Abstract
The biological activity of hyaluronan (HA), a major component of the extracellular matrix in vertebrate tissues, depends on its molecular weight, and thus its degradation is a critical process for HA biological functions. Here, we review the characteristics of newly discovered proteins essential for HA degradation, hyaluronan-binding protein involved in hyaluronan depolymerization (HYBID), also known as cell migration inducing hyaluronidase 1 (CEMIP) and KIAA1199, and transmembrane protein-2 (TMEM2; alias CEMIP2). Human and mouse forms of HYBID exert their HA-degrading activity in special microenvironments including recycling endosomes. Mouse TMEM2 functions as a cell-surface hyaluronidase for HA turnover in local tissues, lymph nodes, and the liver. In contrast, the role of human TMEM2 in HA degradation is the subject of much debate. HYBID expression is upregulated by proinflammatory factors such as histamine and interleukin-6 and downregulated by transforming growth factor-β. HYBID is involved in physiological HA turnover in human skin and joint tissues and plays an important role in their pathological destruction by accelerating HA degradation.

1. Introduction
The biological functions of cells in each organ are strictly regulated by tissue microenvironmental factors, which comprise extracellular matrix (ECM) molecules and their physiologically active interacting factors such as cytokines and growth factors. ECM consists of hyaluronan (HA), proteoglycans, collagens, elastin, and cell-adhesive glycoproteins.1) Among these, HA is most abundant in the skin (400–500 µg/g tissue) accounting for approximately half of total body HA, and it is ubiquitously present as an important constituent in almost all human adult tissues, such as the vitreous of the eye (140–338 µg/g) and synovial fluid (1400–3600 µg/g).2),3) HA was first purified from the vitreous humor of bovine eyes and named “hyaluronic acid” by Meyer and Palmer in 1934.4) They determined that it is a non-sulfated linear glycosaminoglycan (GAG) consisting of repeating β(1,4)-linked D-glucuronic acid and β(1,3)-linked N-acetyl-D-glucosamine units.5) Because HA commonly exists as a sodium salt in tissues, it was called “sodium hyaluronate” but then re-named as “hyaluronan”6), which is now commonly used.
Because of the ability of HA to retain water and cations, it functions as a space-filler, hydrated cushioning agent, and/or molecular filter in tissues under pathophysiological conditions. However, besides these functions, HA not only provides structural and functional integrity to cells and organs but also exerts various biological effects, which include cell-cell and cell-ECM interactions, migration, proliferation, and apoptosis of cells, immune cell adhesion and activation, epithelial to mesenchymal transition, neovascularization and angiogenesis, and stem cell functions through the formation of loose matrices and intracellular signaling.3),7) These diverse biological activities are dependent on the molecular weight size of HA. For example, high-molecular-weight (HMW) HA of 106–107 Da shows anti-inflammatory and anti-angiogenic effects, whereas lower-molecular-weight (LMW) HA of 103–104 Da or less promotes inflammatory and angiogenic reactions.8)–11) Therefore, the generation of HA species with different molecular sizes mainly by enzymatic depolymerization is an important step for exerting their functions in the extracellular milieu.
It is well established that HA is synthesized as HMW-HA at the plasma membrane by specialized membrane proteins named HA synthases (HASs), which include HAS1, HAS2, and HAS3.12),13) In contrast to HA synthesis, molecular mechanisms for HA catabolism have remained controversial. Members of the hyaluronidase (HYAL) family such as HYAL1, HYAL2, and HYAL3 were originally thought to be responsible for HA degradation. However, as described below, these molecules were not sufficient for HA degradation within tissues, and we identified a new molecule involved in HA degradation, designated hyaluronan-binding protein involved in hyaluronan depolymerization (HYBID), in human skin dermal fibroblasts.14) Four years after our report, another molecule, transmembrane protein 2 (TMEM2), which is a type II membrane protein with high homology to human HYBID, was reported as a cell-surface hyaluronidase in mice by searching the database for mammalian proteins that have similarity to HYBID.15)
In this article, we review current knowledge about HA degradation mediated by HYBID and TMEM2 and their differential roles in HA catabolism in human and mouse tissues. We then describe the roles of HYBID and TMEM2 in HA turnover and degradation in pathophysiological conditions by focusing on the skin and joint tissues. Finally, we discuss unanswered problems about HA catabolism and propose the possibility that inhibition of HYBID function and/or expression may be a useful approach to treat pathological conditions.
2. HA synthesis and the classical model for HA degradation
HA is synthesized as HMW-HA in local tissues by the action of HAS1, HAS2, and/or HAS3, which show a high degree of sequence homology (more than 65% amino acid sequence homology among the three isoforms). They contain seven putative membrane-spanning regions, putative glycosyltransferase catalytic sites, and uridine diphosphate (UDP)-binding motifs.3),9),16) Polymerization of HA is considered to occur on the inner face of the plasma membrane and the product is extruded or translocated through the HAS protein complex into the extracellular space.3),9),16) HMW-HA is secreted around cells and interacts with other ECM macromolecules such as sulfated GAGs linked via link proteins and collagen fibers to form an integrated ECM structure (Fig. 1A). Among the three HAS genes, HAS1 and HAS2 seem to be responsible for the production of HMW-HA in various interstitial cells such as skin dermal fibroblasts.17)

The turnover of HA is generally rapid in most tissues, with approximately one-third of the total body HA replaced daily.9),18),19) However, rates of turnover vary between tissues; the metabolic half-life of HA is 1–2 days in the skin and 1–3 weeks in cartilage, whereas HA in the bloodstream is rapidly metabolized with a half-life of 2–5 minutes.20) The classical model explains HA degradation by the action of HYALs, a family of endoglycosidases that hydrolyze the β-1,4 linkages between N-acetyl-hexamines and glucuronic acid found in GAGs.20)–22) Six HYAL-like genes were reported in the human genome: HYAL1, HYAL2, HYAL3, HYAL4, PH-20/SPAM1, and HYALP1.9),18),22),23) Among them, HYALP1 is a pseudogene and the HA-degrading activity of HYAL3 is questionable.18),24) In addition, HYAL4 appears to act as a chondroitinase with a predominant activity toward chondroitin and chondroitin sulfate, and PH-20/SPAM1 functions as the sperm hyaluronidase localized in the acrosome of spermatids.23) Therefore, the classical pathway for HA catabolism in local tissues is that HMW-HA of >106 Da tethered to cell surfaces by binding to HA receptor CD44 is internalized into caveolin-rich lipid rafts and cleaved by HYAL2 into medium-molecular-weight (MMW) HA fragments of 2.5 × 105–106 Da in acidic microenvironments created by the NA+-H+ exchanger.25) Then, the fragments are further degraded into LMW-HA of 104–2.5 × 105 Da and oligosaccharides by HYAL1 together with other lysosomal β-exoglycosidases within lysosomes.20),21) In this model, the entire process is assumed to occur in a single cell3),20),21) (Fig. 1B). On the other hand, it is also known that considerable amounts of HA drain into the lymphatic system for catabolism in regional lymph nodes.3),18),22),26) Then, the HA fragments digested in the lymphatic system are transferred to the blood stream and eliminated in the liver by receptor-facilitated uptake and catabolism in the hepatic sinusoidal endothelial cells and partially in the kidneys and spleen.3),18),22),26) However, enzymes implicated for HA clearance in the lymph nodes and the liver remain elusive, although HYAL1 is expressed in the liver, kidney and spleen.18)
There are several receptors for HA such as CD44, lymphatic vessel endothelial hyaluronic acid receptor 1 and stabilin-2 (also called hyaluronan receptor for endocytosis [HARE]). Among them, CD44 is well known to function as not only a receptor for HA on the cell surface but also as a cofactor for the hyaluronidase activity of HYAL2.24) In addition, another important aspect of this molecule is that CD44 is a cell surface marker for cancer stem cells.27),28) HA binding to CD44 activates signaling pathways such as the Nanog-Stat3, Oct4-Sox2-Nanog, or Src kinase pathways and regulates cancer stem cell survival, self-renewal, maintenance, and chemoresistance.27) The binding may also contribute to cancer stem cell properties by inducing epithelial to mesenchymal transition through activation of transforming growth factor β (TGF-β) receptor 1.27) However, because CD44 acts as a receptor for other ECM molecules such as osteopontin and is a coreceptor for many growth factors and cytokines and promotes programmed death-ligand 1 expression, diverse signaling pathways generated after interactions of CD44 with these molecules may be involved in cancer stem cell functions.27),28)
3. Discovery of HYBID as a molecule involved in HA degradation
Although HA degradation in local tissues was frequently explained based on the aforementioned model, the CD44/HYAL2-mediated step was inapplicable for HA depolymerization by human dermal fibroblasts in the skin. In our experiments, HA-degrading activity was detected using a cell-based assay, in which the degradation of HMW-HA of >106 Da added to culture media of human dermal fibroblasts was shown by the detection of MMW-HA fragments of 104–105 Da by size-exclusion gel filtration chromatography. The fibroblasts expressed CD44 and HYAL2 but not HYAL1. When CD44 and HYAL2 expression was knocked down using siRNAs there were no changes in HA-degrading activity.14) Therefore, to identify new HA-degrading machinery independent of CD44 and HYAL2, we carried out a comprehensive survey using microarray analysis on genes from dermal fibroblasts stimulated with various proinflammatory factors, and we found that the 25 genes were strikingly upregulated by histamine and downregulated by TGF-β1.14) By knocking down the candidate genes one by one with corresponding siRNAs, we determined that HA-degrading activity was lost by knockdown of KIAA1199, which was originally reported as a deafness gene of unknown function.29) We also demonstrated that transfection of KIAA1199 cDNA into HEK293 (human embryonic kidney) and COS-7 (monkey kidney fibroblast) cell lines, neither of which had HA-degrading activity, conferred the ability to catabolize HA at β-endo-N-acetylglucosamine bonds. In addition, KIAA1199-mediated HA degradation was HA specific, because it had binding capability to HA, but not other sulfated GAGs including chondroitin sulfate A, C, and D, dermatan sulfate, heparin, and heparan sulfate. Unlike HYAL2, KIAA1199 was localized to clathrin-coated pits, and siRNA-mediated knockdown of the clathrin heavy chain and α-adaptin subunit of AP-2, an adaptor protein complex functioning as a major organizer of clathrin coats, diminished HA degradation. Thus, our data indicated that KIAA1199 has a key role in HA degradation via the clathrin-coated pit pathway independently from HYAL2/CD44 in human dermal fibroblasts.14)
4. Naming of HYBID and CEMIP for KIAA1199
According to our data on the role of KIAA1199 in HA degradation, we designated this molecule as HYBID.14),30) On the other hand, Evensen et al. reported that KIAA1199 promotes cell migration of breast and colon cancer cells by forming a complex with a molecular chaperone binding immunoglobulin protein in endoplasmic reticulum31) and through hypoxia-induced upregulation by binding of hypoxia-inducible-factor-2α (HIF-2α) to the promoter region, respectively,32) and they named KIAA1199 as cell migration-inducing protein in the latter reference.32) Because two names were proposed for KIAA1199, the HUGO Gene Nomenclature Committee contacted our and the other groups, and after discussion, they decided, despite our disagreement, to name this molecule as cell migration inducing protein, hyaluronan binding, which has been revised as cell migration inducing hyaluronidase protein (CEMIP) 1 (https://www.genenames.org/data/gene-symbol-report/#!/hgnc_id/HGNC:29213). The term “cell migration inducing” of CEMIP does not represent an essential aspect of this molecule, because cell migration is only a non-specific cellular function and can be induced by various kinds of stimulation. Therefore, we prefer to call this molecule HYBID. However, since more than 250 papers using the names of HYBID, CEMIP, and/or KIAA1199 have been so far published according to PubMed, we use the name of HYBID with an annotation of CEMIP and KIAA1199 in this review article.
5. Structures of HYBID and TMEM2
After publication of our paper on HYBID, Yamamoto et al. reported TMEM2 as a cell-surface hyaluronidase in mouse organs.15) Human HYBID (hHYBID) and mouse HYBID (mHYBID) show 91% overall amino acid sequence homology,33) and they are characterized by the presence of an NH2-terminal signal sequence, one eight conserved glycine residues (G8) domain, a pair of two well-conserved glycine residues (GG) domains, and four parallel beta-helix (PbH1) repeats (Fig. 2). Human TMEM2 (hTMEM2), which has 87% amino acid sequence homology with mouse TMEM2 (mTMEM2),34) consists of an NH2-terminal transmembrane domain, one G8 domain, two GG domains, and three PbH1 repeats15),35) (Fig. 2). Thus, they are highly similar in their domain structure with exceptions of the presence or absence of a transmembrane domain and the number of PbH1 repeats (Fig. 2). HYBID and TMEM2 are occasionally referred to as CEMIP or cell migration-inducing hyaluronidase protein 2 (CEMIP2), respectively.
6. Characteristics of HA degradation mediated by HYBID and TMEM2
Recently acquired evidence has demonstrated that HYBID is a molecule indispensable for HA degradation and TMEM2 itself possesses hyaluronidase activity. However, HA degradation mediated by HYBID requires cell organelles and the degree of HA-degrading activity of TMEM2 varies between animal species.
Cells expressing hHYBID show HA-degrading activity, generating MMW-HA fragments of 104–105 Da from HMW-HA of >106 Da, but cell lysates of hHYBID transfectants and recombinant hHYBID protein lack HA-degrading activity.14) We obtained similar results with mHYBID.33) HA-degrading activity mediated by HYBID was exerted within early endosomes after clathrin-dependent endocytosis, indicating that dynamic microenvironments in living cells are required to exert HA-degrading activity.14),33) However, the molecular mechanism by which HYBID-expressing cells exhibit HA-degrading activity remains unclear at present. Because there are no data regarding the co-localization or interaction of HYBID with HYAL2 or TMEM2 when cells exhibit HA-degrading activity, it seems unlikely that HYBID-mediated HA degradation is derived from the activity of HYAL2 or TMEM2. One possible mechanism is that HYBID may undergo conformational changes into an enzymatically active form when it interacts with other cellular molecules, as previously speculated.36),37) In this context, the data shown by Zhang et al. that secreted HYBID (CEMIP) is tethered to the cell membrane of synovial fibroblasts via binding of its G8 domain to annexin A1 (ANXA1) and exhibits HA-degrading activity on the membrane38) appears to be an attractive hypothesis for the HYBID-mediated HA degradation.
mTMEM2 expressed on the cell membrane of living cells and recombinant protein of the mTMEM2 extracellular domain degraded HMW-HA into HA fragments of ∼5 × 103 Da,15) indicating that mTMEM2 is a cell-surface hyaluronidase. However, conflicting results have been reported regarding the HA-degrading activity of hTMEM2. We have shown that unlike mTMEM2, hTMEM2 fails to degrade exogenously added HMW-HA in a cell-based assay using either hTMEM2-expressing human cells including dermal fibroblasts, epidermal keratinocytes, chondrocytes, and TGF-β-stimulated HEK293 cells34),39)–43) or hTMEM2-transfected HEK293T cells.34),39) Like hTMEM2, similar results were obtained with Heterocephalus glaber (naked-mole rat [nmr]) TMEM2.39) This animal is a special mouse species and characterized by the accumulation of extra-large HMW-HA in many organs.44) On the other hand, Narita et al. reported that recombinant hTMEM2 ectodomain and enriched membrane-fractions of HEK293 cells expressing hTMEM2 have definite HA-degrading activity.45) However, in this study, the activity was measured in unusual conditions, i.e. an enzyme/substrate ratio of 10:1 and no activity was shown in a cell-based assay by culturing hTMEM2-expressing HEK293T cells with HMW-HA.45) Nie et al. originally described that the hTMEM2 ectodomain lacks hyaluronidase activity, but recently published a corrigendum that hTMEM2 has very weak hyaluronidase activity when it was incubated with HMW-HA at an enzyme/substrate ratio of 1:1.35) Our semi-quantitative analysis using enriched membrane-fractions prepared from HEK293T cells expressing each TMEM2 species showed that the estimated HA-degrading activity of hTMEM2 and nmrTMEM2 was 14-fold and 20-fold less than that of mTMEM2, respectively,39) suggesting that highly concentrated hTMEM2 on the cell membrane shows weak activity. It was reported that human osteosarcoma cells expressing high levels of hTMEM2 degrade substrate-immobilized HA at focal adhesion sites using in situ HA degradation assays and hTMEM2 deletion inhibits the HA-degrading activity and tumor cell attachment and migration in an HA-rich environment.46) However, in this study, the recovery of diminished HA-degrading activity and changes in cell behavior by siRNA-mediated knockdown of hTMEM2 was shown by transfecting the mTMEM2 gene.46) Thus, the study did not directly prove cellular HA-degrading activity of hTMEM2. In addition, hTMEM2 knockdown in human glioma cells is known to induce the expression of genes related to ECM modulation and cell adhesion such as matrix metalloproteinase (MMP) 2, N-cadherin, and E-cadherin.47) Therefore, these results suggested that the role of hTMEM2-mediated HA degradation in living cells remains obscure. On the other hand, our studies on human dermal fibroblasts and chondrocytes expressing both TMEM2 and HYBID demonstrated that HA-degrading activity was unchanged by knockdown of hTMEM2 but lost after hHYBID knockdown.40)–42) Altogether, although not conclusive, these data show that the degree of TMEM2 activity is dependent on the animal species and suggest that the HA-degrading activity of hTMEM2 at the cellular level is insufficient in human tissues.
7. Role of the GG-1 domain in HA degradation mediated by HYBID and TMEM2
HYBID and TMEM2 share a domain structure composed of one G8 domain, two GG (GG-1 and GG-2) domains, and four or three PbH1 repeats (Fig. 2), but their functional roles remain elusive. Our previous study demonstrated that a deafness missense mutation (R187C and R187H) of the GG-1 domain of hHYBID resulted in the loss of HA-degrading activity, suggesting the involvement of this domain in HA degradation.14) In addition, mutations R265C, D273N, and D286N in mTMEM2, which correspond to amino acid residues within the GG-1 domain of hHYBID, are known to significantly reduce HA-degrading activity.15) Thus, we carried out experiments using HEK293T cells expressing chimeric constructs targeting the GG-1 domain of TMEM2 (Chimeras 1–4) to examine its implication on HA-degrading activity34) (Fig. 3). Like mTMEM2, Chimera 1 composed of human G8 (hG8), mouse GG-1 (mGG-1), and the mouse C-terminal tail containing PbH1 repeats and GG-2 domain was active in HA degradation in a cell-based assay. However, no HA-degrading activity was detected with other chimeras: Chimera 2 composed of hG8, hGG-1, and mouse C-terminal tail; Chimera 3 composed of mG8, mGG-1, and human C-terminal tail; and Chimera 4 comprised of mG8, hGG-1, and human C-terminal tail (Fig. 3). These indicate that the co-existence of mGG-1 and mouse C-terminal tail is critical for the HA-degrading activity.34) The results were consistent with a crystallographic study of hTMEM2, in which the hGG-1 domain was involved in HA binding and one of two Ca2+ binding Glu residues in PbH1 repeats was lacked by comparison with other active glycotransferases.35)

Living cells expressing mHYBID, hHYBID, or mTMEM2 were proven to show HA-degrading activity,14),15),34),39)–42),48) but no activity was detected by expression of hTMEM2 and nmrTMEM2 in a cell-based assay.34),39) Thus, we compared the amino acid sequences of these species’ GG-1 domains.34),39) As shown in Fig. 4, five amino acid residues (His, Val, Ala, Lys, and Ser) in the GG-1 domain were conserved in mHYBID, hHYBID, and mTMEM2 but not in hTMEM2 and nmrTMEM2. Among these, the co-existence of a His residue (His248 for mTMEM2 and His169 for mHYBID and hHYBID) and an Ala residue (Ala303 for mTMEM2 and Ala225 for mHYBID and hHYBID) was critical for HA-degrading activity.34) Double substitution of His248/Ala303 of the mTMEM2 GG-1 domain to Asn248/Phe303 (mTMEM2 GG1-DS1) resulted in the loss of HA-degrading activity (Fig. 3).34) Notably, double substitution of Asn248/Phe303 in the hTMEM2 GG-1 domain in Chimera 2 to His248/Ala303 (Chimera 2 GG1-DS2) resulted in the acquisition of HA-degrading activity (Fig. 3).34),39) These data suggested that both amino acid residues are indispensable for HA-degrading activity, and the loss of cellular HA-degrading activity of hTMEM2 and nmrTMEM2 is due to the replacement of His248 and Ala303 with Asn248 and Phe303 in hTMEM2, and Asn247 and Val302 in mTMEM2, respectively.

The G8 domain was predicted to have a role in extracellular ligand binding,49) and the G8 domain of hHYBID was reported to interact with ANXA1 and colocalize at the cell membrane, possibly contributing to the exhibition of HA-degrading activity.38) In addition, the PbH1 repeats are speculated to function in polysaccharide hydrolysis.14) Although an increasing number of proteins have been reported to interact with HYBID,37) information about the domains responsible for the interactions and/or their relation to HA degradation is still limited. Thus, further studies are needed to clarify the possible roles of the G8 domain and PbH1 repeats in HA degradation.
8. Hypothesis of HA degradation mediated by HYBID and TMEM2 in mice and humans
Induced global mTmem2 knockout (KO) mice show accumulation of a large amount of HA in circulating blood and various organs, suggesting that mTMEM2 has a major role in systemic HA catabolism and turnover in mouse tissues in physiological conditions.50) Indeed, mTmem2 is widely distributed in many mouse organs such as the lung, liver, gastrointestinal tract, kidney, brain, spleen, lymph nodes, and synovium.15) mTMEM2 is also abundantly expressed in endothelial cells of the liver sinusoids and marginal lymphatic sinuses of the lymph node and is prerequisite for the degradation of HA injected into lymphatic and vascular vessels. Thus, mTMEM2 is considered to be responsible for the systemic catabolism and turnover of HA by degradation in the liver and lymphatic system.50) Compared with mTmem2, mHybid seems to be localized to a limited number of organs including the brain, bone, synovium, and skin in mice.11),15),51),52) mHybid KO mice exhibit a mild phenotype showing transiently shorter long bones during development due to impaired endochondral ossification accompanied by local HMW-HA accumulation in the growth plate.11) In the brain, both mTmem2 and mHybid are expressed to a similar extent.15) Analyses of mTmem2 KO mice indicated the importance of mTMEM2-mediated HA catabolism in neural crest cell development and survival,53) whereas mHybid KO mice showed the involvement of mHYBID in increasing dendritic spine density in the dentate gyrus through decreased HA.52) The phenotypes of these KO mice may reflect the different localization of mTMEM2 and mHYBID within the body. In physiological conditions in mouse tissues, it seems likely that mTMEM2 is responsible for the turnover of HA in many tissues through degradation in both local tissues and lymph nodes and liver, and mHYBID plays an important role in endochondral ossification of the developing long bones, in which mHYBID might be upregulated by factors generated in the growth plate.
On the other hand, HA-degrading activity of hTMEM2 in living physiological cells has not been confirmed directly,39)–43) although only weak HA-degrading activity was detected using recombinant human soluble TMEM2 in solution or membrane-fractions of HEK293T cells overexpressing hTMEM2.34),35),39) Therefore, HA-degradation in human cells and tissues seems to be different from the mouse system. Knockdown of hHYBID in human dermal fibroblasts,14) synovial fibroblasts,14),54) or chondrocytes42),48) resulted in the loss of HA degradation without changing hTMEM2 expression, indicating that hHYBID plays a key role in HA degradation in these cells of the skin, synovium, and cartilage. In a sharp contrast, HA degradation was not changed when hTMEM2 was knocked down in chondrocytes42) or synovial fibroblasts (unpublished data). In human dermal fibroblasts, knockdown of hTMEM2 even increased HA degradation, although HA-degrading activity was completely lost by hHYBID knockdown.40) These data strongly suggested that unlike in mouse tissues, hHYBID, but not hTMEM2, contributes to HA degradation in the human tissues such as the skin, synovium, and cartilage (Fig. 5A).
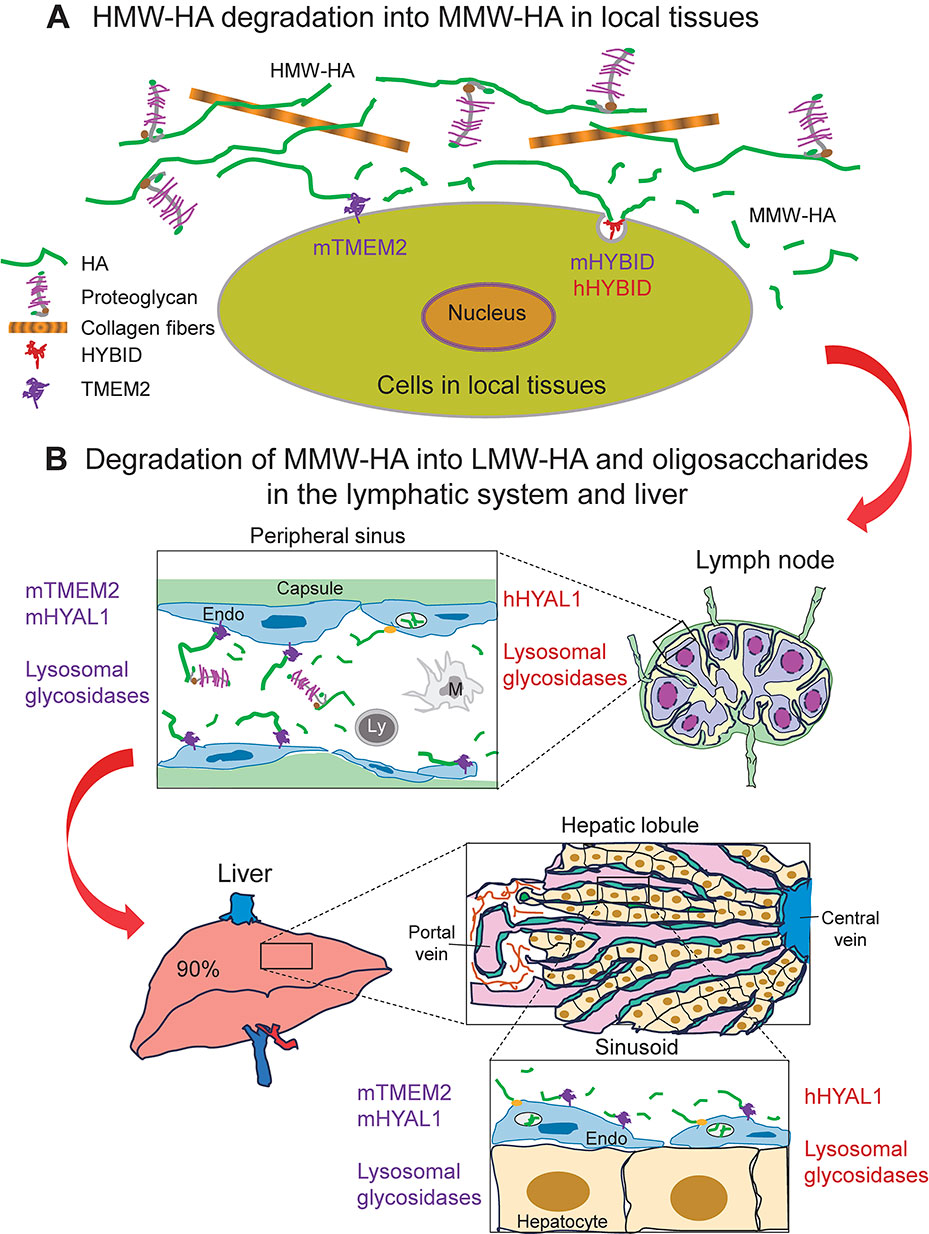
After degradation of HMW-HA into MMW-HA fragments in the local tissues, the HA fragments are released from the ECM, drained into lymphatic vessels and reach the regional lymph nodes, in endothelial cells of which MMW-HA may be further metabolized into LMW-HA by the actions of mTMEM2 and mHYAL1 in mice and hHYAL1 in humans (Fig. 5B). These HA fragments reach the general circulation and are taken up and catabolized predominantly in the liver, in which sinusoidal endothelial cells are responsible for HA uptake via HA receptors such as HARE and HYAL1-dependent degradation in lysosomes, although a minor portion is taken up by the kidney and spleen50) (Fig. 5B). Similar to the process in the lymphatic system, HA fragments in the liver may be further degraded by mTMEM2 and mHYAL1 in mice and hHYAL1 in humans (Fig. 5B). However, information about degradation processes of HA fragments in the lymphatic system and liver is still limited and further detailed studies are needed.
9. Regulation of HYBID and TMEM2 expression by proinflammatory factors and signaling pathways
In inflammatory conditions such as arthritis and skin photoaging, hHYBID is overexpressed compared with corresponding non-inflammatory tissues. In contrast, hTMEM2 is constitutively expressed in inflammatory and normal joint tissues. These data suggested that proinflammatory factors may stimulate the expression of hHYBID but not hTMEM2. In human chondrocytes, interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) upregulated HYBID expression and additively increased the expression through their combined treatment,42),48) although other factors including TGF-β, IL-1, insulin-like growth factor-1 (IGF-1), basic fibroblast growth factor (bFGF), histamine and prostaglandin E2 (PGE2) had negligible effects on expression. In human synovial fibroblasts, IL-6 was also an effective stimulator for HYBID expression.54) Although histamine and TGF-β upregulate and downregulate HYBID expression, respectively, other factors including IL-1, IL-8, TNF-α, vascular endothelial growth factor (VEGF), bFGF, PGE2, and IGF-1 showed negligible effects.54) On the other hand, hTMEM2 expression in chondrocytes and synovial fibroblasts was unchanged by various factors, including TGF-β, IL-6, IL-1α, IL-8, TNF-α, VEGF, bFGF, PGE2, IGF-1, and histamine.42),54) Based on these data, it is evident that HYBID and TMEM2 have the contrasting expression patterns in response to proinflammatory factors and HYBID expression in chondrocytes and synovial fibroblasts is upregulated by inflammatory factors especially IL-6.
In human dermal fibroblasts, platelet-derived growth factor-BB (PDGF-BB), bFGF, epidermal growth factor (EGF), and TGF-β1 are well known to increase HA production by enhancing HAS-mediated HA synthesis.17),55),56) Among these, TGF-β1 almost completely inhibited HYBID expression, whereas PDGF-BB, bFGF and EGF caused mild to moderate suppression of expression in dermal fibroblasts.30) Notably, molecular sizes of newly synthesized HA are controlled by the expression levels of HYBID: Treatment with TGF-β1 resulted in HMW-HA formation, whereas PDGF-BB, bFGF, or EGF causes the production of LMW-HA fragments, which is recovered by knockdown of HYBID.30) Therefore, HYBID is a key regulator for the molecular sizes of newly formed HA by dermal fibroblasts. In addition, histamine increased HA degradation by upregulating HYBID and downregulating HAS2 in dermal fibroblasts and thereby decreased the total amount and sizes of newly produced HA.57) On the other hand, TGF-β1 and IL-1β, but not EGF, bFGF, or PDGF-BB, upregulated hTMEM2 gene expression in human dermal fibroblasts.40),41) Notably, the upregulation of hTMEM2 by TGF-β1 and IL-1β was associated with the downregulation of HYBID expression.40),41) In addition, the knockdown of hTMEM2 increased HYBID expression but decreased HAS2 expression, suggesting that hTMEM2 may be a positive regulator for HMW-HA accumulation. Kobori et al. recently reported supporting data that hTMEM2 knockdown led to decreased HA production in human bronchial cell line BEAS-2B cells.58) Comparative analysis of the 3D structures of the intracellular domains of hTMEM2 and mTMEM2 showed the existence of a clear α-helix structure only in hTMEM2. In addition, a proline-rich sequence (PPPPPP) as a binding site for the SH3 domain and a nuclear migration motif (KQKRHK) were found in the central region of the hTMEM2 intracellular domain and the C-terminus, respectively.34) Thus, these findings suggested that the hTMEM2 intracellular domain may regulate intracellular signaling pathways by interacting with tyrosine kinases of the SH3 domain or may regulate a pathway of genes including HYBID and HAS2.34) However, further detailed studies are needed to clarify this hypothesis.
HYBID expression is also known to involve epigenetic mechanisms, including hHYBID overexpression resulting from hypomethylation of the hHYBID promoter region,59) hypoxia-induced hHYBID expression by binding of HIF-2α to the hypoxia responsible element through increased presence of H3K4me3 in the promoter,32) and regulation of hHYBID expression by several microRNAs.37) In addition, the hHYBID promoter includes several cis-acting elements for transcription factors such as activator protein-1, nuclear factor-κB, and T-cell factor 4. TNF-α-TNF receptor 1-complex I signaling is suggested to be involved in TNF-α-induced hHYBID expression.48) However, hHYBID expression via these epigenetic and transcription factor-mediated signaling pathways may be commonly observed in cancer cells and related to their biological behaviors such as cell migration and proliferation other than HA degradation.32),37),59)–61) On the other hand, TMEM2 is up-regulated in human cancer cell lines46) and embryonic mouse tissues15) through the direct binding of SOX4 to the promoter region of TMEM2. However, the regulatory mechanism of the constitutive expression of TMEM2 in human adult tissues remains to be determined by future studies.
10. Roles of HYBID, TMEM2, and HYALs in HA degradation in pathophysiological conditions
The skin, the biggest organ in the body, is composed of dermis and epidermis, in which HA is present in both as a major ECM component and is metabolized by mechanisms with different regulatory pathways. Joints are defined as structures where two or more bones are connected, and they classified into three different types, i.e. fibrous joint (fixed joint, synarthrosis), cartilaginous joint (slightly movable joint, amphiarthrosis) and synovial joint (freely movable joint, diarthrosis). Among these, synovial joints are the most common type in the body, and the synovial membrane produces a high concentration of HMW-HA, which plays a critical role in joint movement. In this article, we focus on HA metabolism mediated by HYBID and TMEM2 in the skin and synovial joint tissues in physiological and inflammatory conditions.
Many articles have described that HYBID (CEMIP) is overexpressed in various cancers and plays roles in cancer cell proliferation, progression, and metastasis,31),37),61)–66) whereas information about the implication of TMEM2 in cancers is still limited.67),68) The functional roles of HYBID in cancer development and progression are diverse and HYBID influences the tumor microenvironment through various biological phenomena such as hypoxia, angiogenesis, epithelial to mesenchymal transition, and signaling pathway changes as well as hyaluronan degradation. More detailed information on the roles of HYBID in cancers is available in recently published review articles.37),61),66)
10.1. Physiological HA turnover in the human skin.
In human dermis, a large amount of HA is present as HMW-HA (4–6 × 106 Da, 400–500 µg/g tissue) along with collagen and elastin in physiological conditions. Human dermal fibroblasts synthesize HMW-HA predominantly by the action of HAS2 and also express hHYBID to metabolize HMW-HA to MMW-HA (104–105 Da). Although hHYAL2 and hTMEM2, but not hHYAL1, are expressed in dermal fibroblasts, they have a negligible role in HA degradation, and hTMEM2 appears to function as a regulator to increase HMW-HA by suppressing HYBID expression and enhancing HAS2 expression.39)–41) The regulatory function of hTMEM2 is supported by evidence that TGF-β, a stimulator for production of ECM macromolecules including HA, upregulates hTMEM2 and HAS2, whereas it inhibits hHYBID expression.14),17),30),40),41) Therefore, hHYBID and hTMEM2, but not hHYAL1 or hHYAL2, are considered to play a major role in the HA degradation and metabolic regulation, respectively, in human dermis in physiological conditions (Fig. 6). After HYBID-mediated depolymerization of HMW-HA, MMW-HA fragments are transported to the lymphatic system and further degraded to LMW-HA (<104 Da) in the regional lymph nodes and finally in the liver, kidney, and spleen by hHYAL1-dependent degradation in lysosomes, as illustrated in Fig. 5B.69)–71)

In human epidermis, a high concentration of HA (∼2 mg/ml) is known to exist in the intercellular spaces among keratinocytes.72) The epidermis is composed of several layers, i.e. stratum basale, stratum spinosum, stratum granulosum, and stratum corneum consisting of a cornified layer, all of which are formed by the differentiation of basal cells into keratinocytes in upper layers. Basal cells are responsible for the production of HMW-HA of >106 Da in a HAS3-dependent manner, whereas neither HAS1 nor HAS2 is involved in HA production by keratinocytes.43),73)–75) Notably, contrary to dermal fibroblasts, TGF-β and hTMEM2 decrease keratinocyte HA production by downregulating HAS3 expression.43),73) During differentiation, HMW-HA is catabolized to LMW-HA of <104 Da, which is located in the cornified layer and subsequently lost by desquamation76) (Fig. 6). Unlike the dermis, hHYBID is not expressed by human epidermal keratinocytes and thus is not involved in HA degradation in the epidermis. A recent study revealed that human keratinocytes secrete hHYAL1 and degrade HMW-HA in acidic conditions at pH 4.1–4.6 (Fig. 6), which is found in healthy stratum corneum.43) HYAL1 is known to be secreted independently of lysosomes, even in conditions in which the typical lysosomal protease cathepsin D remains present intracellularly.77),78) The data suggest that HA metabolism in the dermis and epidermis is carried out separately by local digestion followed by systemic turnover and surface desquamation, respectively (Fig. 6). However, further studies are needed to provide direct evidence that this degradation process occurs in human skin in vivo and what mechanisms are implicated in hHYAL1 secretion from epidermal keratinocytes. In addition, little information is available for HA degradation in the mouse dermis and epidermis, except for our report on UVB-induced HA depolymerization in mouse epidermis associated with increased Hyal1 gene expression.79)
10.2. Pathological HA degradation in photoaged skin and inflammatory skin diseases.
Chronic ultraviolet irradiation disrupts the homeostasis of the skin and promotes skin photoaging, which is clinically recognized by wrinkles, sagging, increased fragility, rough skin texture, and uneven pigmentation. Major histological changes in advanced photoaged skin are irregular and disorganized collagen fibers and massive accumulation of aberrant elastic materials in the dermis, referred to as solar elastosis (Fig. 7). These abnormal changes in dermal fibrillary components were thought to be caused by both increased degradation of collagen and elastic fibers by ECM-degrading proteinases such as MMPs and de novo synthesis of elastin and its associated molecules.80) However, collagen and elastin fibers in the dermis closely interact with and are embedded in the network structures composed of HA and proteoglycans such as versican. Thus, prior to breakdown of collagen and elastin fibers, HA and proteoglycans in the network seem to be initially degraded, facilitating the susceptibility of these fibrous components to proteinases.80),81)

To overcome the problem in analytical methods of HA data and/or photoaging severity in skin tissue samples,82)–84) we carried out biochemical analysis of the amount and molecular size of HA and histochemical localization of HA in paired biopsy skin specimens from the photoaged corner of the eye and the photoprotected inner arm of the same Japanese women (Fig. 7). As a result, HA was found to be decreased in the superficial papillary dermis of photoaged skin (HA amount, 0.7 µg/mg tissue dry weight; peak HA molecular weight, 980 kDa), in contrast to abundant HA present in the papillary dermis of the photoprotected skin (HA amount, 1.7 µg/mg tissue dry weight; peak HA molecular weight, 1,840 kDa),85) and HA levels in the papillary dermis in the photoaged eye-corners were negatively correlated with skin wrinkling and skin sagging index.85) In addition, HYBID mRNA expression was increased in photoaged skin and was negatively and positively correlated with HA levels in the papillary dermis of photoaged skin and the severity of skin wrinkling and sagging, respectively.85) Because the water-attracting property of HA provides a swelling pressure in the ECM, regulates osmotic pressure and ion flow, and stabilizes dermal structure,2),86) partially degraded and reduced HA in the papillary dermis may weaken the recoil capacity and tensile strength of collagen and elastic fibers. Thus, a reduction in size and level of HA due to accelerated HA degradation mediated by HYBID in the papillary dermis of photoaged skin may lead to the formation of photoaging symptoms such as skin wrinkling and sagging, possibly both through decreased water binding, viscosity and turgidity of HA, and by disruption of the integrity of dermal ECM in association with deteriorated collagen and elastic fibers.
Caucasian women are suggested to have earlier onset and greater skin wrinkling than East Asian women.87),88) Thus, we examined the relationship of HA level and/or HYBID immunohistochemical data with the severity of skin wrinkling at the outer corner of the eye of Caucasian females (mean age, 62.2 years) by conducting skin wrinkling measurement and skin biopsy of the same areas (Fig. 7).89) Similar to Japanese women (mean age, 69.1 years),85) immunostaining of HYBID (ratio of immunoreactive cells to total cells) was positively correlated with the degree of skin wrinkling in Caucasian women, and HA level in the papillary dermis was significantly decreased in skin with high HYBID expression in dermal fibroblasts compared with that with low HYBID expression.89) Our data suggest that aberrantly increased HYBID-mediated HA degradation in the papillary dermis at photoaged eye-corners may have an ethnically common basis with wrinkle formation at least in Caucasian and Japanese women aged over 60 years.
HA plays an important role in wound healing, which is one of the most common pathological conditions of the skin. Although wound healing in adult skin frequently results in a partial recovery with scar formation, fetal skin is known to regenerate without scarring.90) This phenomenon of fetal scarless wound healing is associated with a high amount of HA in the wound area and suggested to be attributed to overproduction of HA by fetal fibroblasts.90) In the early phases of skin wound healing, HA is present mainly as LMW-HA fragments, which promote inflammation and angiogenesis91)–94) and cell migration.95) At later stages, the provisional matrix becomes enriched with synthesized HMW-HA, which contributes to tissue remodeling.90) During these processes, several growth factors and cytokines including PDGF, bFGF, EGF, TGF-β, and IL-6 are overproduced by inflammatory cells, keratinocytes and dermal fibroblasts and involved in HA metabolism observed in wound healing.30),96) LMW-HA fragments are speculated to be generated by the degradation of synthesized HMW-HA by the actions of reactive oxygen species and/or HYALs,90) but no direct data are available for degradation by these factors or the implications of HYBID.
Bacterial infection is another common pathological condition of the skin. In experimental skin infection models using wild-type and Hybid knockout mice, infection by Staphylococcus aureus (S. aureus), a major bacterial pathogen responsible for invasive infections of human skin, is remarkably inhibited by accumulation of HMW-HA, enhanced adipogenesis, increased production of the antimicrobial peptide cathelicidin, and inflammatory reactions in the inoculated site of the skin in Hybid KO mice, and HA degradation by host dermal fibroblast-derived HYBID, which is overexpressed by histamine from mast cells, enables greater proliferation and infection of S. aureus.97) These data suggested a new role for HYBID-mediated HA degradation in the dermal ECM to regulate host defense against S. aureus skin infection.97) HA is also known to increase in the skin of patients with autoimmune diseases such as scleroderma, dermatomyositis, lupus erythematosus, and patients with inflammatory diseases such as psoriasis and atopic dermatitis.90) However, little is known about the molecular sizes of HA and molecules involved in HA degradation such as HYBID in these pathological conditions of the skin. Further studies are needed to elucidate the molecular mechanism of the HA metabolism in these skin diseases.
10.3. Physiological HA turnover in joint tissues.
During embryogenesis, the joint cavity of the synovial joint is formed as HA-filled narrow intercellular clefts in the presumptive joint region, leading to a continuous lumen through their coalescence. Thus, the cavity appears to be formed by the accumulation of HA in the synovial primordium.98) In postnatal joints, the joint cavity is surrounded by articular cartilage and synovium and is filled with synovial fluid that contains a large amount of HA (1400–3600 µg/g) and a protein-rich ultrafiltrate of blood plasma. HA in synovial fluid is synthesized as HMW-HA by the synovial membrane and plays roles in maintaining lubrication for joint movement, shock absorption to mechanical stress and cell signaling via HMW-HA receptors on synovial cells and chondrocytes.99),100) HA is also a major constituent of the ECM in articular cartilage and present as an HA-aggrecan network structure, in which fibrillar collagen fibers are located.101) This well-organized structure of the HA-aggrecan network and collagen fibers is essential to maintaining the integrity of cartilage ECM structure, hydrated cushioning, and resilient articular cartilage.101) Although little information is available, low-level expression of hHYBID in normal synovium54) and articular cartilage42),48) suggests that hHYBID may be involved in the turnover of HA in synovial fluid and articular cartilage. However, the role of mHYBID and mTMEM2 in the murine joint remains unknown.
10.4. HA degradation in the articular cartilage and synovium of arthritic joints.
We reported for the first time that HYBID is overexpressed in synovial cells from patients with osteoarthritis (OA) or rheumatoid arthritis (RA),14) both of which are the two major arthritides. Overproduction of HYBID by synovial cells in patients with RA has been confirmed from data that HYBID levels in the serum and synovial fluid samples are remarkably higher than those in normal subjects.38) OA is the most common joint disease in the elderly and is characterized by progressive degradation of articular cartilage ECM. The initial pathological change of articular cartilage in OA joints is depletion of the HA-aggrecan network, which is followed by degradation of the collagen fibers, causing fibrillation and laceration.102),103) Because the HA-aggrecan network structure has water regain properties, which provide hydrated cushioning and resilience functions to cartilage, degradation of the structure not only causes the destruction of the integrity of cartilage ECM but also increases the susceptibility of cartilage to mechanical stress. The MMP family members with collagenolytic activity such as MMP-1, MMP-13, and MMP-14 are responsible for the degradation of fibrillar collagens,102) and a disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4) and ADAMTS5, which are also referred to as aggrecanase-1 and aggrecanase-2, respectively, appear to play a central role in aggrecan degradation in OA articular cartilage.102) For the degradation of HA in OA cartilage, our studies demonstrated the implication of HYBID, but not HYAL1, HYAL2, or TMEM2.48) mRNA expression levels of HYBID, HYAL1, and HYAL2 were increased compared with normal cartilage, whereas TMEM2 was constitutively expressed, showing similar levels between OA and normal cartilage.48) HYBID immunoreactivity showed a direct correlation with Mankin score (degree of cartilage destruction), whereas TMEM2 was diffusely expressed by chondrocytes in OA and normal cartilage.42) Importantly, HA-degrading activity of OA chondrocytes was lost by knockdown of HYBID but not HYAL1, HYAL2, or TMEM2.48) Accordingly, these data strongly suggested that HA degradation in OA cartilage is ascribed mainly to HYBID expressed by chondrocytes in OA cartilage (Fig. 8).

OA was previously defined as an intrinsic degenerative “wear and tear” disease of articular cartilage. However, recently accumulated data indicated that OA is an inflammatory disease involving many inflammatory mediators produced by cartilage, subchondral bone, and synovium.104) Among the inflammatory processes implicated in OA, synovitis is considered to be an important progression factor for early-stage OA. Our study provided data that HYBID was upregulated in OA synovium and correlated with increased ratios of LMW-HA in synovial fluids from patients with OA.54) We have also shown that HA-degrading activity of OA synovial fibroblasts was abolished by knockdown of HYBID.54) HMW-HA in synovial fluid plays an essential role in not only maintaining the viscosity and shock-absorption function99),100) but also exhibiting anti-inflammatory and anti-angiogenic activity.93),94),105) Therefore, degradation of HMW-HA in OA synovial fluid seems to promote the accelerated damage of articular cartilage mediated by increased mechanical strain on the articular surfaces and persistent synovitis (Fig. 8). The data suggested that destruction of articular cartilage due to HYBID-mediated HA degradation in early-stage OA is carried out through two different pathways: the direct destruction pathway by digestion of HA in the articular cartilage and the indirect destruction pathway by degradation of HA in synovial fluid (Fig. 8).
11. HYBID as a diagnostic and/or therapeutic target
HYBID is a secreted protein; therefore, HYBID can be detected in the circulation when it is overproduced in local tissues, and thus measurement of HYBID might be useful for the diagnosis of diseases. In fact, compared with control healthy subjects, increased levels of HYBID (CEMIP/KIAA1199) in circulating plasma were reported in patients with idiopathic pulmonary fibrosis,106) RA,38) and osteoporosis-associated fracture,107) in all of which HYBID was overexpressed by cells in the affected organs, i.e. the lung, joints, and bone/bone marrow. Although detailed analytical studies with assays for HYBID in plasma samples using a large number of patients and control subjects are needed, HYBID may be a potential biomarker to assess the risks of pulmonary fibrosis, RA, or osteoporotic fracture and/or to monitor disease activities.
HYBID is involved in HA degradation in various inflammatory diseases61) and cancers.37) Therefore, several agents have been developed to inhibit HA-degrading activity and/or the expression of HYBID. Neutralizing monoclonal antibodies against HYBID (KIAA1199/CEMIP) were shown to be effective for improving the clinical and histological scores of collagen-induced arthritis in mice.38) Sulfated HA derivatives were reported to potently inhibit HA degradation mediated by HYBID (CEMIP) in fibroblasts.108) Concerning the inhibition of HYBID expression, various agents such as GSK-J4 (an inhibitor of demethylase), NS398 (cyclooxygenase-2 inhibitor) and HYBID-regulating miRNAs, all of which modulate the epigenetic regulation of HYBID expression, have been shown to downregulate HYBID expression.37) According to transcriptome profiling analyses of patients with idiopathic pulmonary fibrosis treated with pirfenidone (an anti-fibrotic and anti-inflammatory drug for idiopathic pulmonary fibrosis), this drug was reported to downregulate HYBID expression and increase HA levels in the lungs.106) On the other hand, we have discovered that extracts from Geranium thunbergii and Sanguisorba officinalis root, both of which are traditionally used as herbal or folk medicines, not only inhibited HYBID-mediated HA degradation but also downregulated HYBID gene expression in dermal fibroblasts, leading to the production of HMW-HA.109),110) In addition, these extracts showed anti-wrinkle activity in small-scale clinical trials, in which the Japanese women underwent topical application of test lotion formulated with either extract on the outer eye corner on one side of the face and the placebo lotion on the other side. The data suggested that ointment or cosmetics containing these inhibitory agents against HYBID-mediated HA degradation may be a promising remedy to improve photoaging symptoms such as skin wrinkling. IL-6 is a strong stimulator of HYBID expression in OA synovial fibroblasts and chondrocytes42),54) and increased levels of IL-6 were reported in OA synovial membrane111) and synovial fluid;54) therefore, drugs targeting IL-6 including anti-IL6 receptor antibodies may be a good target to suppress HYBID expression in patients with early-stage OA.
12. Conclusions
After discovery of HYBID and TMEM2, research on HA metabolism has progressed extensively. Prospective roles of HYBID and TMEM2 in HA degradation and their relevant cell behaviors in various pathological conditions including inflammation and cancers have been reported. Unlike the previously proposed model for HA degradation mediated by HYAL2/CD44 and HYAL1, growing lines of evidence have shown that hHYBID plays an important role in HA degradation within human tissues in various pathophysiological conditions, and both mHYBID and mTMEM2 are implicated in HA degradation in mouse tissues. However, there are several questions that need to be answered to better understand the mechanisms of HA degradation by HYBID and TMEM2 in vivo. Recombinant hHYBID and mHYBID proteins by themselves do not exhibit HA-degrading activity, but they require interactions with cell organelles such as clathrin-coated pits and/or membrane protein ANXA1 to exert their HA-degrading activity. Therefore, further molecular biological studies on HYBID are necessary to clarify how HA-degrading activity is exerted. mTMEM2 is an efficient HA-degrading enzyme, but debate remains about the HA-degrading activity of hTMEM2. Although recombinant hTMEM2 in solution or enriched membrane-fractions exhibited weak activity, the HA-degrading activity of hTMEM2 expressing on living cells was not detectable in the conditions in which mTMEM2 degrades HA. These data suggested the possibility that mTMEM2 may play a central role in physiological HA turnover in mouse tissues, whereas the role of hTMEM2 in human tissues awaits further investigation. In addition, HA degradation mediated by HYBID and TMEM2 is a cell-associated event, indicating efficient HA degradation around cells. However, it remains unknown how HA located distant from cell surfaces, e.g. HA present in the interterritorial zone of articular cartilage, is degraded and by which enzymes. Furthermore, there are no direct data about whether HA in HA-proteoglycan complexes, such as the HA-aggrecan network, is readily degraded by HYBID and TMEM2. Therefore, further detailed studies are needed to understand the complete story about HA metabolism in local tissues.
Conflict of interest
H.Y. is an employee of Kao Corporation and H.Y. and S.I. are holders of the HYBID patent. The Department of Pathophysiology for Locomotive Diseases, Juntendo University Graduate School of Medicine is supported by funding from 11 companies.
Funding
This study was supported by a Japan Society for the Promotion of Science Grant-in-aid for Scientific Research (JSPS KAKENHI) grant (JP22K07029 to Y. Okada).
Notes
Edited by Kohei MIYAZONO, M.J.A.
Correspondence should be addressed to: Y. Okada, Department of Pathophysiology for Locomotive Diseases, Juntendo University Graduate School of Medicine, 2-1-1 Hongo, Bunkyo-ku, Tokyo 113-8421 Japan (e-mail: ya-okada@juntendo.ac.jp).
References
- 1) Mouw, J.K., Ou, G. and Weaver, V.M. (2014) Extracellular matrix assembly: a multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 15, 771–785.
- 2) Papakonstantinou, E., Roth, M. and Karakiulakis, G. (2012) Hyaluronic acid: A key molecule in skin aging. Dermatoendocrinology 4, 253–258.
- 3) Dicker, K.T., Gurski, L.A., Pradhan-Bhatt, S., Witt, R.L., Farach-Carson, M.C. and Jia, X. (2014) Hyaluronan: a simple polysaccharide with diverse biological functions. Acta Biomater. 10, 1558–1570.
- 4) Meyer, K. and Palmer, J.W. (1934) The polysaccharide of the vitreous humor. J. Biol. Chem. 107, 629–634.
- 5) Meyer, K. (1958) Chemical structure of hyaluronic acid. Fed. Proc. 17, 1075–1077.
- 6) Balazs, E.A., Laurent, T.C. and Jeanloz, R.W. (1986) Nomenclature of hyaluronic acid. Biochem. J. 235, 903.
- 7) Tammi, M.I., Oikari, S., Pasonen-Seppanen, S., Rilla, K., Auvinen, P. and Tammi, R.H. (2019) Activated hyaluronan metabolism in the tumor matrix Causes and consequences. Matrix Biol. 78–79, 147–164.
- 8) Cyphert, J.M., Trempus, C.S. and Garantziotis, S. (2015) Size matters: Molecular weight specificity of hyaluronan effects in cell biology. Int. J. Cell Biol. 2015, 563818.
- 9) Tavianatou, A.G., Caon, I., Franchi, M., Piperigkou, Z., Galesso, D. and Karamanos, N.K. (2019) Hyaluronan: molecular size-dependent signaling and biological functions in inflammation and cancer. FEBS J. 286, 2883–2908.
- 10) Gao, F., Liu, Y., He, Y., Yang, C., Wang, Y., Shi, X. et al. (2010) Hyaluronan oligosaccharides promote excisional wound healing through enhanced angiogenesis. Matrix Biol. 29, 107–116.
- 11) Shimoda, M., Yoshida, H., Mizuno, S., Hirozane, T., Horiuchi, K., Yoshino, Y. et al. (2017) Hyaluronan-binding protein involved in hyaluronan depolymerization controls endochondral ossification through hyaluronan metabolism. Am. J. Pathol. 187, 1162–1176.
- 12) Weigel, P.H. and DeAngelis, P.L. (2007) Hyaluronan synthases: a decade-plus of novel glycosyltransferases. J. Biol. Chem. 282, 36777–36781.
- 13) Hubbard, C., McNamara, J.T., Azumaya, C., Patel, M.S. and Zimmer, J. (2012) The hyaluronan synthase catalyzes the synthesis and membrane translocation of hyaluronan. J. Mol. Biol. 418, 21–31.
- 14) Yoshida, H., Nagaoka, A., Kusaka-Kikushima, A., Tobiishi, M., Kawabata, K., Sayo, T. et al. (2013) KIAA1199, a deafness gene of unknown function, is a new hyaluronan binding protein involved in hyaluronan depolymerization. Proc. Natl. Acad. Sci. U.S.A. 110, 5612–5617.
- 15) Yamamoto, H., Tobisawa, Y., Inubushi, T., Irie, F., Ohyama, C. and Yamaguchi, Y. (2017) A mammalian homolog of the zebrafish transmembrane protein 2 (TMEM2) is the long-sought-after cell-surface hyaluronidase. J. Biol. Chem. 292, 7304–7313.
- 16) Itano, N. and Kimata, K. (2002) Mammalian hyaluronan synthases. IUBMB Life 54, 195–199.
- 17) Sugiyama, Y., Shimada, A., Sayo, T., Sakai, S. and Inoue, S. (1998) Putative hyaluronan synthase mRNA are expressed in mouse skin and TGF-beta upregulates their expression in cultured human skin cells. J. Invest. Dermatol. 110, 116–121.
- 18) Monslow, J., Govindaraju, P. and Pure, E. (2015) Hyaluronan – a functional and structural sweet spot in the tissue microenvironment. Front. Immunol. 6, 231.
- 19) Pandey, M.S., Harris, E.N., Weigel, J.A. and Weigel, P.H. (2008) The cytoplasmic domain of the hyaluronan receptor for endocytosis (HARE) contains multiple endocytic motifs targeting coated pit-mediated internalization. J. Biol. Chem. 283, 21453–21461.
- 20) Stern, R. (2003) Devising a pathway for hyaluronan catabolism: are we there yet? Glycobiology 13, 105R–115R.
- 21) Stern, R. (2004) Hyaluronan catabolism: a new metabolic pathway. Eur. J. Cell Biol. 83, 317–325.
- 22) Petrey, A.C. and de la Motte, C.A. (2014) Hyaluronan, a crucial regulator of inflammation. Front. Immunol. 5, 101.
- 23) Stern, R. and Jedrzejas, M.J. (2006) Hyaluronidases: their genomics, structuresand mechanisms of action. Chem. Rev. 106, 818–839.
- 24) Harada, H. and Takahashi, M. (2007) CD44-dependent intracellular and extracellular catabolism of hyaluronic acid by hyaluronidase-1 and -2. J. Biol. Chem. 282, 5597–5607.
- 25) Bourguignon, L.Y., Singleton, P.A., Diedrich, F., Stern, R. and Gilad, E. (2004) CD44 interaction with Na+-H+ exchanger (NHE1) creates acidic microenvironments leading to hyaluronidase-2 and cathepsin B activation and breast tumor cell invasion. J. Biol. Chem. 279, 26991–27007.
- 26) Fraser, J.R., Laurent, T.C. and Laurent, U.B. (1997) Hyaluronan: its nature, distribution, functions and turnover. J. Intern. Med. 242, 27–33.
- 27) Yan, Y., Zuo, X. and Wei, D. (2015) Concise review: Emerging role of CD44 in cancer stem cells: A promising biomarker and therapeutic target. Stem Cells Transl. Med. 4, 1033–1043.
- 28) Xu, H., Niu, M., Yuan, X., Wu, K. and Liu, A. (2020) CD44 as a tumor biomarker and therapeutic target. Exp. Hematol. Oncol. 9, 36.
- 29) Abe, S., Usami, S. and Nakamura, Y. (2003) Mutations in the gene encoding KIAA1199 protein, an inner-ear protein expressed in Deiters’ cells and the fibrocytes, as the cause of nonsyndromic hearing loss. J. Hum. Genet. 48, 564–570.
- 30) Nagaoka, A., Yoshida, H., Nakamura, S., Morikawa, T., Kawabata, K., Kobayashi, M. et al. (2015) Regulation of hyaluronan (HA) metabolism mediated by HYBID (hyaluronan-binding protein involved in HA depolymerization, KIAA1199) and HA synthases in growth factor-stimulated fibroblasts. J. Biol. Chem. 290, 30910–30923.
- 31) Evensen, N.A., Kuscu, C., Nguyen, H.L., Zarrabi, K., Dufour, A., Kadam, P. et al. (2013) Unraveling the role of KIAA1199, a novel endoplasmic reticulum protein, in cancer cell migration. J. Natl. Cancer Inst. 105, 1402–1416.
- 32) Evensen, N.A., Li, Y., Kuscu, C., Liu, J., Cathcart, J., Banach, A. et al. (2015) Hypoxia promotes colon cancer dissemination through up-regulation of cell migration-inducing protein (CEMIP). Oncotarget 6, 20723–20739.
- 33) Yoshida, H., Nagaoka, A., Nakamura, S., Sugiyama, Y., Okada, Y. and Inoue, S. (2013) Murine homologue of the human KIAA1199 is implicated in hyaluronan binding and depolymerization. FEBS Open Bio 3, 352–356.
- 34) Sato, S., Miyazaki, M., Fukuda, S., Mizutani, Y., Mizukami, Y., Higashiyama, S. et al. (2023) Human TMEM2 is not a catalytic hyaluronidase, but a regulator of hyaluronan metabolism via HYBID (KIAA1199/CEMIP) and HAS2 expression. J. Biol. Chem. 299, 104826.
- 35) Niu, M., McGrath, M., Sammon, D., Gardner, S., Morgan, R.M., Di Maio, A. et al. (2023) Structure of the transmembrane protein 2 (TMEM2) ectodomain and its apparent lack of hyaluronidase activity. Wellcome Open Res. 8, 76.
- 36) Yamaguchi, Y., Yamamoto, H., Tobisawa, Y. and Irie, F. (2019) TMEM2: A missing link in hyaluronan catabolism identified? Matrix Biol. 78–79, 139–146.
- 37) Domanegg, K., Sleeman, J.P. and Schmaus, A. (2022) CEMIP, a promising biomarker that promotes the progression and metastasis of colorectal and other types of cancer. Cancers (Basel) 14, 5093.
- 38) Zhang, W., Yin, G., Zhao, H., Ling, H., Xie, Z., Xiao, C. et al. (2021) Secreted KIAA1199 promotes the progression of rheumatoid arthritis by mediating hyaluronic acid degradation in an ANXA1-dependent manner. Cell Death Dis. 12, 102.
- 39) Sato, S., Mizutani, Y., Abe, M., Fukuda, S., Higashiyama, S. and Inoue, S. (2024) Naked mole-rat TMEM2 lacks physiological hyaluronan-degrading activity. Arch. Biochem. Biophys. 759, 110098.
- 40) Yoshino, Y., Goto, M., Hara, H. and Inoue, S. (2018) The role and regulation of TMEM2 (transmembrane protein 2) in HYBID (hyaluronan (HA)-binding protein involved in HA depolymerization/KIAA1199/CEMIP)-mediated HA depolymerization in human skin fibroblasts. Biochem. Biophys. Res. Commun. 505, 74–80.
- 41) Sato, S., Mizutani, Y., Yoshino, Y., Masuda, M., Miyazaki, M., Hara, H. et al. (2021) Pro-inflammatory cytokines suppress HYBID (hyaluronan (HA)-binding protein involved in HA depolymerization/KIAA1199/CEMIP)-mediated HA metabolism in human skin fibroblasts. Biochem. Biophys. Res. Commun. 539, 77–82.
- 42) Shiozawa, J., de Vega, S., Yoshinaga, C., Ji, X., Negishi, Y., Momoeda, M. et al. (2022) Expression and regulation of recently discovered hyaluronidases, HYBID and TMEM2, in chondrocytes from knee osteoarthritic cartilage. Sci. Rep. 12, 17242.
- 43) Abe, M., Masuda, M., Mizukami, Y., Inoue, S. and Mizutani, Y. (2024) Epidermal keratinocytes regulate hyaluronan metabolism via extracellularly secreted hyaluronidase 1 and hyaluronan synthase 3. J. Biol. Chem. 300, 107449.
- 44) Tian, X., Azpurua, J., Hine, C., Vaidya, A., Myakishev-Rempel, M., Ablaeva, J. et al. (2013) High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 499, 346–349.
- 45) Narita, T., Tobisawa, Y., Bobkov, A., Jackson, M., Ohyama, C., Irie, F. et al. (2023) TMEM2 is a bona fide hyaluronidase possessing intrinsic catalytic activity. J. Biol. Chem. 299, 105120.
- 46) Irie, F., Tobisawa, Y., Murao, A., Yamamoto, H., Ohyama, C. and Yamaguchi, Y. (2021) The cell surface hyaluronidase TMEM2 regulates cell adhesion and migration via degradation of hyaluronan at focal adhesion sites. J. Biol. Chem. 296, 100481.
- 47) Gao, L., Tong, S., Liu, J., Cai, J., Ye, Z., Zhou, L. et al. (2023) TMEM2 induces epithelial-mesenchymal transition and promotes resistance to temozolomide in GBM cells. Heliyon 9, e16559.
- 48) Shimizu, H., Shimoda, M., Mochizuki, S., Miyamae, Y., Abe, H., Chijiiwa, M. et al. (2018) Hyaluronan-binding protein involved in hyaluronan depolymerization is up-regulated and involved in hyaluronan degradation in human osteoarthritic cartilage. Am. J. Pathol. 188, 2109–2119.
- 49) He, Q.Y., Liu, X.H., Li, Q., Studholme, D.J., Li, X.W. and Liang, S.P. (2006) G8: a novel domain associated with polycystic kidney disease and non-syndromic hearing loss. Bioinformatics 22, 2189–2191.
- 50) Tobisawa, Y., Fujita, N., Yamamoto, H., Ohyama, C., Irie, F. and Yamaguchi, Y. (2021) The cell surface hyaluronidase TMEM2 is essential for systemic hyaluronan catabolism and turnover. J. Biol. Chem. 297, 101281.
- 51) Yoshino, Y., Ishisaka, M., Tsuruma, K., Shimazawa, M., Yoshida, H., Inoue, S. et al. (2017) Distribution and function of hyaluronan binding protein involved in hyaluronan depolymerization (HYBID, KIAA1199) in the mouse central nervous system. Neuroscience 347, 1–10.
- 52) Yoshino, Y., Shimazawa, M., Nakamura, S., Inoue, S., Yoshida, H., Shimoda, M. et al. (2018) Targeted deletion of HYBID (hyaluronan binding protein involved in hyaluronan depolymerization/ KIAA1199/CEMIP) decreases dendritic spine density in the dentate gyrus through hyaluronan accumulation. Biochem. Biophys. Res. Commun. 503, 1934–1940.
- 53) Inubushi, T., Nakanishi, Y., Abe, M., Takahata, Y., Nishimura, R., Kurosaka, H. et al. (2022) The cell surface hyaluronidase TMEM2 plays an essential role in mouse neural crest cell development and survival. PLoS Genet. 18, e1009765.
- 54) Shiozawa, J., de Vega, S., Cilek, M.Z., Yoshinaga, C., Nakamura, T., Kasamatsu, S. et al. (2020) Implication of HYBID (hyaluronan-binding protein involved in hyaluronan depolymerization) in hyaluronan degradation by synovial fibroblasts in patients with knee osteoarthritis. Am. J. Pathol. 190, 1046–1058.
- 55) Yamada, Y., Itano, N., Hata, K., Ueda, M. and Kimata, K. (2004) Differential regulation by IL-1beta and EGF of expression of three different hyaluronan synthases in oral mucosal epithelial cells and fibroblasts and dermal fibroblasts: quantitative analysis using real-time RT-PCR. J. Invest. Dermatol. 122, 631–639.
- 56) Ellis, I., Banyard, J. and Schor, S.L. (1997) Differential response of fetal and adult fibroblasts to cytokines: cell migration and hyaluronan synthesis. Development 124, 1593–1600.
- 57) Yoshida, H., Aoki, M., Komiya, A., Endo, Y., Kawabata, K., Nakamura, T. et al. (2020) HYBID (alias KIAA1199/CEMIP) and hyaluronan synthase coordinately regulate hyaluronan metabolism in histamine-stimulated skin fibroblasts. J. Biol. Chem. 295, 2483–2494.
- 58) Kobori, Y., Tachizaki, M., Imaizumi, T., Tanaka, Y., Seya, K., Miki, Y. et al. (2024) TMEM2 suppresses TLR3-mediated IFN-beta/ISG56/CXCL10 expression in BEAS-2B bronchial epithelial cells. Mol. Biol. Rep. 51, 417.
- 59) Kuscu, C., Evensen, N., Kim, D., Hu, Y.J., Zucker, S. and Cao, J. (2012) Transcriptional and epigenetic regulation of KIAA1199 gene expression in human breast cancer. PLoS One 7, e44661.
- 60) Shostak, K., Zhang, X., Hubert, P., Goktuna, S.I., Jiang, Z., Klevernic, I. et al. (2014) NF-kappaB-induced KIAA1199 promotes survival through EGFR signalling. Nat. Commun. 5, 5232.
- 61) Spataro, S., Guerra, C., Cavalli, A., Sgrignani, J., Sleeman, J., Poulain, L. et al. (2023) CEMIP (HYBID, KIAA1199): structure, function and expression in health and disease. FEBS J. 290, 3946–3962.
- 62) Matsuzaki, S., Tanaka, F., Mimori, K., Tahara, K., Inoue, H. and Mori, M. (2009) Clinicopathologic significance of KIAA1199 overexpression in human gastric cancer. Ann. Surg. Oncol. 16, 2042–2051.
- 63) Jami, M.S., Hou, J., Liu, M., Varney, M.L., Hassan, H., Dong, J. et al. (2014) Functional proteomic analysis reveals the involvement of KIAA1199 in breast cancer growth, motility and invasiveness. BMC Cancer 14, 194.
- 64) Fink, S.P., Myeroff, L.L., Kariv, R., Platzer, P., Xin, B., Mikkola, D. et al. (2015) Induction of KIAA1199/CEMIP is associated with colon cancer phenotype and poor patient survival. Oncotarget 6, 30500–30515.
- 65) Rodrigues, G., Hoshino, A., Kenific, C.M., Matei, I.R., Steiner, L., Freitas, D. et al. (2019) Tumour exosomal CEMIP protein promotes cancer cell colonization in brain metastasis. Nat. Cell Biol. 21, 1403–1412.
- 66) Ko, Y.G., Jo, J.H., Song, S.Y. and Lee, H.S. (2025) The crucial role of CEMIP in cancer metastasis: Mechanistic insights and clinical implications. FASEB J. 39, e70284.
- 67) Lee, H., Goodarzi, H., Tavazoie, S.F. and Alarcon, C.R. (2016) TMEM2 is a SOX4-regulated gene that mediates metastatic migration and invasion in breast cancer. Cancer Res. 76, 4994–5005.
- 68) Kudo, Y., Sato, N., Adachi, Y., Amaike, T., Koga, A., Kohi, S. et al. (2020) Overexpression of transmembrane protein 2 (TMEM2), a novel hyaluronidase, predicts poor prognosis in pancreatic ductal adenocarcinoma. Pancreatology 20, 1479–1485.
- 69) Tammi, R., Agren, U.M., Tuhkanen, A.L. and Tammi, M. (1994) Hyaluronan metabolism in skin. Prog. Histochem. Cytochem. 29, 1–81.
- 70) Banerji, S., Day, A.J., Kahmann, J.D. and Jackson, D.G. (1998) Characterization of a functional hyaluronan-binding domain from the human CD44 molecule expressed in Escherichia coli. Protein Expr. Purif. 14, 371–381.
- 71) Zhou, B., Weigel, J.A., Fauss, L. and Weigel, P.H. (2000) Identification of the hyaluronan receptor for endocytosis (HARE). J. Biol. Chem. 275, 37733–37741.
- 72) Tammi, R., MacCallum, D., Hascall, V.C., Pienimaki, J.P., Hyttinen, M. and Tammi, M. (1998) Hyaluronan bound to CD44 on keratinocytes is displaced by hyaluronan decasaccharides and not hexasaccharides. J. Biol. Chem. 273, 28878–28888.
- 73) Sayo, T., Sugiyama, Y., Takahashi, Y., Ozawa, N., Sakai, S., Ishikawa, O. et al. (2002) Hyaluronan synthase 3 regulates hyaluronan synthesis in cultured human keratinocytes. J. Invest. Dermatol. 118, 43–48.
- 74) Sayo, T., Sakai, S. and Inoue, S. (2004) Synergistic effect of N-acetylglucosamine and retinoids on hyaluronan production in human keratinocytes. Skin Pharmacol. Physiol. 17, 77–83.
- 75) Sayo, T., Sugiyama, Y. and Inoue, S. (2013) Lutein, a nonprovitamin A, activates the retinoic acid receptor to induce HAS3-dependent hyaluronan synthesis in keratinocytes. Biosci. Biotechnol. Biochem. 77, 1282–1286.
- 76) Sakai, S., Yasuda, R., Sayo, T., Ishikawa, O. and Inoue, S. (2000) Hyaluronan exists in the normal stratum corneum. J. Invest. Dermatol. 114, 1184–1187.
- 77) Puissant, E., Gilis, F., Dogne, S., Flamion, B., Jadot, M. and Boonen, M. (2014) Subcellular trafficking and activity of Hyal-1 and its processed forms in murine macrophages. Traffic 15, 500–515.
- 78) Puissant, E. and Boonen, M. (2016) Monocytes/macrophages upregulate the hyaluronidase HYAL1 and adapt its subcellular trafficking to promote extracellular residency upon differentiation into osteoclasts. PLoS One 11, e0165004.
- 79) Tobiishi, M., Sayo, T., Yoshida, H., Kusaka, A., Kawabata, K., Sugiyama, Y. et al. (2011) Changes in epidermal hyaluronan metabolism following UVB irradiation. J. Dermatol. Sci. 64, 31–38.
- 80) Lee, H., Hong, Y. and Kim, M. (2021) Structural and functional changes and possible molecular mechanisms in aged skin. Int. J. Mol. Sci. 22, 12489.
- 81) Yoshida, H. and Okada, Y. (2019) Role of HYBID (hyaluronan binding protein involved in hyaluronan depolymerization), alias KIAA1199/CEMIP, in hyaluronan degradation in normal and photoaged skin. Int. J. Mol. Sci. 20, 5804.
- 82) Takahashi, Y., Ishikawa, O., Okada, K., Kojima, Y., Igarashi, Y. and Miyachi, Y. (1996) Disaccharide analysis of human skin glycosaminoglycans in sun-exposed and sun-protected skin of aged people. J. Dermatol. Sci. 11, 129–133.
- 83) Tzellos, T.G., Klagas, I., Vahtsevanos, K., Triaridis, S., Printza, A., Kyrgidis, A. et al. (2009) Extrinsic ageing in the human skin is associated with alterations in the expression of hyaluronic acid and its metabolizing enzymes. Exp. Dermatol. 18, 1028–1035.
- 84) Hasegawa, K., Yoneda, M., Kuwabara, H., Miyaishi, O., Itano, N., Ohno, A. et al. (2007) Versican, a major hyaluronan-binding component in the dermis, loses its hyaluronan-binding ability in solar elastosis. J. Invest. Dermatol. 127, 1657–1663.
- 85) Yoshida, H., Nagaoka, A., Komiya, A., Aoki, M., Nakamura, S., Morikawa, T. et al. (2018) Reduction of hyaluronan and increased expression of HYBID (alias CEMIP and KIAA1199) correlate with clinical symptoms in photoaged skin. Br. J. Dermatol. 179, 136–144.
- 86) Manuskiatti, W. and Maibach, H.I. (1996) Hyaluronic acid and skin: wound healing and aging. Int. J. Dermatol. 35, 539–544.
- 87) Tsukahara, K., Fujimura, T., Yoshida, Y., Kitahara, T., Hotta, M., Moriwaki, S. et al. (2004) Comparison of age-related changes in wrinkling and sagging of the skin in Caucasian females and in Japanese females. J. Cosmet. Sci. 55, 351–371.
- 88) Nouveau-Richard, S., Yang, Z., Mac-Mary, S., Li, L., Bastien, P., Tardy, I. et al. (2005) Skin ageing: a comparison between Chinese and European populations. A pilot study. J. Dermatol. Sci. 40, 187–193.
- 89) Yoshida, H., Komiya, A., Ohtsuki, R., Kusaka-Kikushima, A., Sakai, S., Kawabata, K. et al. (2018) Relationship of hyaluronan and HYBID (KIAA1199) expression with roughness parameters of photoaged skin in Caucasian women. Skin Res. Technol. 24, 562–569.
- 90) Anderegg, U., Simon, J.C. and Averbeck, M. (2014) More than just a filler - the role of hyaluronan for skin homeostasis. Exp. Dermatol. 23, 295–303.
- 91) Noble, P.W. (2002) Hyaluronan and its catabolic products in tissue injury and repair. Matrix Biol. 21, 25–29.
- 92) Stern, R., Asari, A.A. and Sugahara, K.N. (2006) Hyaluronan fragments: an information-rich system. Eur. J. Cell Biol. 85, 699–715.
- 93) West, D.C., Hampson, I.N., Arnold, F. and Kumar, S. (1985) Angiogenesis induced by degradation products of hyaluronic acid. Science 228, 1324–1326.
- 94) Rooney, P., Kumar, S., Ponting, J. and Wang, M. (1995) The role of hyaluronan in tumour neovascularization (review). Int. J. Cancer 60, 632–636.
- 95) Sugahara, K.N., Murai, T., Nishinakamura, H., Kawashima, H., Saya, H. and Miyasaka, M. (2003) Hyaluronan oligosaccharides induce CD44 cleavage and promote cell migration in CD44-expressing tumor cells. J. Biol. Chem. 278, 32259–32265.
- 96) Barrientos, S., Stojadinovic, O., Golinko, M.S., Brem, H. and Tomic-Canic, M. (2008) Growth factors and cytokines in wound healing. Wound Repair Regen. 16, 585–601.
- 97) Dokoshi, T., Zhang, L.J., Li, F., Nakatsuji, T., Butcher, A., Yoshida, H. et al. (2020) Hyaluronan degradation by Cemip regulates host defense against staphylococcus aureus skin Infection. Cell Rep. 30, 61–68.e64.
- 98) Okada, Y., Nakanishi, I. and Kajikawa, K. (1981) Ultrastructure of the mouse synovial membrane. Development and organization of the extracellular matrix. Arthritis Rheum. 24, 835–843.
- 99) Uebelhart, D. and Williams, J.M. (1999) Effects of hyaluronic acid on cartilage degradation. Curr. Opin. Rheumatol. 11, 427–435.
- 100) Tamer, T.M. (2013) Hyaluronan and synovial joint: function, distribution and healing. Interdiscip. Toxicol. 6, 111–125.
- 101) Heinegard, D. and Saxne, T. (2011) The role of the cartilage matrix in osteoarthritis. Nat. Rev. Rheumatol. 7, 50–56.
- 102) Okada, Y. (2017) Proteinases and Matrix Degradation. In Kelley and Firestein’s Texbook of Rheumatology, 10th Edition (eds. Firestein, G.S., Budd, R.C., Gabriel, S.E., McInnes I.B. and O’Dell J.R.). Elsevier, Inc., Philadelphia, pp. 106–125.
- 103) Primorac, D., Molnar, V., Rod, E., Jelec, Z., Cukelj, F., Matisic, V. et al. (2020) Knee osteoarthritis: A review of pathogenesis and state-of-the-art non-operative therapeutic considerations. Genes (Basel) 11, 854.
- 104) Sellam, J. and Berenbaum, F. (2010) The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol. 6, 625–635.
- 105) Rooney, P., Kumar, S., Ponting, J. and Wang, M. (1995) The role of hyaluronan in tumour neovascularization (review). Int. J. Cancer 60, 632–636.
- 106) Kwapiszewska, G., Gungl, A., Wilhelm, J., Marsh, L.M., Thekkekara Puthenparampil, H., Sinn, K. et al. (2018) Transcriptome profiling reveals the complexity of pirfenidone effects in idiopathic pulmonary fibrosis. Eur. Respir. J. 52, 1800564.
- 107) Chen, L., Shi, K., Ditzel, N., Qiu, W., Figeac, F., Nielsen, L.H.D. et al. (2023) KIAA1199 deficiency enhances skeletal stem cell differentiation to osteoblasts and promotes bone regeneration. Nat. Commun. 14, 2016.
- 108) Schmaus, A., Rothley, M., Schreiber, C., Moller, S., Rosswag, S., Franz, S. et al. (2022) Sulfated hyaluronic acid inhibits the hyaluronidase CEMIP and regulates the HA metabolism, proliferation and differentiation of fibroblasts. Matrix Biol. 109, 173–191.
- 109) Yoshida, H., Yamazaki, K., Komiya, A., Aoki, M., Nakamura, T., Kasamatsu, S. et al. (2019) Inhibition of HYBID (KIAA1199)-mediated hyaluronan degradation and anti-wrinkle effect of Geranium thunbergii extract. J. Cosmet. Dermatol. 18, 1052–1060.
- 110) Yoshida, H., Yamazaki, K., Komiya, A., Aoki, M., Kasamatsu, S., Murata, T. et al. (2019) Inhibitory effects of Sanguisorba officinalis root extract on HYBID (KIAA1199)-mediated hyaluronan degradation and skin wrinkling. Int. J. Cosmet. Sci. 41, 12–20.
- 111) Zhang, Y.H., Lin, J.X., Yip, Y.K. and Vilcek, J. (1988) Enhancement of cAMP levels and of protein kinase activity by tumor necrosis factor and interleukin 1 in human fibroblasts: role in the induction of interleukin 6. Proc. Natl. Acad. Sci. U.S.A. 85, 6802–6805.
Non-standard abbreviation list
3Dthree-dimensional
ADAMTSa disintegrin and metalloproteinase with thrombospondin motifs
ANXA1annexin A1
bFGFbasic fibroblast growth factor
CEMIPcell migration-inducing hyaluronidase protein
COS-7monkey kidney fibroblast
ECMextracellular matrix
EGFepidermal growth factor
G8eight conserved glycine residues
GAGglycosaminoglycan
GGtwo well-conserved glycine residues
GlcUDP-glucuronic acid
GlcNAcUDP-N-acetylglucosamine
HAhyaluronan
HAREhyaluronan receptor for endocytosis
HAShyaluronan synthase
HEK293human embryonic kidney
HIF-2αhypoxia-inducible-factor-2α
HMWhigh-molecular-weight
HYALhyaluronidase
HYBIDhyaluronan-binding protein involved in hyaluronan depolymerization
IGF-1insulin-like growth factor-1
ILinterleukin
LMWlow-molecular-weight
KOknockout
MMPmatrix metalloproteinase
MMWmedium-molecular-weight
nmrnaked-mole rat
OAosteoarthritis
PbH1parallel beta-helix
PDGF-BBplatelet-derived growth factor-BB
PGE2prostaglandin E2
RArheumatoid arthritis
S. aureusStaphylococcus aureus
TGF-βtransforming growth factor beta
TMtransmembrane
TMEM2transmembrane protein 2
TNF-αtumor necrosis factor-α
UDPuridine diphosphate
VEGFvascular endothelial growth factor
Profile

Hiroyuki Yoshida was born in Osaka Prefecture, Japan, in 1973 and graduated from Kyoto University in 1996. He majored in microbial biotechnology at the Graduate School of Agriculture, Kyoto University and graduated in 1998. After he joined Kanebo, Co. Ltd., he was engaged in research on hyaluronan metabolism in the skin and discovered HYBID, on which he wrote his thesis for his Ph.D. from Tokyo University of Pharmacy and Life Sciences in 2014. Then, he joined Kao Corporation and worked at Americas Research Laboratories, Kao USA Inc. from 2016 to 2019. He became Manager in 2020 and Director in 2024 of Biological Science Research, Kao Corporation, and he is working as Director for Skin Care Products Research, Kao Corporation. In addition to hyaluronan metabolism, he has studied the mechanism of extracellular matrix metabolism and the regulation of skin barrier function in various skin conditions. For his accomplishments, he received Excellence Award of the Japanese Society for Matrix Biology and Medicine (2013), Otaka Prize of the Japanese Society for Matrix Biology and Medicine (2015), Excellent Paper Award of the Japanese Cosmetic Science Society (2022), and Presentation Awards of the Society of Cosmetic Chemists of Japan (2022).

Shintaro Inoue was born in Hyogo Prefecture, Japan, in 1952 and graduated from the Faculty of Engineering at Osaka University in 1975. After studying yeast genetics for two years in the master’s program at the same university, he joined Kanebo, Co. Ltd. and was involved in the development of new drugs in the pharmaceutical research laboratory for 10 years. During the following five years, he was engaged in research on amino acid metabolism at the Department of Medical Chemistry, Kyoto University Faculty of Medicine. Then, he worked at Biochemistry Lab., Kanebo Co. Ltd., and Basic Research Lab. and Innovative Beauty Science Lab., Kanebo Cosmetics Inc. (1987–2011). He received his Ph.D. from Tokyo University of Pharmacy and Life Sciences in 2005. He has served as Director of the Basic Research Laboratory (2004–2009) and Innovative Beauty Science Lab. (2009–2011), Executive Officer (Head of R&D) (2009–2012), Kanebo Cosmetics, Inc., and Vice President of Global R&D Beauty, Kao Corporation (2010–2012). He has been a Research Professor of the Department of Cosmetic Health Science, Gifu Pharmaceutical University since 2016. He has studied skin cell functions to improve beauty and health and has been focusing on skin tight junction barrier, regulation of hyaluronan metabolism, and recently on the roles of the glycoprotein GPNMB in vitiligo.

Yasunori Okada was born in Toyama prefecture, Japan, in 1949 and graduated from the School of Medicine, Kanazawa University in 1974. He majored in Pathology at the Graduate School of Medicine, Kanazawa University and received his Ph.D. degree in 1978. After working for the Department of Pathology, School of Medicine, Kanazawa University from 1978 to 1983, he obtained a Fogarty International Research Fellowship and joined the lab of Dr. Edward D. Harris, Jr. and Dr. Hideaki Nagase at the University of Medicine and Dentistry of New Jersey-Rutgers Medical School from 1984 to 1986. He became a Professor at the Cancer Research Institute, Kanazawa University in 1994 and a Professor at the Department of Pathology, School of Medicine, Keio University in 1997. After retirement from Keio University in 2015, he is working as a Professor for Juntendo University Graduate School of Medicine. He has studied the roles of extracellular matrix- degrading enzymes including MMPs, ADAMs/ADAMTSs, and HYBID in various pathological conditions such as arthritides and cancers by focusing on the mechanisms of tissue destruction and remodeling through metabolism of tissue microenvironmental factors such as extracellular matrix, cytokines/growth factors, and membrane proteins. For his accomplishments, he received the Novartis Rheumatisms Award (1998), Pathology Award of the Japanese Society of Pathology (2002), The Commendation for Science and Technology by the Minister of Education, Culture, Sports, Science and Technology (2010), Keio University Gijuku Award (2015), and Distinguished Investigator Award of the Japanese Society for Matrix Biology and Medicine (2016).

