Abstract
Neuroendocrine tumors (NETs) are neoplasms that originate from cells of the endocrine and nervous systems, and are commonly found in the gastrointestinal and respiratory tracts. Primary intracranial NETs are extremely rare and have been the focus of only a few studies thus far. Herein, we report the case of a primary intracranial NET of the skull base complicated with tension pneumocephalus after radiotherapy. An 84-year-old woman visited a local hospital for a head injury, and CT revealed a skull base tumor. MRI showed that the tumor was located mainly on the clivus and extended into the paranasal sinuses and nasal cavity. We biopsied the tumor via the nasal cavity, and the pathological diagnosis was NET, WHO grade 2. We subsequently administered focal intensity-modulated radiation therapy, but the patient developed tension pneumocephalus 1 year after radiotherapy. We therefore performed endoscopic transnasal cerebrospinal fluid leak closure with a nasoseptal flap. The postoperative course was successful, and the patient returned home but died of an unknown cause 2 years after discharge. The optimal postoperative management of primary intracranial NETs remains controversial. Tension pneumocephalus related to radiotherapy is a rare complication. Assessing skull bone erosion before radiotherapy and performing regular radiological follow-up examinations are essential to prevent this rare complication.
Introduction
Neuroendocrine tumors (NETs) are neoplasms that originate from cells of the endocrine and nervous systems. They can arise in many different organs but are most commonly found in the gastrointestinal and respiratory tracts. In the central nervous system (CNS), metastatic brain lesions frequently arise from primary poorly differentiated lung NETs, but the incidence of patients with NETs who develop brain metastases is <5%.1) There are only a few reports of NETs arising primarily from the CNS, and the incidence of NETs of intracranial origin is extremely low.2) Here we report an extremely rare case of an elderly woman in whom there was an incidental finding of a primary intracranial NET of the skull base extending to the paranasal sinuses and nasal cavity. She received focal radiotherapy but developed tension pneumocephalus 1 year later because of tumor shrinkage.
Clinical Presentation
An 84-year-old woman visited a local hospital for a head injury, and CT demonstrated a clival tumor. Initially, she underwent only radiological follow-up, but tumor biopsy was performed 2 years later due to tumor extension into the intranasal space through the sphenoid sinuses. The pathological diagnosis of the intranasal tumor tissue was olfactory neuroblastoma. The local hospital referred the patient to our institute for detailed examination. Radiological studies showed that the tumor was located mainly on the clivus, but it extended laterally to the cavernous sinuses where it involved the bilateral internal carotid arteries, superiorly to the sella turcica adjacent to the pituitary gland, and anteriorly to the posterior nasal aperture inside the sphenoid sinuses. MRI depicted the tumor as an isointense area on T1-weighted sequences (Fig. 1A), an iso- to high-intensity area on T2-weighted sequences (Fig. 1B), and a homogeneously enhanced area with gadolinium contrast (Fig. 1C–E). CT revealed that the tumor extended from the clivus to the ethmoidal sinuses and posterior nasal aperture, and that it eroded the sphenoid bone (Fig. 2). Whole-body F-18 fluorodeoxyglucose (18F-FDG) positron emission tomography (PET) detected no other neoplastic lesions. Endocrinological examinations showed the following serum basal levels of anterior pituitary hormones; growth hormone (GH) 1.11 ng/ml, adrenocorticotropic hormone (ACTH) 17.30 pg/ml, thyroid-stimulating hormone (TSH) 0.46 μIU/ml, free triiodothyronine 2.65 pg/ml, free thyroxine 1.30 ng/dl, prolactin (PRL) 10.49 ng/ml, follicle-stimulating hormone (FSH) 72.83 mIU/ml, and luteinizing hormone (LH) 18.14 mIU/ml.

Fig. 1
Head MR reveals a clival tumor extending to the paranasal sinuses and left nasal cavity. The lesion appears as an isointense area on T1-weighted imaging (A), an iso- to high-intensity area on T2-weighted imaging (B), and homogeneous enhancement on T1-weighted imaging with gadolinium contrast (C: axial, D: coronal, E: sagittal).
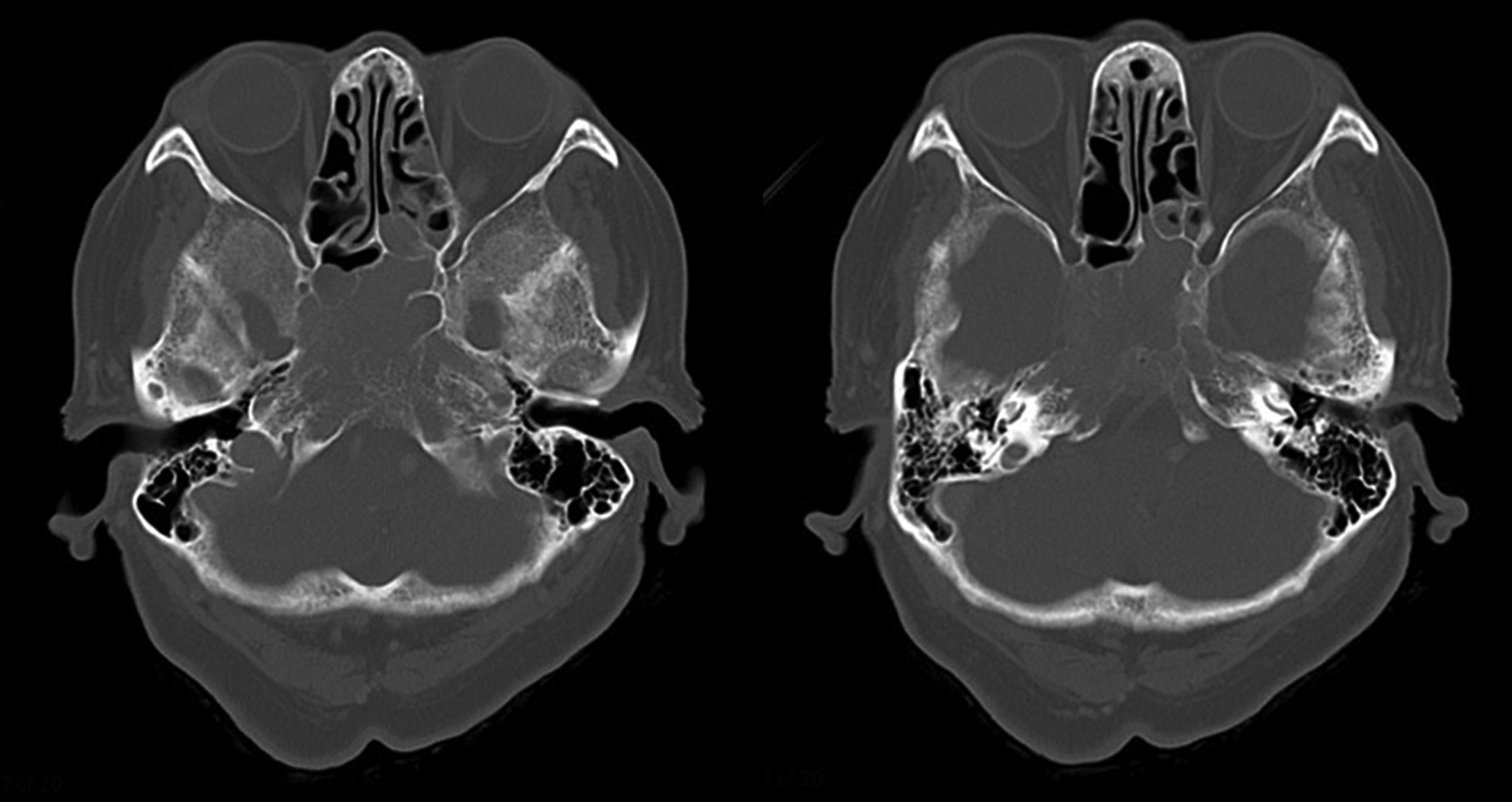
Fig. 2
Axial skull CT shows that the tumor extends from the clivus to the ethmoidal sinuses and posterior nasal aperture, and that it erodes the sphenoid bone.
We performed a repeat biopsy of the intranasal tumor tissue to confirm the pathological diagnosis because the radiological findings differed from those of typical olfactory neuroblastoma. The pathological diagnosis at our institute was a NET, WHO grade 2. Histological examination revealed that the tumor was composed of oval atypical cells with uniform nuclei and scant cytoplasm arranged in sheets or a lobular pattern (Fig. 3A and 3B). The specimen was immunohistochemically positive for cytokeratins AE1/AE3, which are epithelial markers, and for synaptophysin (Fig. 3C), chromogranin A (Fig. 3D), and CD56, which are neuroendocrine markers. Furthermore, on immunochemical analysis, the tumor cells showed weak, patchy expression of ACTH (Fig. 3E), but were negative for GFAP, S-100, Pit-1, SF-1, GH, TSH, PRL, FSH, and LH. The mitotic count was approximately three per 10 high-power fields, and the Ki-67 labeling index was 1%–5%. Electron microscopy revealed the presence of scattered cytoplasmic neuroendocrine granules (Fig. 3F).

Fig. 3
Histological examination of the tumor cells reveals oval atypical cells with uniform nuclei and scant cytoplasm arranged in sheets or a lobular pattern (A: ×100 and B: ×400, hematoxylin and eosin stain). The tumor cells are immunohistochemically positive for synaptophysin (C: ×400) and chromogranin A (D: ×400), and demonstrate weak, patchy expression of ACTH (E: ×400). Electron microscopy reveals the presence of scattered cytoplasmic neuroendocrine granules (F: bar = 2.0 μm). ACTH: adrenocorticotropic hormone.
Due to her extremely advanced age (84 years), the patient received only radiotherapy and not surgical removal or chemotherapy. Specifically, focal radiotherapy (50.1 Gy/30 fr) with intensity-modulated radiation therapy (IMRT) was performed, and MRI with gadolinium contrast at the end of radiotherapy revealed no significant change in the size of the clival tumor. We discharged the patient, but 10 months after radiotherapy she became aware of cerebrospinal fluid (CSF) rhinorrhea and returned to our institute. CT revealed pneumocephalus in the quadrigeminal cistern. We performed transnasal endoscopy to determine the cause of the CSF rhinorrhea and identified a firm, reddish tumor in the sphenoid sinus, but with no pooling of CSF. We advised the patient to undergo hospitalization for further examinations, but she refused. Two months later, she required transportation to the hospital due to disturbed consciousness, and CT revealed massive pneumocephalus around the basal cistern (Fig. 4A–C). The clival tumor had shrunk and a little air had entered the interpeduncular cistern (Fig. 4D and 4E). We performed emergent endoscopic transnasal CSF leak closure with a nasoseptal flap. After removing the firm, reddish tumor in the sphenoid sinuses, we were able to visualize the thin sellar floor. Both the sellar floor and the clivus were partially destroyed, but the tumor was not attached to the sellar dura and had not invaded the sella turcica. On the other hand, the tumor had invaded the interpeduncular cistern, tearing the dura mater. We packed the cavity with abdominal subcutaneous fat tissue, applied a nasoseptal flap using fibrin glue, and finally inserted a sinus balloon into the nasal cavity.

Fig. 4
Axial head CT reveals massive pneumocephalus around the basal cistern (A–C). CT reveals that clival tumor shrinkage and a little air intrusion into the interpeduncular cistern (D, E).
The patient’s postoperative course was successful and her CSF rhinorrhea resolved. Postoperative MRI with gadolinium contrast revealed tumor shrinkage and a reduction in pneumocephalus size. The patient returned home but did not visit our institute afterward, and therefore did not undergo radiological follow-up. Two years after discharge she developed cardiopulmonary arrest and was transported to the emergency department of a local hospital where she died that day of an unknown cause.
Discussion
NETs originate from the cells of the diffuse neuroendocrine system that are responsible for amine precursor uptake and decarboxylation.3) Neuroendocrine cells are involved in the coordination of the neurotransmitter-initiated synthesis and release of biologically active substances into the blood.4) Primary intracranial NETs are extremely rare entities, and to date, only 12 cases have been reported in the literature.1,4–13) Most were located in the skull base and sellar regions, except for one case each in the third ventricle,1) frontal convexity,6) parietal lobe,9) and cerebellum.13)
Based on the pathological findings of the present case, we consider that the differential diagnosis of NETs should include esthesioneuroblastoma and ectopic sparsely granulated corticotroph adenoma (SGCA). In fact, the patient was initially diagnosed with olfactory neuroblastoma. Esthesioneuroblastoma usually comprises neuroblasts with uniform circular nuclei and longitudinal cerebromedullary tubes in the dendrites, but NETs originate from the epithelium and lack neural features.6–8,10,11) On electron microscopy, verification of the presence of intracytoplasmic neuroendocrine granules is highly valuable for the differential diagnosis of NETs.10) Intraoperative findings in this case indicated that the sellar dura was preserved and there was no continuity between the tumor and the pituitary gland. On immunohistochemical examination, the tumor cells showed weak, patchy expression of ACTH (Fig. 3E). However, the cells had an atypical oval morphology with uniform nuclei and scant cytoplasm (Fig. 3A and 3B), characteristics that differ from the typical pathological features of SGCA, which is composed of weakly basophilic or chromophobe cells.14) Furthermore, we could not confirm the “honeycomb-Golgi” appearance that is a distinctive electron microscopic finding in silent corticotroph adenomas.15) However, it is very difficult to distinguish NETs from pituitary adenomas due to their similar pathological features; therefore, the members of the International Pituitary Pathology Club proposed the use of the term “pituitary NET” rather than pituitary adenoma to define adenohypophyseal tumors.16)
NETs are clinically classified as either functional or non-functional depending on their ability to secrete active hormones.4) Some authors described ACTH-secreting NETs that were immunochemically positive for ACTH and were located in the liver,17) pancreas,18) lung,19) kidney,20) and thymus.21) However, there have been few reports of functional primary intracranial NETs. Liu et al. reported a primary intracranial NET with ectopic ACTH syndrome.22) The patient’s high serum ACTH level and bilateral adrenal gland hyperplasia improved after complete removal of the tumor, but in contrast to the present case, the tumor was immunonegative for ACTH.
Regarding imaging studies, NETs show no specific findings by CT or MR. However, CT may be useful to evaluate the extension and degree of skull bone erosion, especially in tumors located in the skull base or sellar region. On MR, NETs usually appear as a hypointense area on T1-weighted sequences, a hyperintense area on T2-weighted sequences, and a homogeneous enhanced area with gadolinium contrast.4) Whole-body 18F-FDG PET is useful to detect primary lesions and metastasis, but increased FDG uptake may not be apparent in well-differentiated NETs because of their low cellular proliferative activity and high differentiation rate.8,11) Recently, several NET-specific tracers were developed for use in PET imaging. For example, PET using 11C-5-hydroxytryptophan has been shown to be a sensitive method to detect small NET lesions.11) In the present case, whole-body 18F-FDG PET before biopsy did not identify primary extracranial neoplastic lesions, which is a major reason why the tumor was thought to have an intracranial origin. However, we could not perform repeat follow-up PET studies or further detailed examinations such as gastroscopy and colonoscopy because the patient was elderly and she refused further examinations. Considering our limited workup, we cannot rule out the possibility that the tumor was an intracranial metastasis of an extracranial primary lesion that was undetectable by 18F-FDG PET.
The optimal treatment of primary intracranial NETs seems to be surgical resection. However, patients who receive comprehensive treatment, including a full course of chemotherapy, were shown to have longer tumor-free survival than patients who received only surgery.10,22,23) Platinum-based chemotherapy using a combination of cisplatin, ifosfamide, and etoposide is suggested for high-grade neuroendocrine neoplasms (NENs).1) In the present case, we performed focal radiotherapy with IMRT after confirmation of the pathological diagnosis, but the patient developed tension pneumocephalus 1 year after radiotherapy. Some authors reported success with postoperative treatment consisting of radiotherapy, including IMRT or fractioned stereotactic radiotherapy, with or without chemotherapy.1,8,9,11) Radiotherapy seems to be more effective in poorly differentiated NENs.10) Given the advanced age of our patient, we performed only tumor biopsy via the nasal cavity, followed by focal radiotherapy; this led to a marked reduction in tumor size, which led to the development of tension pneumocephalus. Intraoperative findings revealed that the tension pneumocephalus was due to intermittent CSF rhinorrhea induced by a connection between the nasal cavity and the interpeduncular cistern. These observations support the CT findings of clival tumor shrinkage and air intrusion into the interpeduncular cistern (Fig. 4D and 4E). There have been no reports of tension pneumocephalus as a complication in primary skull base NETs. Tension pneumocephalus secondary to radiotherapy is rare, with only five previous reports of cases involving nasopharyngeal carcinomas.24–28) Unlike in our case, all of those patients underwent high-dose irradiation (more than 60 Gy). Tension pneumocephalus developed between 2 and 4 years after radiotherapy, except in one patient in whom it developed during radiotherapy.24) Three of five patients had a fatal clinical course,24,27,28) but the remaining two patients experienced a favorable clinical course after surgical repair of CSF fistulas.25,26) It is important to confirm the degree and extension of skull bone erosion by CT before radiotherapy, as well as to perform repeat radiological follow-up examinations during and after radiotherapy.
Conclusion
We report an extremely rare case of a primary intracranial NET of the skull base that extended to the paranasal sinuses and nasal cavity. Because the patient was elderly, she underwent only tumor biopsy via the nasal cavity followed by focal radiotherapy with IMRT. However, she was transported to our hospital due to disturbed consciousness 1 year after radiotherapy, and CT revealed tension pneumocephalus due to tumor shrinkage. We performed endoscopic transnasal CSF leak closure with a nasoseptal flap. Her postoperative course was successful, and she returned home but died of an unknown cause 2 years after discharge. Assessing skull bone erosion by CT before radiotherapy and performing repeat radiological follow-up examinations are essential to prevent tension pneumocephalus related to radiotherapy. Once tension pneumocephalus develops, emergent surgical repair of the CSF fistula seems to result in a favorable clinical course.
Acknowledgment
The authors appreciate Dr. Naoko Inoshita, Department of Pathology, Tokyo Metropolitan Geriatric Hospital and Institute of Gerontology, for her valuable advice on the pathological diagnosis.
Conflicts of Interest Disclosure
None of the authors have conflicts of interest.
References
- 1) Reed CT, Duma N, Halfdanarson T, Buckner J: Primary neuroendocrine carcinoma of the brain. BMJ Case Rep 12: e230582, 2019
- 2) Faggiano A, Mansueto G, Ferolla P, et al.: Diagnostic and prognostic implications of the World Health Organization classification of neuroendocrine tumors. J Endocrinol Invest 31: 216–223, 2008
- 3) Volante M, Rindi G, Papotti M: The grey zone between pure (neuro)endocrine and non-(neuro)endocrine tumours: a comment on concepts and classification of mixed exocrine-endocrine neoplasms. Virchows Arch 449: 499–506, 2006
- 4) Joshi VV, Thombare P, Verma M, Kamat N, Patkar D: Primary skull base neuroendocrine carcinoma-a case report and review of literature. Radiol Case Rep 15: 1071–1077, 2020
- 5) Porter DG, Chakrabarty A, McEvoy A, Bradford R: Intracranial carcinoid without evidence of extracranial disease. Neuropathol Appl Neurobiol 26: 298–300, 2000
- 6) Deshaies EM, Adamo MA, Qian J, DiRisio DA: A carcinoid tumor mimicking an isolated intracranial meningioma. Case report. J Neurosurg 101: 858–860, 2004
- 7) Ibrahim M, Yousef M, Bohnen N, Eisbruch A, Parmar H: Primary carcinoid tumor of the skull base: case report and review of the literature. J Neuroimaging 20: 390–392, 2010
- 8) Hood B, Bray E, Bregy A, Norenberg M, Weed D, Morcos JJ: Primary carcinoid tumor of the cavernous sinus. World Neurosurg 81: 202.e9–13, 2014
- 9) Tamura R, Kuroshima Y, Nakamura Y: Primary neuroendocrine tumor in brain. Case Rep Neurol Med 2014: 295253, 2014
- 10) Liu H, Wang H, Qi X, Yu C: Primary intracranial neuroendocrine tumor: two case reports. World J Surg Oncol 14: 138, 2016
- 11) Nasi D, Perano D, Ghadirpour R, Iaccarino C, Servadei F, Romano A: Primary pituitary neuroendocrine tumor: case report and literature review. Surg Neurol Int 8: 101, 2017
- 12) Jayme EM, Morimoto TP, Lozano TM, Campos ZMS, de Castro CC: Sphenoid sinus neuroendocrine carcinoma. BJR Case Rep 3: 20150334, 2017
- 13) Vernieri C, Femia D, Pusceddu S, et al.: Primary cerebellar neuroendocrine tumors: chimeras or real entities? A case report with a 6-year follow-up. Case Rep Oncol 9: 432–439, 2016
- 14) Larkin S, Ansorge O, Boyce A, et al.: Pathology and pathogenesis of pituitary adenomas and other sellar lesions. Endotext. South Dartmouth (MA): MDText.com, Inc., 2000-2017 Feb 15.
- 15) Sano T, Mader R, Asa SL, Qian ZR, Hino A, Yamada S: “Honeycomb Golgi” in pituitary adenomas: not a marker of gonadotroph adenomas. Endocr Pathol 14: 363–368, 2003
- 16) Asa SL, Casar-Borota O, Chanson P, et al.: From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): an International Pituitary Pathology Club proposal. Endocr Relat Cancer 24: C5–C8, 2017
- 17) El Zein M, Vali R, Charron M, Manson D, Perlman K, Shammas A: Neuroendocrine tumor in liver with positive ACTH receptor: a case report. J Pediatr Hematol Oncol 36: e1–4, 2014
- 18) Surace A, Ferrarese A, Benvenga R, et al.: ACTH-secreting neuroendocrine pancreatic tumor: a case report. Int J Surg 12 Suppl 1: S222–S224, 2014
- 19) Takashi Y, Kinoshita Y, Makita N, et al.: Rapid recovery of hypothalamic-pituitary axis after successful resection of an ACTH-secreting neuroendocrine tumor. Inter Med 54: 2201–2205, 2015
- 20) Kocher NJ, Loloi J, Warrick J, Burns AS, Decter RM: Ectopic ACTH-secreting neuroendocrine tumor: a rare etiology of a pediatric solid renal mass. Can J Urol 26: 9956–9959, 2019
- 21) Okumura T, Takayama S, Nishio SI, et al.: ACTH-producing thymic neuroendocrine tumor initially presenting as psychosis: A case report and literature review. Thorac Cancer 10: 1648–1653, 2019
- 22) Liu H, Zhang M, Wang X, Qu Y, Zhang H, Yu C: Primary intracranial neuroendocrine tumor with ectopic adrenocorticotropic hormone syndrome: A rare and complicated case report and literature review. Mol Clin Oncol 5: 99–102, 2016
- 23) Kolesnikova GS, Lapshina AM, Voronkova IA, et al.: Comparative analysis of clinical, hormonal and morphological studies in patients with neuroendocrine ACTH-producing tumors. Int J Endocrinol 2013, 659232, 2013.
- 24) Jimenez-Jimenez E, Martí SS, Villas MV: Tension pneumocephalus related to radiotherapy for nasopharyngeal carcinoma. Case Rep Oncol Med 2014: 327380, 2014
- 25) Wang HC, Hwang JC, Peng JP, Hsieh CH, Liliang PC: Tension pneumocephalus--a rare complication of radiotherapy: a case report. J Emerg Med 31: 387–389, 2006
- 26) Wu CT, Lee ST: Delayed spontaneous tension pneumocephalus caused by radionecrosis of the skull base. Br J Neurosurg 13: 214–216, 1999
- 27) Kiu MC, Wan YL, Ng SH, Lee ST, Hao SP: Pneumocephalus due to nasopharyngeal carcinoma: case report. Neuroradiology 38: 70–72, 1996
- 28) Ng WF, Fung KH, Sham JS: Tension pneumocephalus--a rare complication of radiotherapy in nasopharyngeal carcinoma. Pathology 27: 204–208, 1995