2. Historical background and principles of photoredox catalysis
2.1. Father of organic photochemistry: Ciamician.
Giacomo Luigi Ciamician,4) an Italian chemist, is widely regarded as the pioneer who initiated the deliberate use of light energy for the conversion of organic compounds, earning him the title of the “father of organic photochemistry”. Because no strong artificial light sources were available during his time, he conducted his experiments on the roof of the chemistry building at his department (Fig. 1). He discovered many fundamental organic photoreactions induced by UV light in sunlight. In a thought-provoking article published in Science in 1912,5) he clearly demonstrated the potential utility of solar energy as an alternative to fossil fuels, envisioning the concept of green chemistry.
2.2. Principles of photoredox catalysis.
The electronic configuration of the photo-excited species, which differs from that in the ground state (Fig. 2a; path a), leads to unique reactions that cannot be achieved under thermal reaction conditions.6) Typical reactions include bond dissociation (e.g., Norrish type-I and -II reactions), cycloaddition (e.g., [2+2]-cycloaddition and Paterno–Büchi reaction), coupling (e.g., photochemical pinacol coupling), and pericyclic reactions,7) some of which were originally discovered by Ciamician.

Recently, sunlight has garnered increasing attention as a green energy source, as exemplified by solar power generation.8) Solar energy can also be harnessed to drive organic reactions. However, many organic compounds are colorless and thus cannot absorb visible light (the main component of sunlight) to become activated (Fig. 2a; path b). Therefore, alternative photochemical techniques that can activate colorless organic compounds using visible light needed to be developed. The conventional photochemical reactions described above are driven by UV light, which has a higher energy than visible light.6)
How can transparent organic compounds be activated with visible light? The use of a photosensitizer as a catalyst (PC) is one effective way to solve this problem.9) Photosensitizers are colored dyes that can absorb visible light and become photo-activated (path c). Although the excited state of the photosensitizer (PC*) has a very short lifetime (on the order of microseconds), it can interact with external molecules in two key ways: electron transfer and energy transfer.3)
In the electron transfer system, the excited PC* may donate an electron (e) to an external organic molecule or accept an electron from an external molecule to the hole (f) (path d), leading to the formation of a reactive organyl radical species (path g). As a result, organic molecules, regardless of their coloration, can be converted into versatile and reactive radical intermediates (R·) for various organic transformations (path c → d → g → h).
Conversely, the energy released when the excited species deactivates can be captured by a nearby molecule, activating it via an energy transfer process (path i). Both electron and energy transfer processes have been employed in catalytic organic transformations; however, the discussion in this article focuses on electron transfer processes, which is the key principle of photoredox catalysis.10)–13)
Photo-induced electron transfer, the core concept of photoredox catalysis,10) is illustrated in Figs. 2b and 2c. The irradiation of a molecule (paths j and j′ in Figs. 2b, c) leads to the excited state involving two half-occupied orbitals: the highest occupied system orbital (HOSO) and the lowest unoccupied system orbital (LUSO). For example, consider a situation in which the reduction potential of an acceptor (A) lies between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of a donor molecule (D) (Fig. 2b). In the ground state, an electron transfer from D’s HOMO to A is thermodynamically infeasible (path k) because it is an uphill process. However, upon irradiation, the energy level of the excited electron in the HOSO is sufficiently raised, enabling a downhill electron transfer (path l) to generate the anionic radical (A·−) and the oxidized donor (D°+) (· and ° stand for electron and hole, respectively.). Similarly, as shown in Fig. 2c, although an uphill electron transfer from a donor (D′) to an acceptor (A′) is not feasible (path k′), electron transfer from D′ to the hole in the LUSO of the excited state is possible (path l′), leading to the formation of the cationic radical D′°+ and the anionic radical A′·−. In this way, the apparent uphill electron transfer processes (paths k and k′) become feasible through the photo-excited states generated by photoirradiation (paths j and j′). The redox powers of D (Fig. 2b) and A′ (Fig. 2c) are enhanced by the HOMO-LUMO gap, i.e., the energy gained through photoirradiation. The compound that mediates these electron transfer processes (D in Fig. 2b and A′ in Fig. 2c) is called a photosensitizer. Thus, colorless external molecules that cannot be activated by visible light can be activated by a colored photosensitizer.
When these electron transfer processes are combined with organic reactions, photoredox catalysis is achieved, where the photosensitizer serves as a photocatalyst (PC) (Fig. 3).10) In the presence of an acceptor A, the electron in the HOSO of the photo-excited state of the photosensitizer can be transferred to A (as shown in Fig. 2b), generating the reduced anionic radical A·− and the oxidized catalyst PC°+ (step 1). The electron transfer from an electron donor (D) in the reaction system to PC°+ regenerates the ground-state catalyst PC and produces the cationic radical D°+ (step 2). This enables the photocatalyst (PC) to undergo sequential reduction-oxidation processes with external substrates A and D upon irradiation, generating the radical species, A·− and D°+. Since step 1 involves PC oxidation, this catalytic cycle is referred to as the oxidative quenching cycle (OQC), with the external substrate being reduced. Conversely, in the reductive quenching cycle (RQC), the hole in the LUSO of the excited state can accept an electron from a donor molecule D′ (Fig. 2c), leading to the 1e-oxidation of D′ (step 1′) and the 1e-reduction of A′ (step 2′) to generate the corresponding radical species D′°+ and A′·−. If these systems are integrated into organic transformations requiring these redox functions, the reactions can be driven by simple irradiation with visible light. The balance between the HOSO and LUSO energy levels of PC* and the redox potentials of the external molecules is key to the success of these reactions. The most notable feature of photoredox catalysis is the generation of organic radical species, such as A·− and D°+, under mild reaction conditions, which contrasts with conventional radical generation methods, as discussed below.

The systems discussed above, however, are not atom-economical. For instance, in the OQC cycle (Fig. 3), when an external organic compound is reduced in step 1, the resulting catalytic species (PC°+) must be returned to its ground state (PC) by the action of an electron donor (D) in step 2 to close the catalytic cycle. However, the generated species D°+ is usually not useful and is thus discarded as waste. Such reagents are referred to as sacrificial reagents. While the four redox processes in Fig. 3 (steps 1, 1′, 2, and 2′) can be used to reduce or oxidize external substrates, each process is paired with its counterpart redox process (path 1 vs. path 2 and path 1′ vs. path 2′), which requires a sacrificial reagent. However, for example, for an OQC cycle, if the resulting anionic radical (A·−) can be functionalized and then subjected to the subsequent reduction step (step 2), as indicated by the arrow ($ \Rightarrow $) in Fig. 3, the catalytic cycle can proceed without the need for a sacrificial electron donor. This would make the catalytic system atom-economical (green). Such systems are considered redox-neutral. As shown by the arrows, dashed arrows, and double-line arrows indicating the flow of holes and electrons, the electrons in the OQC and the holes in the RQC remain within the catalytic system and return to the photocatalyst (PC). Examples of such systems are provided in the following sections.
2.3. A brief history of photoredox catalysis.
Figure 4 shows the year-on-year changes in the number of publications on photoredox catalysis. The first report on photoredox catalysis was published by the Kellogg group as early as 1978, nearly half a century ago, although its importance was not fully recognized at the time.14) The reaction catalyzed by eosin involved the reductive desulfurization of sulfonium salts to yield the corresponding hydrogenated compounds. This pioneering work was followed by sporadic publications, including those by Fukuzumi and Tanaka (H-transfer),15) Pac (H-transfer),16) and Deronzier (photo-Pschorr reaction).17)

In parallel, inorganic, photochemical, and electrochemical studies of the [Ru(bipy)3]2+ salts revealed their unique redox properties in the photo-excited states (Fig. 2d).18) Upon photoirradiation, the orange-red color [Ru(bipy)3]2+ salts, now used as a standard photoredox catalyst, undergo a metal-to-ligand charge transfer (MLCT) transition to form the excited species, where the electron and the hole are virtually localized on the bipy ligand and the metal center, respectively, enabling redox functions.
The reaction reported by Deronzier was the first example of a redox-neutral reaction, and in 1990, Okada reported a general method for alkyl radical generation from the corresponding N-(acyloxy)phthalimides.19) It is worth noting that chemists from Osaka University, including Fukuzumi, Tanaka, Pac, and Okada, made significant contributions to the development of photoredox catalysis.
Although the field seemed ripe for explosive development, the time had not yet arrived until MacMillan,20) Yoon,21) Stephenson,22) and we23) published their papers in the late 2000s (Fig. 5).
In the 1980s and 1990s, considerable investments were made in energy and highly reactive reagents to improve the efficiency and selectivity of chemical transformations. However, in the 21st century, with the advent of green chemistry,24) the fact that organic reactions can be promoted by visible light, including sunlight, began to attract significant attention. Photoredox catalysis must have seemed especially attractive to chemists concerned with energy and environmental issues.
Meanwhile, the development of radical technology was essential.25) Radical reactions form the core processes of industrial olefin polymerization, and systematic studies using organotin reagents revealed many valuable aspects of radical reactions. Reaction mechanisms were well understood by the 1980s and 1990s. Although organic radicals can be generated and used under fairly mild conditions, concerns about the toxicity of frequently used organotin reagents and the explosive nature of radical initiators (e.g., BPO and AIBN) remained. Heating or UV light irradiation was still required. Thus, the development of methods for generating radical species under milder conditions was highly anticipated. This gap was filled by the rise of photoredox catalysis, which promotes one-electron transfer processes, blooming all at once in the late 2000s. As a result, the number of publications related to photoredox catalysis increased exponentially (Fig. 4).
In the late 2000s, several research groups, including ours, reported a series of reactions mediated by photoredox catalysis (Fig. 5).20)–23) Although the reactions differed, [Ru(bipy)3]2+ salts and visible light were used as a common catalyst and energy source, respectively. Since then, this research has experienced explosive growth, as shown in Fig. 4. In particular, David MacMillan, who won the Nobel Prize in Chemistry in 2021, helped ignite a boom in this field and coined the term “photoredox catalysis”.20) However, this growth should be seen as a “Renaissance” of photoredox catalysis, since the foundational concepts were already established in the 1980s, as discussed above.
4. Photoredox catalysis: Generation of organyl radicals under mild conditions
During the study of the binuclear catalysis mentioned above, we recognized the unique redox properties of [Ru(bipy)3]2+, specifically its ability to undergo oxidation/reduction of organic compounds in its photo-excited state.
Conventional methods for generating organyl radicals (R·) involve the homolysis of the R–LG bond (LG: leaving group) under thermal or photochemical conditions, which often require harsh conditions such as high temperatures or UV irradiation. These conditions can deteriorate the reaction substrates (Fig. 7).

On the other hand, radicals (R·) can be interconverted with cationic (R+) and anionic species (R−) via one-electron redox processes (Fig. 7). It is conceivable that applying the one-electron redox processes in a photoredox system could lead to the one-electron reduction of the cation (R+) (via OQC; path a) or the one-electron oxidation of the anion (R−) (via RQC; path b), resulting in the generation of the corresponding radical species. The ionic species, R+ and R−, are typically unstable, but if stable electron-rich or electron-deficient adducts (R–X and R–Y) with auxiliary leaving groups (X, Y) are available, they can serve as precursors. The cationic (R–X+) and anionic (R–Y−) species are more reactive toward redox processes than their corresponding neutral species. A key feature of photoredox catalysis is that by simply irradiating with visible light (including sunlight), organyl radicals can be generated under very mild reaction conditions, even at low temperatures.
The photoredox reactions developed in our laboratory are summarized in Table 1, where the metal photoredox catalysts are also shown along with the reagents for trifluoromethylation (see section 4.2). The reactions are classified according to the quenching cycles. Our study began with the development of reactions via RQC, but later, reactions via OQC became the primary focus, as described below.
Table 1. Representative photoredox catalytic reactions developed in the author’s laboratory. Metal photoredox catalysts and trifluoromethylating reagents are also included. The numbers in parentheses in the “entry” columns refer to the corresponding references.

Trialkylamines are frequently used as sacrificial reducing agents for OQC to quench the photo-excited catalyst PC* (step 1 in Fig. 3). Although the aminium radical cation (R3N°+) formed concomitantly could serve as a versatile synthon, it received little attention. We then examined the trapping of the aminium radical cation by the stable oxygen-centered radical TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl) (Fig. 8; entry I-1 in Table 1).23) When a mixture of aldehyde, piperidine, and TEMPO dissolved in acetonitrile was irradiated with visible light (using a Xe lamp) in the presence of 2 mol% of [Ru(bipy)3]2+ Ru, α-oxylated aldehyde 10 was obtained. The first event involves the condensation of aldehyde with morpholine to form enamine 11, a functionalized trialkylamine that undergoes one-electron oxidation by the photo-excited Ru* species to generate the cationic aminium radical 12. The radical species is trapped by TEMPO at the α-position, followed by hydrolysis to provide the α-oxylated product 10. The reduced Ru(I) species should be oxidized by adventitious oxygen to regenerate the ground-state catalyst. It is noteworthy that in this system, organocatalysis (enamine formation) and photoredox catalysis (oxylation) are combined.

Oxyamination follows the RQC mechanism, and we have reported several examples of reactions that follow RQC. The aminium radicals derived from enamines coupled with electron-rich silyl enol ethers in the presence of 2 equivalents of a one-electron acceptor to furnish 1,4-diketones via C–C coupling (entry I-2).29) Furthermore, we developed a versatile method for generating organyl radicals from organoborates such as [R–BF3]− via the one-electron oxidation process included in RQC (Fig. 9).25e),25f) This method was applied to the redox-neutral Giese reaction (entry I-3).30) Although the generation of organyl radicals from organoborates via one-electron oxidation was known, previous methods required special equipment or generated large amounts of metal waste. In contrast, our method requires only visible-light irradiation, providing a clean and green alternative widely used in organic synthesis.
4.2. Trifluoromethylative difunctionalization of unsaturated hydrocarbons.31),32)
Fluorine is the most electronegative element, and fluorine-containing groups are prevalent in pharmaceutical and agrochemical compounds as well as in functional organic materials.33) In particular, the fluorine-containing groups in drugs exert significant effects on biological activity, metabolic stability, drug absorption, and distribution. It is noteworthy that fluorine-containing drugs now account for approximately 25% of small-molecule drugs in the pharmaceutical market.34) Furthermore, of the 55 approved drugs by the US FDA (Food and Drug Administration) in 2023, 33 were small molecules, 12 of which contained fluorine atoms, representing 20% of the newly approved drugs. Therefore, the pharmaceutical industry is seeking new ways to introduce fluorine atoms into organic structures, such as by replacing hydrogen atoms with fluorine or introducing fluorine-containing functional groups. We first investigated the trifluoromethylation reactions promoted by photoredox catalysis.
Fluorine-containing compounds are electron-deficient because they contain highly electronegative fluorine atoms. We hypothesized that CF3-containing reagents, such as those shown in Table 1 and Fig. 10 (Umemoto reagent U,35) Yagupolskii reagent Y36) (cationic sulfonium salts), and Togni reagent T (with hypervalent iodine center)),37) could be electrophilic enough to be reduced by the one-electron reduction step in OQC (path a in Fig. 7), allowing for the generation of CF3 radicals under mild reaction conditions via photoredox catalysis. This concept can be extended to other fluorine-containing alkyl radicals, as described below (section 4.4). CF3 radicals can also be accessed by one-electron oxidation (RQC) of electron-rich precursors like CF3SO2Na (Langlois reagent).38)
4.2.1. Oxy-trifluoromethylation.
Irradiation of a mixture of styrene and Umemoto reagent U dissolved in an acetone-water mixture for 2 h with blue LED lamps in the presence of the iridium catalyst Ir(ppy)3 (Ir1, 0.5 mol%) led to complete conversion of styrene to the oxy-trifluoromethylated product, 1-phenyl-3,3,3-trifluoropropanol (13a), with an almost quantitative yield (97%), as estimated by 1H-NMR analysis (entry II-1 in Table 1; Fig. 11a).39) When alcohol or carboxylic acid was used instead of water, the corresponding ethers (13b–i) or esters (13j,k) were formed analogously. The olefin double functionalization reaction, involving concomitant C–C and C–O bond formation, proved to be regiospecific, with the CF3 and OH groups attached to the β- and α-positions of styrene, respectively.
We propose a plausible mechanism via OQC, as shown in Fig. 11b, based on the following data:
1. Cyclic voltammetry (CV) data indicate that U is readily reduced by the photo-excited catalyst Ir1* (Eox(U) = −0.75 V. (vs. FeCp2) $ \gg$ E*ox(Ir1*) = −2.14 V).
2. Stern–Volmer plots showed that the photo-excited Ir catalyst (Ir1*) was quenched by U but not by styrene.
3. Ring opening of a strained bicyclic compound (β-pinene) indicated the participation of a radical or cationic intermediate.
The electron-deficient Umemoto reagent U is reduced by 1e transfer from the photo-excited catalyst Ir1*, inducing C–S bond homolysis and generating the CF3 radical, along with the formation of dibenzothiophene and the oxidized catalyst Ir1+. The CF3 radical adds to the β-position of styrene, forming the more stable benzyl radical intermediate 14 in a regioselective manner. This radical intermediate 14 is oxidized by Ir1+ to form cationic intermediate 15, which is subsequently trapped by an oxygen nucleophile to afford the difunctionalized product 13 after deprotonation. Simultaneously, the ground-state catalyst Ir1 is regenerated.
The CF3-containing cationic intermediate 15 could, in principle, be generated by the addition of a CF3 cation to the olefin, but such a process has not been reported, presumably due to the instability of the cation (Fig. 11c). In this system, the process is achieved by combining (1) the addition of the CF3 radical, generated by the 1e-reduction of the electron-deficient precursor, and (2) the 1e-oxidation of the resulting radical intermediate, utilizing both the redox functions of photoredox catalysis to make the reaction redox-neutral.
To demonstrate the effective use of sunlight energy, we conducted the reaction outdoors on a sunny day and found that it proceeded with an efficiency comparable to that observed under LED-lamp irradiation. Although we did not test all the photoredox reactions under sunlight, the reactions we examined proceeded successfully.
The reaction was extended to the regio- and diastereoselective cyclization of ene-carboxylic acids (entry II-2 in Table 1),40) ene-amides (entry II-3),41),42) and further to allenes (entry II-4).43)
Alkynes were also successfully oxy-trifluoromethylated. In this case, the triflate salt of the Yagupolskii reagent (Y) also acted as the oxygen nucleophile to yield trifluoromethylated alkenyl triflate (16, entry II-5) in a stereoselective manner (>90% selectivity).44) These triflate esters (16) are suitable substrates for Pd-catalyzed cross-coupling reactions (Suzuki–Miyaura and Sonogashira couplings),45) and CF3-containing tetrasubstituted trans-alkenes (17, Fig. 12), which are difficult to access via conventional methods, were readily obtained with retention of the stereochemistry.
4.2.2. Amino-trifluoromethylation.
The oxy-trifluoromethylation described above (reaction 1 in Fig. 12) involved solvolysis. Therefore, we expected that the reaction in the presence of other nucleophiles would capture cationic intermediate 15, leading to the formation of trifluoromethylated products bearing the corresponding functional groups (Fig. 13).

The reaction in nitrile, in the presence of 1 equivalent of water, furnished the corresponding amino-trifluoromethylated product 18 via a Ritter-type reaction (entry II-6 in Table 1; reaction (2) in Fig. 13; Fig. 14). The cationic intermediate 15, formed in a manner similar to oxy-trifluoromethylation (Fig. 11a), was trapped by nitrile. Subsequent hydrolysis of the resulting species 20 yielded product 18.46) The amount of added water should be exactly in a 1:1 ratio; otherwise, an excess of water leads to the oxy-trifluoromethylated compound 13 as a byproduct. The reaction was tolerant to various functional groups, including the reactive Bpin, CHO, and Br groups. The reaction was extended to β-substituted styrenes, although the diastereoselectivity was not satisfactory.
4.2.3. Keto-trifluoromethylation.
The solvolytic reaction in DMSO produced α-trifluoromethylated ketones 19 (entry II-7 in Table 1; reaction 3 in Fig. 13; Fig. 15).47) The reaction pathways were dependent on the trifluoromethylating reagents as well as the photoredox catalysts (A and B).
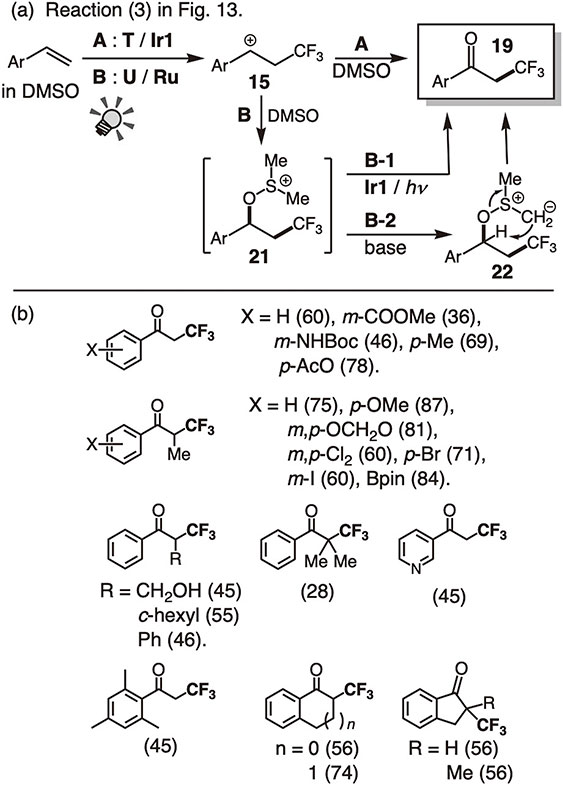
Reaction of styrene derivatives with Togni reagent T in the presence of the Ir catalyst Ir1 (A) gave α-trifluoromethylated ketones 19. On the other hand, the reaction with Umemoto reagent U in the presence of the Ru catalyst Ru (B) formed a sulfonium salt 21, which was characterized by 1H-NMR and MS analysis. The salt 21 was converted to 19 by further irradiation with the Ir catalyst Ir1 (B-1) or treatment with a base (B-2), suggesting the occurrence of a two-step reaction via 21. The photochemical reaction (B-1) may involve the 1e reduction of 21 by Ir1* followed by C–O bond homolysis. In the case of B-2, deprotonation with a base should form the ylide intermediate 22, which undergoes Kornblum-type oxidation to give 19. In the reaction with T and Ir1 (A), the m-iodobenzoate formed from T after ·CF3 transfer may serve as the base (B-2) to give 19 directly.
Thus, styrene derivatives can be converted to various CF3-substituted compounds with OH, NHCOR, and =O groups at the α-position by simply changing the nucleophile (solvent) (Fig. 13). These reactions are redox-neutral. The reactions described here are regarded as prototypes for the photoredox-catalyzed fluoroalkylative difunctionalization of olefins. Subsequently, many related reactions such as hydro-, carbo-, and halo-trifluoromethylation have been reported.48)
4.2.4. Trifluoromethylation of olefins.
The cationic intermediate 15 formed during difunctionalization (Fig. 11a) may undergo deprotonation to afford CF3-substituted olefin 23 (Fig. 16). While simple olefins 24 were unaffected by the CF3 radical, presumably due to the low electron density of the C=C moiety (reaction 1), alkenylborate 25 underwent substitution to afford 23 (reaction 2; entry II-8 in Table 1), where the BF3 group worked as both an electron-donating and a directing group in addition to a leaving group (L).49) In the case of 1,1-diarylethene 26 (reaction 3; entry II-9 in Table 1), substitution proceeded without activation to give 27 because the intermediate 28 resulting from the ·CF3-addition could be stabilized by conjugation with the two aryl groups.50) Furthermore, α-substituted styrene derivatives 29 afforded the isomerized substituted olefins 30 as major products (reaction 4; entry II-10 in Table 1).51)
4.3. Introduction of N–, O–, S–, and C–functional groups.
4.3.1. Oxy-amination of olefins.
The successful photoredox-catalyzed trifluoromethylation reactions prompted us to investigate the generation of other radicals following path a, as shown in Fig. 7. The design of electron-deficient precursors is key to success.
For oxy-amination, we designed the cationic aminopyridinium salt 31, which should generate an N-centered radical upon 1e-reduction followed by the elimination of pyridine. We performed the difunctionalization of styrene under conditions similar to those of the oxy-trifluoromethylation discussed above (Fig. 17; entry II-11 in Table 1).52) The reactivity of the aminopyridinium salt 31 was dependent on the protecting group P, and other N-centered radical sources, such as chloramine-T and N-tosyloxyphthalimide, were ineffective. The tosyl (31b) and pentafluorobenzoyl derivatives (31c) afforded the oxy-aminated products 32 in good yields, whereas the parent salt (31a) and carbobenzoxy (Cbz; 31d) and trifluoroacetyl derivatives (31e) were sluggish. The parent aminopyridinium salt 31a could be activated upon the addition of Sc(OTf)3, a Lewis acid.53)

The reaction mechanism is similar to that of oxy-trifluoromethylation (Fig. 11). The 1e-reduction of 31, followed by the elimination of pyridine, generates the aminyl radical 33, which adds to styrene to give the β-amino-substituted benzyl radical intermediate like 14. Subsequent 1e oxidation followed by the nucleophilic addition of water to the resulting cationic intermediate 15 furnishes 2-aminoalcohol 32 after deprotonation.
While amino-hydroxylation has been achieved by the Sharpless Os-catalyst system,54) the present catalyst system provides the products in a regiospecific manner, although the diastereoselectivity can be poorly controlled.
4.3.2. Generation of oxygen-centered radicals.
We also explored the installation of oxygen functional groups using acyloxypyridinium salts 35, which were readily synthesized by reacting acyl chlorides with pyridine N-oxide (Fig. 18). Although oxygen-centered radicals 36 were generated under photoredox conditions, the reaction was only partially successful due to a side reaction involving decarboxylation, which produced the corresponding alkyl radical 37. When the benzoyloxy derivative 35a was reacted, a mixture of the desired diol derivative 38a and the oxy-alkylated byproduct 39a was obtained in low yield. In contrast, the acetoxy derivative exclusively produced 39b in moderate yield. The formation of these products likely follows a mechanism similar to that shown in Fig. 11, with the product ratio determined by the decarboxylation rate of radical 36. Stephenson successfully applied the decarboxylation process to trifluoromethylation with a trifluoroacetyl derivative.55)

To suppress decarboxylation and achieve selective oxygenation, electron-withdrawing substituents were introduced. As expected, the acyloxy radical 36c, bearing the 3,5-bis(trifluoromethyl)phenyl group, generated from 35c underwent hydrogen substitution on various arenes to form 40 (Fig. 18; entry II-12 in Table 1). Because the acyloxy radical 36c is electrophilic, it attacks the o- and p-positions of electron-donating groups such as methoxy group. By blocking the p-positions, o-selective oxygenation was achieved.56)
4.3.3. Generation of sulfur- and carbon-centered radicals.
Photoredox-promoted radical generation was also applied to sulfur- and carbon-centered radicals. Trifluoromethylthio radical (CF3S·; entry II-13 in Table 1)57) and pentafluorosulfanylphenyl radical (F5S-C6H4·; entry II-14)58) were successfully generated from their respective electron-deficient precursors, N-(trifluoromethylthio)succinimide 41 and pentafluorosulfanylphenyliodonium salts 42, leading to the formation of oxy-thiolated (43) and oxy-phenylated products (44) (Fig. 19). The SF5 group, regarded as a “super trifluoromethyl” group due to its strong electron-withdrawing inductive effect, lipophilicity, and chemical stability, has garnered increasing attention in pharmaceutical and materials science.59)

As demonstrated above, our concept allows the generation of various organyl radicals via the 1e-reduction of electron-deficient precursors using photoredox catalysis (path a in Fig. 7). An alternative pathway (path b in Fig. 7) for generating organyl radicals was also explored,60) although it was not extensively studied.
4.4. Fluoroalkylation.
The trifluoromethylation process described in section 4.2 has been extended to related fluoroalkylation reactions such as difluoromethylation (CF2H) and monofluoromethylation (CFH2). However, simply replacing the trifluoromethylating reagents with the corresponding fluoroalkylating reagents did not result in successful reactions. For instance, replacing the most electronegative fluorine atom(s) in the fluoromethylating reagents with hydrogen atom(s) results in significant negative shifts in the reduction potentials (>1 V), preventing reduction by conventional photoredox catalysts (Fig. 20). In some cases, we had to use neutral arenesulfonyl precursors (e.g., H2), which are harder to reduce than cationic Umemoto-type reagents. Therefore, the development of photoredox catalysts stronger than conventional ruthenium and iridium catalysts (e.g., Ir1) was essential.
4.4.1. Design of stronger organic photoredox catalysts.
Conventional catalysts such as Ru and Ir complexes are primarily designed to have long excited-state lifetimes, which increase the probability of collisions between the excited catalyst and reactants. These catalysts often contain heavy elements, such as transition metals (e.g., Ru and Ir), as well as bromine (e.g., eosin Y), which promote intersystem crossing, leading to the triplet excited state. The triplet excited species has a much longer lifetime (in the microsecond range) than the singlet excited species (nanosecond range). However, when considering the energy levels, the singlet excited state (S1) is always higher than the triplet excited state (T1), as illustrated in the Jablonski diagram (Fig. 21a). This implies that the singlet excited species exhibits stronger reducing power. Although Ir(ppy)3 (Ir1) is considered a strong reducing catalyst in its triplet state, as shown in Fig. 20, it is insufficient to reduce neutral fluoromethylating reagents, such as the Hu-type reagents H1 and H2.61) To address this, we turned our attention to polyaromatic hydrocarbon (PAH) catalysts in the singlet excited state. Metal-free catalytic systems are appealing from environmental and pharmaceutical perspectives because residual trace metals are a concern.

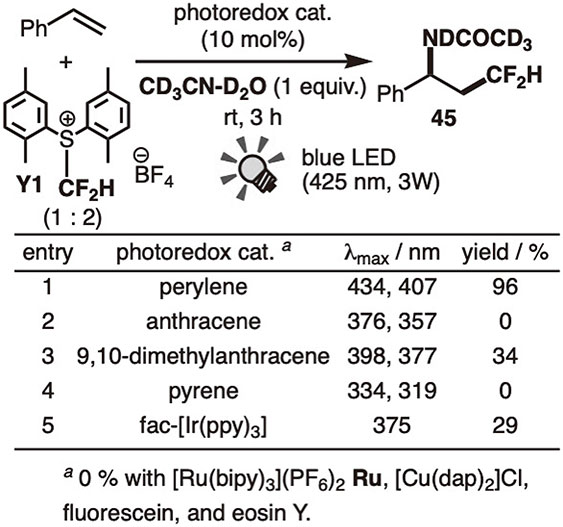
We first investigated amino-difluoromethylation.62) The previously reported sulfonium-type difluoromethylating reagents were problematic due to thermal instability and stickiness; therefore, we developed the thermally stable crystalline solid reagent Y2 (E*ox = −1.7 V). Among the PAH catalysts, perylene (per) exhibited the highest catalytic activity, yielding product 45 in nearly quantitative yield when irradiated with 425 nm of visible light (entry 1 in Fig. 22). The other PAH catalysts showed moderate (entry 3) or no catalytic activity (entries 2 and 4). Notably, metal catalysts such as Ir(ppy)3 (Ir1) (entry 4) and [Ru(bipy)3]2+ (Ru) (entry 5) were sluggish. The very short lifetime of the perylene’s singlet excited state (8.2 ns) may be compensated by its highly emissive quantum yield (per: 94%, compared to Ir1: 38%) to promote the single-electron transfer (SET) process. The neutral Hu reagent H1 did not work, likely because of its very low reduction potential (Fig. 20).63)
The successful outcome of the perylene catalyst encouraged us to improve its catalytic activity, but the derivatization of perylene is not always straightforward. We then turned to anthracene, another PAH system. Although anthracene showed some activity, it deteriorated rapidly, presumably due to attack at the 9- and 10-positions, as also evidenced by the irreversible CV wave. Taking into account the photophysical properties of the substituent groups, we designed and synthesized the 9,10-bis(di-p-t-butylphenylamino) derivative (BDA), which blocks the deteriorative reaction pathways (Fig. 23).64) BDA was easily prepared by Suzuki–Miyaura cross-coupling, and the t-butyl groups were introduced to improve the solubility in organic solvents and enhance the stability. BDA displayed a reversible CV wave and proved effective for solvolytic difluoromethylation with Y1, in addition to trifluoromethylation (CF3), 2,2-difluoroethylation (CH2CF2H), and 2,2,2-trifluoroethylation (CH2CF3) with the corresponding sulfonium-type fluoroalkylating reagents as well as trifluoromethylation with T and CF3SO2Cl. For the amino-trifluoroethylation of styrene in acetonitrile with the Y-type CH2CF3 reagent Y3 (Ered = −1.60 V) yielding product 46, the BDA catalyst (78% yield) was superior to other catalysts such as Ir1 (43%) and perylene (63%).

The estimated reducing power of BDA (E*ox = −1.88 V) was significantly stronger than that of Ru, although it was still weaker than that of Ir1 (Fig. 20). Stern–Volmer plots indicated that the mechanism involved static quenching, where the BDA catalyst interacts with the fluoroalkylating reagents in their ground state (prior to irradiation), as opposed to the dynamic quenching observed in the reactions described above.
We then attempted a more challenging reaction—monofluoromethylation—which required an even stronger reducing catalyst. The reducing power of a photo-excited catalyst (E*ox) can be estimated using the equation:
| \begin{equation*}
{E^{*}}_{\text{ox}} = E_{\text{ox}} - E_{0,0}
\end{equation*}
|
where Eox is the oxidation potential of the ground-state catalyst, as determined by the CV measurements, and E0,0 is the 0,0-excitation energy, as obtained from the fluorescence measurements (E0,0 = hc/λem; h: Planck constant, c: speed of light, λem: emission maximum) (Fig. 21b). There are two ways to enhance the reducing power: one is to maximize E0,0, and the other is to raise the S0 energy level, i.e., by minimizing Eox. We opted for the first approach, introducing bathochromic shifts in the emission maximum (λem) by reducing the size of the central arene rings.65)
As expected, the emission maxima (λem) shifted to higher energies, significantly enhancing the reducing power from −1.94 V (BDA) to −2.40 V (BDN) and −2.95 V (BDB) (Fig. 24; see also Fig. 20). Notably, the photo-excited benzene derivative BDB exhibited a reducing power that was even stronger than that of magnesium metal, although UVA irradiation was required.
4.4.3. Monofluoromethylation.
With stronger organic photoredox catalysts in hand, we examined oxy-monofluoromethylation.65) The reaction with the Yagupolskii-type sulfonium reagent Y2 and the BDN catalyst afforded the desired oxy-monofluoromethylated product 47 in a low yield (36%), along with oxy-arylated byproducts. These byproducts formed via the transfer of aryl groups from Y2 rather than the desired CFH2 group. We then chose the sulfoxyimine-type Hu reagent H2.61) Although the neutral reagent H2 was harder to reduce than the cationic sulfonium reagent Y2, the naphthalene catalyst BDN was strong enough to reduce H2 (Figs. 20 and 25). This method was tolerant to various functional groups and was also effective for late-stage monofluoromethylation of bioactive compounds such as flavonoids and steroids.

BDN (in acetone) showed blue fluorescence (λem = 449 nm) with a very high quantum yield (Φ = 0.99) while its excited-state lifetime was very short (t = 9 ns), indicating that the photo-excited species with a nanosecond span of lifetime (singlet species) can act as an efficient photoredox catalyst. The visible absorption band assigned to the HOMO–LUMO transition results from the interchromophoric conjugation derived from the twisted arrangements of the diarylamino groups with respect to the central arene group. These orbital features lead to efficient intramolecular charge transfer, extending the absorption band into the visible region, and contributing to the high quantum yield.
The transient absorption spectroscopic analyses revealed that the excited species BDN* in the presence of H2 began to diminish in approximately 100 ps, suggesting very fast electron transfer processes. The oxy-monofluoromethylation of styrene was not significantly affected by triplet quenchers such as O2 (53%), and 1,3,5,7-cyclooctatetraene (77%), indicating that singlet catalytic species play a key role. These results lead to an unconventional concept for the design of catalysts, where a long lifetime of the excited state is not always a requisite for efficient photoredox catalysts.66)
Design concepts for strongly reducing organic photoredox catalysts, such as PhenX (X = O, S, N (NPh); Fig. 20), were also proposed by Hawker and Miyake, who applied these catalysts to radical olefin polymerization.67)
To our surprise, however, the strongest reducing catalyst, BDB, was not effective for oxy-monofluoromethylation. The low catalytic activity can be interpreted as follows: Although the reducing power of BDB* is strong, as indicated by its E*ox value, completing the catalytic cycle requires the oxidized catalyst (PC°+) to oxidize a reductant in the reaction system to regenerate the ground-state catalyst (PC, step 2 in Fig. 3). The oxidizing power of PC°+ (BDB+) is lower due to the catalyst design—its frontier orbitals are higher in energy—making PC°+ a less efficient oxidant to close the catalytic cycle.
4.4.4. Generation of (fluoro)alkyl radicals from the corresponding benzoate esters with BDB catalyst.68)
The strong reducing ability of BDB* was demonstrated in the generation of (fluoro)alkyl radicals.
Organyl radical species are useful and reactive intermediates, but methods for generating them from versatile precursors under mild conditions are still highly sought after (see, for example, Fig. 9). Alcohols (R–OH) are ubiquitous organic molecules, but C–O homolysis is thermodynamically inaccessible. Instead, organic radicals can be generated via the corresponding ester derivatives (Fig. 26a). A classical method developed by Barton and Macombie involves converting alcohols into xanthate esters, followed by reaction with a thiophilic tin radical.69) Recently, Overman and MacMillan reported radical generation from oxalate esters, particularly cesium salts, formed in one-pot,70a) and more recently, an effective method mediated by benzoxazolium salts was developed by MacMillan.70b)–70f)

In light of this, we attempted to generate organic radicals from readily accessible carboxylic acid esters using the strongly reducing BDB catalyst.71) It should be noted that the oxalate method follows the RQC mechanism, whereas the carboxylate method follows the OQC mechanism.
Markó and Lam reported the generation of radicals from toluate esters with the aid of a samarium reductant or via an electrochemical method, and the resulting radicals were hydrogenated or used for C–C coupling reactions.72) Later, Reiser reported a photoredox version (Fig. 26b),73) in which electron-deficient benzoic acid derivatives bearing two m-CF3 groups were employed. While the sacrificial electron donor enhanced the reducing power of the iridium catalyst, the generated radicals were trapped by the oxidized sacrificial reagent, leading to hydrogenated or mono-functionalized products. We hypothesized that our strongly reducing catalyst system could reduce esters under redox-neutral conditions to enable olefin difunctionalization.
We first examined oxy-fluoroalkylation with the benzoate ester of 1,2-dimethyl-2,2,2-trifluoroethyl alcohol 48a (Fig. 27a) considering its higher reduction potential (Fig. 27b) and the stability of the resulting tertiary alkyl radical.71) The yield of product 49a was highly dependent on the reducing power (E*ox) of the photoredox catalysts. The strongly reducing catalysts BDB and PhenS afforded product 49a in good yields, in contrast to the low reactivity of BDN, Ir1, and BDA. UV irradiation was essential for BDB and PhenS because of their absorption maxima in the UVA region.

For the structure of the alkoxy moiety, the fluoroalkyl derivatives 49a–c were obtained in higher yields than the corresponding alkyl derivatives 49e,f. Furthermore, the tertiary derivatives 49a,e were obtained in higher yields than the corresponding secondary derivatives 49b,c,f. Notably, no reaction occurred for the primary alkyl esters 48d,g despite their higher reduction potentials. Therefore, the reduction potentials and stability of the resulting radicals influence the efficiency of radical generation.
The mechanism (Fig. 28) may be similar to that of oxy-trifluoromethylation (Fig. 11). In this case, a stoichiometric amount of benzoic acid was formed as a byproduct (Fig. 27a). The acid promotes protonation of 1e-reduced anionic intermediate 50 (path a), resulting in protonated neutral intermediate 51, which generates radical R with elimination of benzoic acid. The autocatalytic effect of protonation was supported by (1) the acceleration of the photoreaction in the presence of a Brønsted acid and (2) the retardation upon the addition of a base, although the addition of benzoic acid did not significantly improve the yield. Protonation prevented back electron transfer from 50 (path b), and a proton-coupled electron transfer (PCET) mechanism involving protonation before electron transfer (path c) was also suggested by the enhancement of the quenching rate of BDB* in the presence of benzoic acid.

In summary, we developed a method for generating tertiary and secondary alkyl radicals from readily accessible benzoate esters. The use of the strongly reducing photoredox catalyst BDB was essential, while the generation of primary alkyl radicals remained to be solved.
5. Photoredox catalysis in water
Water is a unique solvent. Because it is environmentally benign, organic synthesis in water promoted by visible-light (sunlight) photoredox catalysis should contribute to the development of green and sustainable chemical production systems (Fig. 29). In addition, water is highly polar, and the O–H bond in water is much stronger than the C–H bonds, making it compatible with processes such as C–H bond activation. Previous examples of water-soluble photoredox catalysts have been reported.74) For instance, Lipschutz demonstrated the oxy-sulfonylation promoted by a water-soluble catalyst 52.74b) However, its synthesis is tedious, and if modification of the catalyst is required, the ligand must be prepared from the beginning and combined with the metal species. Our strategy involves the preparation of water-soluble catalysts via supramolecular interactions without the formation of covalent bonds.

A part of our research activity has focused on supramolecular chemistry, and we developed a V-shaped meta-terphenyl compound with two sulfonate tethers, 53.75) Planar polyaromatic hydrocarbons often stack with each other, making them insoluble in solvents. However, the orthogonally oriented bulky peripheral pentamethylphenyl groups in the meta-terphenyl skeleton of 53 prevent this stacking, making the compound water-soluble by attaching the sulfonate tethers. Furthermore, in water, 53 forms a micelle structure (M) that can incorporate aromatic substances, including C60. We hypothesized that micelle M could incorporate PAH catalysts (PC) (section 4.4.2) to form the composite M$ \supset $PC, which would be soluble in water.
We first examined the phenoxazine catalyst PhenOH developed by Miyake (Fig. 30).67b)–67d) However, it showed very weak visible absorption although it was solubilized in water. We then switched the catalyst to the biphenyl-tethered derivative PhenOBP, and the resulting composite M$ \supset $PhenOBP exhibited an intense absorption in the visible region. DLS measurements revealed the formation of particles with an average diameter of 4 nm. A conventional surfactant such as sodium dodecyl sulfate (DDS) sparingly dissolved the aromatic catalyst. This method allows for the creation of a new catalyst simply by changing the content of the micelle (PC: PhenOH → PhenOBP) without the need for a long synthetic route involving covalent bond formation processes.

As a test reaction, we examined the pinacol coupling of benzaldehyde to yield benzopinacol 54 in the presence of triethylamine as a sacrificial electron donor (Fig. 31a), based on a reaction previously reported by Rueping, which was promoted by the Ir catalyst Ir4.76) Initially, we conducted the reaction in water with conventional water-soluble catalysts, Ru (chloride salt) and eosin Y. However, no reaction occurred because their reducing powers were insufficient to reduce benzaldehyde (Ered = −2.11 V vs. FeCp2). The organic phenoxazine catalyst with biphenyl tethers, PhenOBP, showed some catalytic activity in organic solvents (entries 4–7), but the micelle catalyst M$ \supset $PhenOBP afforded the coupling product 54 in nearly quantitative yield in water. Furthermore, although Rueping reported that the Ir catalyst Ir4 deteriorated in air due to quenching by dioxygen,76) the micelle catalyst was unaffected and gave product 54 in 96% yield, indicating that the catalytic system was not disturbed by air.

The reaction was extended to substituted benzaldehydes (52a–e,g) and even ketones such as acetophenone (54f). Additionally, solid, water-insoluble substrates such as benzophenone (54g) were converted to coupling products, although a longer reaction time was required (Fig. 31b). Figure 31c shows images of the benzaldehyde reaction. Initially, the substrate is dispersed as droplets. After 1 h of irradiation, the product precipitated from the micelle as white solids, which were extracted with ether. The remaining aqueous layer could be recycled multiple times without a loss in catalytic activity (1st run: 86%; 2nd run: 91%; 3rd run: 92%).
We conducted several experiments to clarify the reaction mechanism. In an organic solvent like DMF, the organic catalyst PhenOBP showed an oxidation peak in differential pulse voltammetry (DPV) measurements, whereas the micelle catalyst M$ \supset $PhenOBP in water did not show the corresponding peak, suggesting that the inside of the micelle is electrochemically isolated from the outer aqueous phase. In addition, the luminescence quenching of the micelle catalyst was accelerated by the addition of benzaldehyde, indicating that benzaldehyde enters the micelle and quenches the excited catalyst. Furthermore, the reaction was not hindered by air, as mentioned above.
Based on these results, we propose the following reaction mechanism (Fig. 32): First, the photocatalyst PC is incorporated into micelle M to form the supramolecular water-soluble catalyst M$ \supset $PC, which also incorporates the substrate ArCHO to form the composite M$ \supset $(PC·ArCHO). Subsequent visible-light irradiation generates the excited species M$ \supset $(PC*·ArCHO), which undergoes a 1e-transfer to yield a pair of anionic radical and an oxidized cationic catalyst within the micelle (M$ \supset $(PC+·ArCHO·−)). The resulting anionic radical ArCHO·− (55) is expelled from the micelle and protonated by water, and subsequent dimerization leads to benzopinacol 54. The oxidized catalyst PC+ within the micelle is reduced by NR3, a sacrificial reductant, to regenerate the ground-state catalyst M$ \supset $PC. In this way, we successfully developed a water-soluble catalyst based on the supramolecular interaction between the aromatic micelle M and the polyaromatic hydrocarbon catalyst in water.

The supramolecular catalyst system was extended to the carbocyclization of the amide compound 56 (Fig. 33a). 1e-reduction of 56 causes elimination of the iodine atom to generate the radical intermediate 58, which undergoes hydrogen atom transfer followed by cyclization to afford 57 (Noto, N., Koike, T. and Akita, M., unpublished results). Trifluoromethylation of a nucleic acid derivative, N,N-dimethyluracil 59, was also possible (Fig. 33a, b; Noto, N., Koike, T. and Akita, M., unpublished results). For these reactions, the catalyst containing phenoxazine PhenOBP 67b)–67d) was used, but we recently reported that the modified bis(diarylamino)naphthalene catalyst (BDN′) performed better for removing the alkoxy groups in Weinreb amides 60 (Fig. 33c).77b)
Recently, photoredox catalysis in water has gained increasing attention, and micelles and surfactants have been used to dissolve catalysts.74)



;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/PJA10105B-02tab01.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)










