A Case of Suprasellar Papillary Glioneuronal Tumor Mimicking Craniopharyngioma
論文ID: cr.2019-0163
この記事には本公開記事があります。

詳細
論文ID: cr.2019-0163
Papillary glioneuronal tumor (PGNT) is a low-grade biphasic neoplasm with astrocytic and neuronal differentiation. This tumor occurs most commonly in the frontal and temporal lobes, close to the ventricles, and rarely in the cerebellum, brainstem, and pineal gland. However, there has been no report of this tumor in the suprasellar region to date. In this paper, we report a case of PGNT in the suprasellar region in a 16-year-old girl. Magnetic resonance imaging (MRI) revealed a cystic tumor with calcification that progressed from the anterior skull base to the suprasellar and temporal regions. Preoperatively distinguishing this tumor from craniopharyngioma was difficult because of the patient’s age, localization of the tumor, and neuroimaging results. This case showed a backward shift of the chiasma, which is observed in only 4.7% of craniopharyngioma, as well as normal endocrine findings. Endocrinological examination and an MRI evaluation of the chiasmal shift may be useful for discrimination.
Papillary glioneuronal tumor (PGNT) is a low-grade biphasic neoplasm with astrocytic and neuronal differentiation, which was first reported in 1998 by Komori et al.1) PGNTs are very rare, and account for less than 0.02% of intracranial tumors. The tumor usually occurs in the supratentorial region in children and young adults.2) Common symptoms at presentation include headache, seizures, and weakness. In this paper, we report a case of PGNT in the suprasellar region with the symptom of blurred vision in a 16-year-old girl.
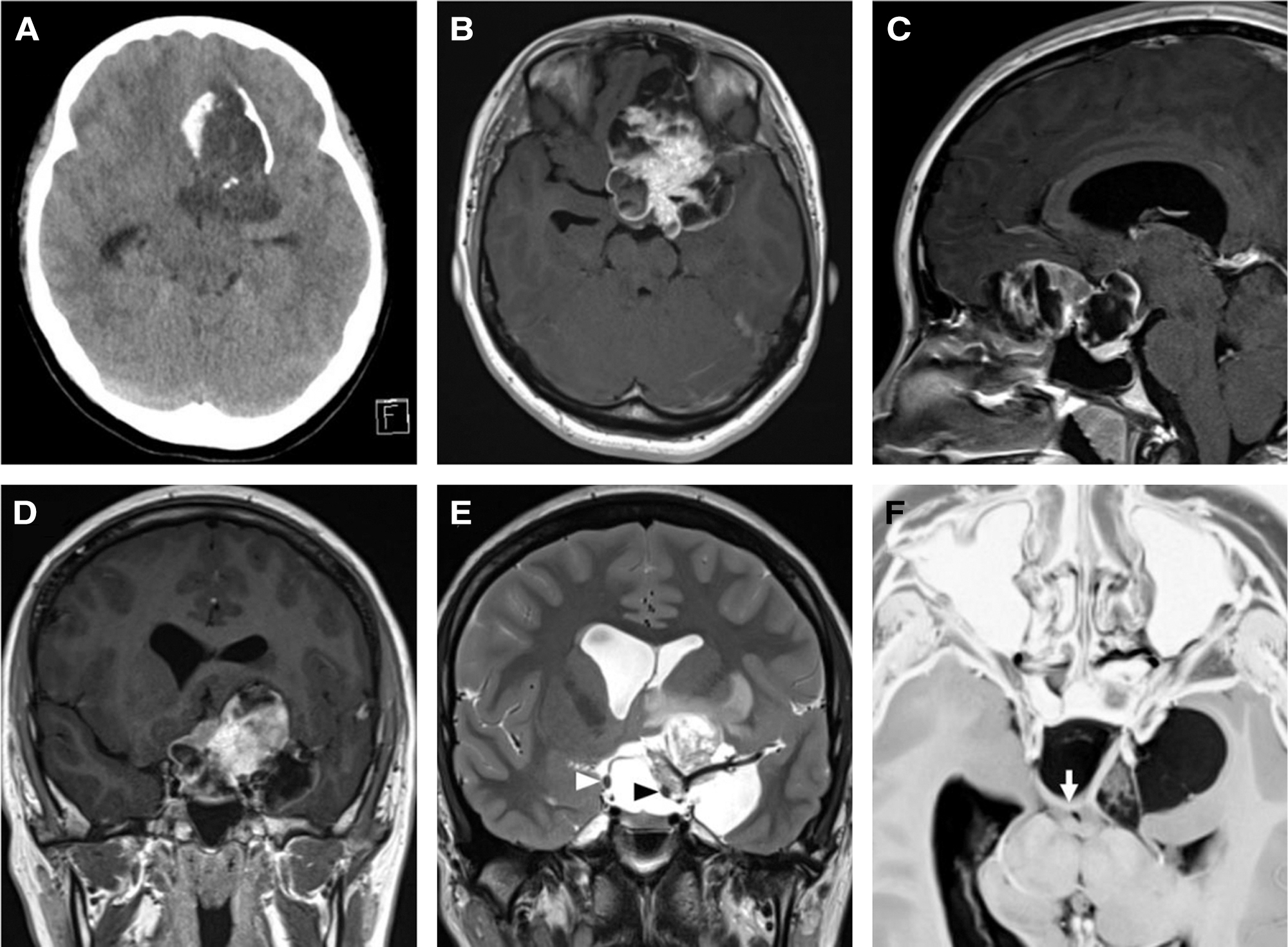
A 16-year-old girl was admitted to Kanazawa Medical University Hospital (Ishikawa, Japan) because of blurred vision lasting 1 year. Neurological examination revealed bitemporal hemianopia with normal visual acuity. The fundus test showed stasis papillae in both eyes. The basal serum levels of adenohypophyseal hormones, such as growth hormone, thyrotropin, prolactin, corticotropin, luteinizing hormone, and follicle-stimulating hormone were within normal levels. Computed tomography (CT) (Fig. 1A) and magnetic resonance imaging (MRI) (Figs. 1B–1D) revealed a cystic tumor with calcification that extended from the anterior skull base to the suprasellar and temporal regions. The tumor showed inhomogeneous gadolinium contrast and mild perifocal edema. It involved the major vessels and the left optic nerve (Fig. 1E). A heavily T2-weighted MRI parallel slice of the optic chiasma clearly showed a posteriorly shifted chiasma (Fig. 1F). The tumor did not extend to the ventricle on MRI. The patient underwent surgery with bifrontal craniotomy. The left sylvian fissure was opened, and the cyst was confirmed. The cyst was punctured, following which the pale-yellow contents of the cyst were discharged. The left optic nerve, oculomotor nerve, and internal carotid artery were confirmed. These structures were not adhered to the tumor but were completely buried inside the tumor. The tumor at the frontal base was then removed. There was no adhesion between the tumor and the frontal base, and it reached the sella turcica. The tumor around the optic nerve was removed and the chiasma was exposed. The calcified components from the inferior median part of the left frontal lobe to the hypothalamus were strongly adhered to the surrounding brain. The tumor removal was limited to only 70% to prevent direct hypothalamic damage and major vessel perforator damage due to extraction.
Histological findings revealed that the tumor contained two major components (Figs. 2A and 2B). The first component consisted of pseudopapillary proliferation of cuboidal cells with small round nuclei around hyalinized vessels. The second component consisted of sheets of polygonal cells with large round nuclei and prominent nucleoli. There were also ganglion-like cells, gemistocytic cells, and calcification without pleomorphic cells, mitosis, and necrosis.

Immunohistochemistry results revealed that cuboidal cells, which formed pseudopapillary patterns, were strongly positive for glial fibrillary acidic protein (GFAP) (Fig. 2C); focally positive for S-100 protein; and negative for synaptophysin and neuronal nuclei (NeuN), which was consistent with a glial component. In addition, the polygonal cells with prominent nucleoli were positive for neuronal markers, such as synaptophysin (Fig. 2D), GFAP, S-100 protein, and NeuN. Gemistocytic cells were positive for glial markers and neuronal markers, GFAP, S-100 protein, synaptophysin, and NeuN. Tumor cells were negative for epithelial membrane antigen (EMA) and isocitrate dehydrogenase (IDH)-1 R132H. The Ki-67 labeling index was 4.1%.
Intraoperative cytological specimens also presented a pseudopapillary pattern of astrocytes, with hyalinized vessels and sheets of neuronal cells (Fig. 2E). The histopathological findings allowed us to classify this tumor as a typical PGNT.
Postoperative images showed residual tumor in the region from the inferior median part of the left frontal lobe up to the hypothalamus. The brain was decompressed, and the pituitary stalk was depicted away from the tumor (Fig. 3). After surgery, her visual dysfunction improved. She was followed without postoperative therapy, and no tumor progression was observed for 1 year.

The radiological features of PGNT are as follows: (1) the tumor is typically located in the cerebral hemispheres, close to the ventricles;2) (2) presents as a demarcated, contrast-enhanced mass; (3) shows cyst formations (in 86% of tumors)3); and (4) calcification, the frequency of which varies. This tumor occurs most commonly in the frontal and temporal lobes and rarely in the cerebellum, brainstem, and pineal gland; however, there has been no previous report of this tumor in the suprasellar region.4) In this case, there was no adhesion to the anterior skull base and pituitary gland; however, there was strong adhesion in the region from the inferior median part of the left frontal lobe up to the hypothalamus. So, this case is likely to have occurred from the hypothalamus or the surrounding brain.
In this patient, craniopharyngioma was initially considered as the preoperative diagnosis because of the patient’s age, cyst formation, calcification, and the tumor’s location in the suprasellar region. Prieto et al.5) classified six patterns of optic chiasma distortion in craniopharyngioma. Based on their classification, only 4.7% of tumors have a stretched-backward pattern. Endocrinological abnormalities are observed in about 85% of craniopharyngioma patients.6) Consequently, a diagnosis of craniopharyngioma was not appropriate for this case.
Pathological features of PGNT are pseudopapillary proliferation of GFAP-positive glial cells and sheets synaptophysin-positive neuronal cells. Both of these features were observed in the present case, and the tumor was thus diagnosed as a typical PGNT. In this case, typical PGNT findings were observed in smear preparations of intraoperative cytological specimens, which were useful for intraoperative pathological diagnosis. Although not evaluated in this case, Bridge and colleagues recently identified a translocation, t(9;17)(q31;q24) which results in a fusion of solute carrier family 44 choline transporter member 1 (SLC44A1) and protein kinase C alpha (PRKCA) in PGNT.7) This fusion product is currently recognized only in PGNT and rosette-forming glioneuronal tumors.8)
In the World Health Organization 2016 guidelines for classifying tumors2) and in the report by Ahmed et al.,4) a Ki-67-positive rate of 5% or less and maximal surgical resection are positive prognostic factors. Postoperative therapy is used rarely and on a case-by-case basis in the above report.4) In this patient, the Ki-67 value was low, and so she did not receive postoperative therapy. However, her tumor was partially extracted, and she continues to be followed carefully.
We reported a case of PGNT in the suprasellar region, which was difficult to distinguish preoperatively from other suprasellar tumors, such as craniopharyngioma, because of the patient’s age, localization of the tumor, and neuroimaging results. However, endocrinological examination and an MRI evaluation of the chiasmal shift may assist in differential diagnosis.
The authors have no conflicts of interest.