.
2000;
53(7):
550-564.
- [33]
Armishaw
CJ
,
Daly
NL
,
Nevin
ST
,
Adams
DJ
,
Craik
DJ
,
Alewood
PF
.
α-selenoconotoxins, a new class of potent α7 neuronal nicotinic receptor antagonists.
J. Biol. Chem.
2006;
281(20):
14136-14143.
- [34]
Walewska
A
,
Zhang
MM
,
Skalicky
JJ
,
Yoshikami
D
,
Olivera
BM
,
Bulaj
G
.
Integrated oxidative folding of cysteine/selenocysteine containing peptides: improving chemical synthesis of conotoxins.
Angew. Chem. Int. Ed.
2009;
48(12):
2221–2224.
- [35]
Muttenthaler
M
,
Nevin
ST
,
Grishin
AA
,
Ngo
ST
,
Choy
PT
,
Daly
NL
,
Hu
SH
,
Armishaw
CJ
,
Wang
CIA
,
Lewis
RJ
,
Martin
JL
,
Noakes
PG
,
Craik
DJ
,
Adams
DJ
,
Alewood
PF
.
Solving the α-conotoxin folding problem: efficient selenium-directed on-resin generation of more potent and stable nicotinic acetylcholine receptor antagonists.
J. Am. Chem. Soc.
2010;
132(10):
3514–3522.
- [36]
Gowd
KH
,
Yarotskyy
V
,
Elmslie
KS
,
Skalicky
JJ
,
Olivera
BM
,
Bulaj
G
.
Site-specific effects of diselenide bridges on the oxidative folding of a cystine knot peptide, ω-selenoconotoxin GVIA.
Biochemistry.
2010;
49(12):
2741–2752.
- [37]
de Araujo
AD
,
Callaghan
B
,
Nevin
ST
,
Daly
NL
,
Craik
DJ
,
Moretta
M
,
Hopping
G
,
Christie
MJ
,
Adams
DJ
,
Alewood
PF
.
Total synthesis of the analgesic conotoxin MrVIB through selenocysteine-assisted folding.
Angew. Chem. Int. Ed.
2011;
50(29):
6527–6529.
- [38]
Rajarathnam
K
,
Sykes
BD
,
Dewald
B
,
Baggiolini
M
,
Clark
LI
.
Disulfide bridges in interleukin-8 probed using non-natural disulfide analogues: Dissociation of roles in structure from function.
Biochemistry.
1999;
38(24):
7653-7658.
- [39]
Walewska
A
,
Jaśkiewicz
A
,
Bulaj
G
,
Rolka
K
.
Selenopeptide analogsof EETI-II retain potent trypsin inhibitory activities.
Chem. Biol. Drug Des.
2011;
77(1):
93–97.
- [40]
Arai
K
,
Takei
T
,
Okumura
M
,
Watanabe
S
,
Amagai
Y
,
Asahina
Y
,
Moroder
L
,
Hojo
H
,
Inaba
K
,
Iwaoka
M
.
Preparation of selenoinsulin as a long-lasting insulin analogue.
Angew. Chem. Int. Ed.
2017;
56(20):
5522-5526.
- [41]
Arai
K
,
Okumura
M
,
Lee
YH
,
Katayama
H
,
Mizutani
K
,
Lin
Y
,
Park
SY
,
Sawada
K
,
Toyoda
M
,
Hojo
H
,
Inaba
K
,
Iwaoka
M
.
Diselenide-bond replacement of the external disulfide bond of insulin increases its oligomerization leading to sustained activity.
Commun. Chem.
2023;
6(1):
258-267.
- [42]
Weil
KO
,
Rege
N
,
Lansky
S
,
Shalev
DE
,
Shoham
G
,
Weiss
MA
,
Metanis
N
.
Substitution of an internal disulfide bridge with a diselenide enhances both foldability and stability of human insulin.
Chem. Eur. J.
2019;
25(36):
8513-8521.
- [43]
Weil
KO
,
Dhayalan
B
,
Chen
YS
,
Weiss
MA
,
Metanis
N
.
Se-Glargine: Chemical synthesis of a basal insulin analogue stabilized by an internal diselenide bridge.
ChemBioChem.
2024;
25(5):
e202300818. DOI:
10.1002/cbic.202300818.
- [44]
Takei
T
,
Tanaka
H
,
Okumura
N
,
Takao
T
,
Moroder
L
,
Hojo
H
.
Chemical synthesis of per-selenocysteine human epidermal growth factor.
J. Pep. Sci.
2023;
29(5):
e3464. DOI:
10.1002/psc.3464
- [45]
Dawson
P
,
Muir
TW
,
Clark
LI
,
Kent
SB
.
Synthesis of proteins by native chemical ligation.
Science.
1994;
266(5186):
776-779.
- [46]
Wieland
T
,
Bokelmann
E
,
Bauer
L
,
Lang
HU
,
Lau
H
.
Über peptidsynthesen. 8. Mitteilung bildung von S-haltigen peptiden durch intramolekulare wanderung von Aminoacylresten.
Justus Liebigs Ann. Chem.
1953;
583(1):
129-149.
- [47]
Yan
LZ
,
Dawson
PE
.
Synthesis of peptides and proteins without cysteine residues by native chemical ligation combined with desulfurization.
J. Am. Chem. Soc.
2001;
123(4):
526–533.
- [48]
Hoffmann
FW
,
Ess
RJ
,
Simmons
TC
,
Hanzel
RS
.
The desulfurization of mercaptans with trialkyl phosphites.
J. Am. Chem. Soc.
1956;
78(24):
6414.
- [49]
Wan
Q
,
Danishefsky
SJ
.
Free-radical-based, specific desulfurization of cysteine: a powerful advance in the synthesis of polypeptides and glycopolypeptides.
Angew. Chem. Int. Ed.
2007;
46(48):
9248-9252.
- [50]
Crich
D
,
Banerjee
A
.
Native chemical ligation at phenylalanine.
J. Am. Chem. Soc.
2007;
129(33):
10064–10065.
- [51]
Haase
C
,
Rohde
H
,
Seitz
O
.
Native chemical ligation at valine.
Angew. Chem. Int. Ed.
2008;
47(36):
6807–6810.
- [52]
Harpaz
Z
,
Siman
P
,
Kumar
KSA
,
Brik
A
.
Protein synthesis assisted by native chemical ligation at leucine.
ChemBioChem.
2010;
11(9):
1232–1235.
- [53]
Chen
J
,
Wang
P
,
Zhu
JL
,
Wan
Q
,
Danishefsky
SJ
.
A program for ligation at threonine sites: application to the controlled total synthesis of glycopeptides.
Tetrahedron.
2010;
66(13):
2277–2283.
- [54]
Ding
H
,
Shigenaga
A
,
Sato
K
,
Morishita
K
,
Otaka,
A
.
Dual kinetically controlled native chemical ligation using a combination of sulfanylproline and sulfanylethylanilide peptide.
Org. Lett.
2011;
13(20):
5588-5591.
- [55]
Malins
LR
,
Cergol
KM
,
Payne
RJ
.
Peptide ligation-desulfurization chemistry at arginine.
ChemBioChem.
2013;
14(5):
559–563.
- [56]
Thompson
RE
,
Chan
B
,
Radom
L
,
Jolliffe
KA
,
Payne
RJ
.
Chemoselective peptide ligation-desulfurization at aspartate.
Angew. Chem. Int. Ed.
2013;
52(37):
9723-9727.
- [57]
Pasunooti
KK
,
Yang
R
,
Banerjee
B
,
Yap
T
,
Liu
CF
.
5-Methylisoxazole-3-carboxamide-directed palladium-catalyzed γ-C (sp3)–H acetoxylation and application to the synthesis of γ-mercapto amino acids for native chemical ligation.
Org. Lett.
2016;
18(11):
2696-2699.
- [58]
Hojo
H
,
Aimoto
S
.
Polypeptide synthesis using the S-alkyl thioester of a partially protected peptide segment. Synthesis of the DNA-binding domain of c-Myb protein (142-193)-NH2.
Bull. Chem. Soc. Jpn.
1991;
64(1):
111-117.
- [59]
Blake
J
.
Peptide segment coupling in aqueous medium: silver ion activation of the thiolcarboxyl group.
Int. J. Pep. Protein Res.
1981;
17(2):
273-274.
- [60]
Hojo
H
,
Murasawa
Y
,
Katayama
H
,
Ohira
T
,
Nakahara
Y
,
Nakahara
Y
.
Application of a novel thioesterification reaction to the synthesis of chemokine CCL27 by the modified thioester method.
Org. Biomol. Chem.
2008;
6(10):
1808-1813.
- [61]
Asahina
Y
,
Kamitori
S
,
Takao
T
,
Nishi
N
,
Hojo
H
.
Chemoenzymatic synthesis of the immunoglobulin domain of Tim-3 carrying a complex-type N-glycan by using a one-pot ligation.
Angew. Chem. Int. Ed.
2013;
52(37):
9733-9737.
- [62]
Asahina
Y
,
Kanda
M
,
Suzuki
A
,
Katayama
H
,
Nakahara
Y
,
Hojo
H
.
Fast preparation of an N-acetylglucosaminylated peptide segment for the chemoenzymatic synthesis of a glycoprotein.
Org. Biomol. Chem.
2013;
11(41):
7199-7207.
- [63]
Asahina
Y
,
Komiya
S
,
Ohagi
A
,
Fujimoto
R
,
Tamagaki
H
,
Nakagawa
K
,
Sato
T
,
Akira
S
,
Takao
T
,
Ishii
A
,
Nakahara
Y
,
Hojo
H
.
Chemical synthesis of O-glycosylated human interleukin-2 by the reverse polarity protection strategy.
Angew. Chem. Int. Ed.
2015;
54(28):
8226-8230.
- [64]
Hojo
H
,
Takei
T
,
Asahina
Y
,
Okumura
N
,
Takao
T
,
So
M
,
Suetake
I
,
Sato
T
,
Kawamoto
A
,
Hirabayashi
Y
.
Total synthesis and structural characterization of caveolin-1.
Angew. Chem. Int. Ed.
2021;
60(25):
13900-13905.
- [65]
Hojo
H
,
Suetake
I
.
Chemical synthesis of palmitoylated histone H4.
ARKIVOC.
2021;
2021(4):
186-197.
- [66]
Takei
T
,
Andoh
T
,
Takao
T
,
Hojo
H
.
One-pot four-segment ligation using seleno- and thioesters: synthesis of superoxide dismutase.
Angew. Chem. Int. Ed.
2017;
56(49):
15708-15711.
- [67]
Takei
T
,
Ando
T
,
Takao
T
,
Ohnishi
Y
,
Kurisu
G
,
Iwaoka
M
,
Hojo
H
.
Chemical synthesis of ferredoxin with 4 selenocysteine residues using a segment condensation method.
Chem. Commun.
2020;
56(91):
14239-14242.
- [68]
Hojo
H
,
Onuma
Y
,
Akimoto
Y
,
Nakahara
Y
,
Nakahara
Y
.
N-Alkyl cysteine-assisted thioesterification of peptides.
Tetrahedron Lett.
2007;
48(1):
25-28.
- [69]
Quaderer
R
,
Sewing
A
,
Hilvert
D
.
Selenocysteine-mediated native chemical ligation.
Helv. Chim. Acta.
2001;
84(5):
1197-1206.
- [70]
Hondal
RJ
,
Nilsson
BL
,
Raines
RT
.
Selenocysteine in native chemical ligation and expressed protein ligation.
J. Am. Chem. Soc.
2001;
123(21):
5140-5141.
- [71]
Gieselman
MD
,
Xie
L
,
Van Der Donk
WA
.
Synthesis of a selenocysteine-containing peptide by native chemical ligation.
Org. Lett.
2001;
3(9):
1331-1334.
- [72]
Dery
S
,
Reddy
PS
,
Dery
L
,
Mousa
R
,
Dardashti
RN
,
Metanis
N
.
Insights into the deselenization of selenocysteine into alanine and serine.
Chem. Sci.
2015;
6(11):
6207-6212.
- [73]
Malins
LR
,
Mitchell
NJ
,
McGowan
S
,
Payne
RJ
.
Oxidative deselenization of selenocysteine: applications for programmed ligation at serine.
Angew. Chem. Int. Ed.
2015;
54(43):
12716-12721.
- [74]
Metanis
N
,
Keinan
E
,
Dawson
PE
.
Traceless ligation of cysteine peptides using selective deselenization.
Angew. Chem. Int. Ed.
2010;
49(39):
7049-7053.
- [75]
Malins
LR
,
Mitchell
NJ
,
Payne
RJ
.
Peptide ligation chemistry at selenol amino acids.
J. Pep. Sci.
2014;
20(2):
64-77.
- [76]
Gieselman
MD
,
Xie
L
,
van Der Donk
WA
.
Synthesis of a selenocysteine-containing peptide by native chemical ligation.
Org. Lett.
2001;
3(9):
1331-1334.
- [77]
Shimodaira
S
,
Takei
T
,
Hojo
H
,
Iwaoka
M
.
Synthesis of selenocysteine-containing cyclic peptides via tandem N-to-S acyl migration and intramolecular selenocysteine-mediated native chemical ligation.
Chem. Commun.
2018;
54(83):
11737-11740.
- [78]
Berry
SM
,
Gieselman
MD
,
Nilges
MJ
,
van der Donk
WA
,
Lu
Y
.
An engineered azurin variant containing a selenocysteine copper ligand.
J. Am. Chem. Soc.
2002;
124(10):
2084–2085.
- [79]
Mitchell
NJ
,
Malins
LR
,
Liu
X
,
Thompson
RE
,
Chan
B
,
Radom
L
,
Payne
RJ
.
Rapid additive-free selenocystine-selenoester peptide ligation.
J. Am. Chem. Soc.
2015;
137(44):
14011−14014.
- [80]
Chisholm
TS
,
Kulkarni
SS
,
Hossain
KR
,
Cornelius
F
,
Clarke
RJ
,
Payne
RJ
.
Peptide ligation at high dilution via reductive diselenide-selenoester ligation.
J. Am. Chem. Soc.
2020;
142(2):
1090-1100.
- [81]
Kulkarni
SS
,
Watson
EE
,
Premdjee
B
,
Conde
FKW
,
Payne
RJ
.
Diselenide–selenoester ligation for chemical protein synthesis.
Nat. Protoc.
2019;
14(7):
2229-2257.
- [82]
Liczner
C
,
Hanna
CC
,
Payne
RJ
,
Wilds
CJ
.
Generation of oligonucleotide conjugates via one-pot diselenide-selenoester ligation–deselenization/alkylation.
Chem. Sci.
2022;
13(2):
410-420.
- [83]
Dery
L
,
Reddy
PS
,
Dery
S
,
Mousa
R
,
Ktorza
O
,
Talhami
A
,
Metanis
N
.
Accessing human selenoproteins through chemical protein synthesis.
Chem. Sci.
2017;
8(3):
1922-1926.
- [84]
Korotkov
KV
,
Novoselov
SV
,
Hatfield
DL
,
Gladyshev
VN
.
Mammalian selenoprotein in which selenocysteine (Sec) incorporation is supported by a new form of Sec insertion sequence element.
Mol. Cell Biol.
2002;
22(5):
1402-1411.
- [85]
Nunes
LGA
,
Cain
A
,
Comyns
C
,
Hoffmann
PR
,
Krahn
N
.
Deciphering the role of selenoprotein M.
Antioxidants (Basel).
2023;
12(11):
1906-1922.
- [86]
Blanco
CJB
,
Dawson
PE
.
An efficient Fmoc-SPPS approach for the generation of thioester peptide precursors for use in native chemical ligation.
Angew. Chem. Int. Ed.
2008;
47(36):
6851-6855.
- [87]
Blanco
CJB
,
Nardone
B
,
Albericio
F
,
Dawson
PE
.
Chemical protein synthesis using a second-generation N-acylurea linker for the preparation of peptide-thioester precursors.
J. Am. Chem. Soc.
2015;
137(22):
7197-7209.
- [88]
Whanger
PD
.
Selenoprotein expression and function—Selenoprotein W.
Biochim. Biophys. Acta.
2009;
1790(11):
1448-1452.
- [89]
Kim
H
,
Lee
K
,
Kim
JM
,
Kim
MY
,
Kim
JR
,
Lee
HW
,
Chung
YW
,
Shin
HI
,
Kim
T
,
Park
ES
,
Rho
J
,
Lee
SH
,
Kim
N
,
Lee
SY
,
Choi
Y
,
Jeong
D
.
Selenoprotein W ensures physiological bone remodeling by preventing hyperactivity of osteoclasts.
Nat. Commun.
2021;
12(1):
2258-2270.
- [90]
Miao
Z
,
Wang
W
,
Miao
Z
,
Cao
Q
,
Xu
S
.
Role of Selenoprotein W in participating in the progression of non-alcoholic fatty liver disease.
Redox Biol.
2024;
71:
103114-103128.
- [91]
Mitchell
NJ
,
Sayers
J
,
Kulkarni
SS
,
Clayton
D
,
Goldys
AM
,
Rozada
JR
,
Pereira
PJB
,
Chan
B
,
Radom
L
,
Payne
RJ
.
Accelerated protein synthesis via one-pot ligation-deselenization chemistry.
Chem.
2017;
2(5):
703-715.
- [92]
Liao
P
,
Liu
H
,
He
C
.
Chemical synthesis of human selenoprotein F and elucidation of its thiol-disulfide oxidoreductase activity.
Chem. Sci.
2022;
13(21):
6322-6327.
- [93]
Dardashti
RN
,
Laps
S
,
Gichtin
JS
,
Metanis
N
.
The semisynthesis of nucleolar human selenoprotein H.
Chem. Sci.
2023;
14(44):
12723-12729.
- [94]
Cheng
R
,
Liu
J
,
Daithankar
V
,
Rozovsky
S
.
Applying selenocysteine-mediated expressed protein ligation to prepare the membrane enzyme selenoprotein S.
Methods Enzymol.
2022;
662:
159-185.
- [95]
Kildahl
NK
.
Bond Energy Data Summarized.
J. Chem. Educ.
1995;
72(5):
423–424.
- [96]
Kang
X
,
Yuan
Y
,
Xu
H
,
Chen
Y
.
Thermal-and Light-driven Metathesis Reactions Between Different Diselenides.
Chem. Res. Chinese U.
2022;
38(2):
516-521.
- [97]
Anugrah
DSB
,
Ramesh
K
,
Kim
M
,
Hyun
K
,
Lim
KT
.
Near-infrared light-responsive alginate hydrogels based on diselenide-containing cross-linkage for on demand degradation and drug release.
Carbohydr. Polym.
2019;
223(1):
115070-115079.
- [98]
Han
X
,
Yu
F
,
Song
X
,
Chen
L
.
Quantification of cysteine hydropersulfide with a ratiometric near-infrared fluorescent probe based on selenium–sulfur exchange reaction.
Chem. Sci.
2016;
7(8):
5098-5107.
- [99]
Nomoto
A
,
Higuchi
Y
,
Kobiki
Y
,
Ogawa
A
.
Synthesis of selenium compounds by free radical addition based on visible-light-activated Se-Se bond cleavage.
Mini Rev. Med. Chem.
2013;
13(6):
814-823.
- [100]
Xia
J
,
Zhao
P
,
Zheng
K
,
Lu
C
,
Yin
S
,
Xu
H
.
Surface modification based on diselenide dynamic chemistry: towards liquid motion and surface bioconjugation.
Angew. Chem. Int. Ed.
2019;
58(2):
542-546.
- [101]
Cheng
X
,
Li
H
,
Sun
X
,
Xu
T
,
Guo
Z
,
Du
X
,
Li
S
,
Li
X
,
Xing
X
,
Qiu
D
.
Visible-light-induced diselenide-crosslinked polymeric micelles for ROS-triggered drug delivery.
Molecules.
2024;
29(16):
3970-3986.
- [102]
Waliczek
M
,
Pehlivan
Ö
,
Stefanowicz
P
.
Light-driven diselenide metathesis in peptides.
ChemistryOpen.
2019;
8(9):
1199-1203.
- [103]
He
Y
,
Takei
T
,
Moroder
L
,
Hojo
H
.
Unexpected diselenide metathesis in selenocysteine-substituted biologically active peptides.
Org. Biomol. Chem.
2024;
22(30):
6108-6114.
- [104]
Ji
S
,
Cao
W
,
Yu
Y
,
Xu
H
.
Dynamic diselenide bonds: exchange reaction induced by visible light without catalysis.
Angew. Chem. Int. Ed.
2014;
53(26):
6781-6785.
- [105]
Chen
S
,
Lu
W
,
Zhang
J
,
He
H
,
Cang
Y
,
Pan
X
,
Zhu
J
.
Thermally driven diselenide metathesis: polarization process vs radical process.
ACS Macro Lett.
2022;
11(2):
264-269.
