2. Establishment of metabolomics and integrated omics platform for decoding plant metabolome
2.1. General concept of phytochemical genomics.
Metabolomics-based functional genomics provides a fundamental approach for unveiling the chemical diversity of plants through comprehensive metabolic investigations, including untargeted metabolic profiling.10) This approach, combined with genomic or transcriptomic information, allowed us to uncover the principles behind the production and its regulation of plant metabolites at the genome and metabolome levels.11),12) Moreover, their biological roles can be addressed within an organism and trace the evolutionary history of the biosynthetic pathways and genes involved. These insights can pave the way for genome-based biotechnological applications. Such a fundamental understanding could significantly impact the fields of plant science and those such as biotechnology, agriculture, bioorganic chemistry, and pharmaceutical science, contributing to the development of sustainable resources.
The general concept of phytochemical genomics is shown in Fig. 1, outlining the research pipeline of multiomics data-driven systems biology and subsequent functional genomics for biotechnological applications such as engineering biology. First, omics data from the genome, transcriptome, and metabolome of a plant were obtained. Through the integration of omics data, data-driven systems biology focusing on plant metabolites and their production can be carried out using informatics. In this context, systems biology, which interconnects the components of each omics layer and analyzes the behavior of entire systems, primarily contributes to the generation of testable hypotheses. Next, the generated hypotheses here are verified as functional genomics through reverse genetics, reverse biochemistry, and mathematical modeling. These steps of hypothesis generation and validation are reversible as needed. The verified hypotheses are applied to biotechnology through transgenics, genome editing, and engineering biology. Thus, a comprehensive framework has been consistently developed to promote research in phytochemical genomics.6),7)

Genomics and transcriptomics are represented by four bases (four digits) of nucleic acids, making it relatively easy to automate their analytical processes. In recent years, the development of ultrahigh-throughput DNA sequencing has accelerated data acquisition. However, the automation of processes, such as sequencing of nucleic acids, for metabolomics is extremely difficult, and possibly unlikely even in the future as well, due to the diversity and complexity of the chemical structures and properties of the metabolites. We developed fundamental technologies to decode plant metabolomes by combining mass spectrometry (MS) and chemo/bioinformatics.
The plant metabolome encompasses a large chemical space composed of numerous compound properties, such as molecular weight and hydrophilicity (Fig. 2). To maximize the coverage of the analyzed metabolites, we set up an MS-based analytical platform combining gas chromatography-mass spectrometry (GC-MS), liquid chromatography-mass spectrometry (LC-MS), capillary electrophoresis-mass spectrometry (CE-MS), and Fourier-transform ion cyclotron resonance mass spectrometry combined with liquid chromatography (LC-FT-ICR-MS). Almost all plant metabolites can be analyzed by combining these different separation technologies and mass spectrometry, as we established.13),14)

In metabolomic analysis, there is generally a trade-off relationship between coverage or novelty and sensitivity or quantitativeness (Fig. 3). As metabolomics represents the non-bias analysis of entire metabolites in a given organism, non-target analysis, a narrow definition of “metabolomics”, is the preferred option. Due to the high coverage of metabolites detected using this method, novel compounds could be identified. Thus, this method can be adopted for the “discovery phase”. Wide-target and targeted analyses diminish the coverage of metabolites but increase their sensitivity and quantitativeness. In our metabolomic study, we primarily focused on developing multi-platform-based non-target analysis13),15),16) and then extended it to wide-target17),18) and target19) analyses according to the requirements of particular investigations.

Together with setting up cutting-edge MS equipment that can cover a wide range of metabolites, we have also developed databases and software to facilitate metabolomic analysis. These informatics tools have been all stored on the website of our laboratory in RIKEN Center for Sustainable Resource Science, named “PRIMe” <http://prime.psc.riken.jp/>, which stands for Platform for RIKEN Metabolomics.20),21) Table 2 summarizes the contents of the PRIMe website. Several tools and databases are currently available on the website, and some old tools have been transferred to other sites or archives. The PRIMe website is highly recognized by the scientific community, with approximately 45,000 viewing records per year. In addition to this internal lab-based activity for setting up a metabolomics database and toolbox, we have also been involved in the community-based construction of a large MS spectral database, MassBank <https://massbank.jp/MassBank/>.22) MassBank is the largest publicly available MS database and extremely powerful for identifying and characterizing peaks in MS spectra obtained from any MS-based chemical analysis.
Table 2. Content of RIKEN PRIMe website <
https://prime.psc.riken.jp/>
| Name |
Description |
Status |
PlanMAXdb
(Plant Metabolite Alliance
eXplorer database) |
Contains mass spectral molecular correlations from 434 plant samples,
encompassing 157 species across 108 genera and 25 plant orders |
Since 2024 |
CompMS
(Computational Mass
Spectrometry) |
Computational MS tools including MS-DIAL, MS-FINDER, and
MRMPROBES |
Since 2020; transferred to
Hiroshi Tsugawa Laba) |
| RIKEN LIPIDOMICS |
Lipidomics data depository of RIKEN |
Since 2020 |
RIKEN Plant Metabolome
MetaDatabase |
Integrated plant metabolomic data repository based on the semantic
web |
Since 2019 |
PlaSMA
(Plant Specialized Metabolome Annotation) |
Fully 13C-labeled plant metabolomic data sets across 12 plant species,
carefully curated for comprehensive structural characterization |
Since 2018; in 2024 integrated
into PlanMAXdb |
HIFI
(Heteroatom-containing Ion
FInder) |
Tool for LC-FT-ICR-MS with search and visualization functions to
predict heteroatom-containing ions |
Since 2017; closed in 2021 |
| PASMet |
Web-based tool for prediction and modelling metabolic systems |
Since 2015; closed in 2019 |
| MetBoard |
Information board of standard compounds provided from collaborators |
Since 2015; closed in 2021 |
Targeted and Non-targeted
Analysis Software |
Toolbox of standalone software for targeted and non-targeted analysis |
Since 2013; moved to
CompMS in 2020 |
| PRIMeLink |
Integration of databases in PRIMe and searchable function between
gene and metabolite |
Since 2012, archived in 2024 |
MeKO
(Metabolite Profiles for Knock-
Out Mutants) |
GC-MS datasets of knock-out mutants in A. thaliana |
Since 2012; closed in 2020 |
| AtMetExpress |
LC-MS based phytochemical atlas of A. thaliana |
Since 2011; closed in 2020 |
| ReSpect |
Data collection from literature and in-house MSn spectra |
Since 2010; archived in 2024 |
MS/MS spectral tag (MS2T)
library |
MS2 spectral data of plant metabolites aquired by an automatic MS2
acquisition of LC-ESI-Q-TOF/MS |
Since 2009; closed in 2020 |
| PRIMe LC-MS Branch |
Toolbox of LC-MS branch of PRIMe |
Since 2008; closed in 2020 |
| Introduction of KNApSAcK |
Comprehensive species-metabolite relationships database |
Since 2007; closed in 2024
(KNApSAcK is still activeb)) |
| SpinAssign |
Tool for spectral assignment for NMR |
Since 2006; moved to Jun
Kikuchi Labc) |
| Standard Spectrum Search |
Database of the standardized spectra of GC-MS and LC-MS, along with
NMR for representative metabolites |
Since 2006; closed in 2022 |
| Cluster Cutting |
Tool for cutting hierarchical clustering data of AtGenExpress |
Since 2006; closed in 2017 |
| Correlated Gene Search |
Search tool for finding correlated gene pairs in AtGenExpress data with
correlation-viewer |
Since 2006; closed in 2017 |
| Simple BL-SOM (mirror site) |
Mirror site for a batch-learning self-organizing mapping |
Since 2006; archived in 2024 |
a) https://systemsomicslab.github.io/compms/index.html
b) https://www.knapsackfamily.com/KNApSAcK_Family/
c) https://integbio.jp/dbcatalog/en/record/nbdc00633
Identification or annotation of unknown peaks generated from any MS analysis has been challenging in non-target metabolomics. We have addressed this issue through two approaches: empirical and informatics-based methods. In empirical approach, we performed non-biased isolation of metabolites from model plants species, A. thaliana23) and rice.24) The availability of these actual standard compounds, many of which are not commercially available, serves as the foundation for the reliable identification and annotation of MS peaks. For informatics approach, our colleagues developed several advanced chemo-informatics tools for computational MS peak annotation, such as MS-DIAL25) and MS-FINDER.26) By integrating these empirical and informatics resources with fully-13C labeled plant materials, we extensively annotated MS peaks of 31 tissues across 12 plant species, assigning 1,092 structures and 344 formulae to 3,604 carbon-determined metabolite ions.27) This constitutes one of the most reliable and widely used analytical pipelines in plant metabolomics.14)
2.3. General principle for integrated omics for functional genomics.
The integration of metabolomics with transcriptomics and genomics is remarkably powerful for identifying the functions of genes that produce particular metabolites through association analyses of the elements in each omics. Based on a general “guilt-by-association” principle, a set of genes and metabolites involved in each biological process are expected to be co-regulated and thus co-expressed or co-accumulated. Therefore, if an unknown gene or metabolite is co-expressed/co-accumulated with known genes/metabolites under a particular biological condition or, more practically, in a particular metabolic pathway, this unknown gene/metabolite is likely involved in the same process or pathway. This approach has been widely used to predict unknown gene functions using transcriptome co-expression analysis, often with web tools, such as ATTED-II <https://atted.jp/> and GENEVESTIGATOR <https://genevestigator.com/>, using public datasets of holistic gene expression.28),29)
The “guilt-by-association” principle that successfully identified gene functions using the transcriptome dataset alone was extended to integrate metabolite accumulation patterns in metabolomic and transcriptome data as shown in Fig. 4. If a pathway is co-regulated in terms of gene expression and metabolite accumulation, the patterns of both gene expression and metabolite accumulation should be coordinated as represented co-expression/co-accumulation relationships. Using this combined strategy of metabolomic and transcriptomic co-expression/co-accumulation, a comprehensive prediction of gene and metabolite functions involved in a particular process or pathway can be performed, as exemplified by the AtMetExpress project.30),31) Although their relationship is occasionally nonlinear or ambiguous, the correlation between metabolites and transcripts is useful for decoding the function of genes and metabolites, as exemplified in the following sections.

If whole-genome sequence information for a given plant species is available, additional genome-wide strategies can be employed for an integrated analysis. All genes categorized into particular gene families were identified from the genome assembly information. From comparative genomics and metabolomics of closely related plant species, insight can be obtained into the genomic principle that causes metabolic differences, if any, in these distinct species. Together with genomic resources, metabolomic quantitative loci and metabolomic genome-wide association studies can be used to identify genomic regions responsible for producing particular metabolites,32),33) as described in the following sections.
Using the platforms that we developed, we applied phytochemical genomics strategies to identify novel genes, metabolites, and their networks, ultimately leading to biotechnological applications. These efforts began in the late 1990s at Chiba University, and the functional metabolomics platform was formally launched in 2005 at RIKEN, Japan’s nationally recognized platform for plant metabolomics. Since then, the platform has functioned as a world-leading platform and has become available to the wider research community. With this platform, phytochemical genomics of the model plant A. thaliana, crops, and medicinal plants have been performed, leading to the discovery of many new key genes, metabolites, and their networks.
3. Metabolomics and functional genomics in a model plant Arabidopsis
A. thaliana, an annual plant of the Brassicaceae family, is widely used as a model plant in research because of its numerous advantages, such as small genome size, short generation time, easy indoor cultivation, susceptibility to genetic transformation, and massive production of seeds. Since its genome sequence was the first decoded in a higher plant in 2000,34) a novel strategy, the reverse genetics approach, has become possible. Gene functions can be identified by analyzing the traits determined by genes of unknown function, whose nucleotide sequence are known but lack functional annotation. This is in contrast to traditional forward genetics, in which mutated gene functions are identified from their modified traits. To implement the reverse genetics strategy efficiently, several research resources must be established, including gene annotations, tagged mutant panels, full-length cDNA collections, and databases containing this information. Fortunately, thanks to the international efforts of the Arabidopsis research community, these resources, e.g., The Arabidopsis Information Resource <https://www.arabidopsis.org/>, have gradually become publicly available since 2000. Considering this research environment, we decided to use A. thaliana for integrated functional genomic research based on metabolomics.
3.1. Sulfur metabolism in A. thaliana.
Sulfur is an essential macronutrient for plant growth. Many plant metabolites containing sulfur exhibit unique biological activities and play crucial roles in plant cells.35),36) Additionally, some sulfur-containing plant metabolites have beneficial health-promoting effects on humans.37) We identified novel enzymes and the genes, regulatory networks, and mechanisms involved in sulfur metabolism on a genome-wide scale in A. thaliana.
Key enzymes and genes related to sulfur assimilatory metabolism, such as multiple cysteine synthase (and its homolog β-cyanoalanine synthase),38)–44) serine acetyltransferases,45)–49) sulfate ion transporters,50)–55) ATP sulfurylase,56),57) adenosine-5′-phosphosulfate kinase,58) and glutathione-related metabolic enzymes,59),60) were identified primarily in A. thaliana (Fig. 5) and other plant species such as spinach. Some of these were the first molecular clones from the plant kingdom. In addition, the biosynthetic enzymes of serine, the precursor of cysteine in the phosphorylation pathway, were first isolated from the plant kingdom.61)–65) By applying a metabolomics strategy, we elucidated the functions and regulatory mechanisms of these genes in a pioneering manner compared with their global competitors. We also succeeded in identifying novel regulatory factors involved in sulfur metabolism, including transcription factors66) and microRNAs.67),68) These factors control the sulfur assimilation pathway and the breakdown of sulfur-containing metabolites, such as glucosinolates.

In addition to the functional analyses of individual genes involved in sulfur metabolism, we carried out integrated omics studies to investigate cellular responses to sulfur deficiency, including transcriptome and metabolome analyses,69),70) together with proteome analysis in some cases.71) This integrated omics study on sulfur metabolism is the first proof-of-concept investigation of the aforementioned “phytochemical genomics” study, which has been further extended to a variety of metabolic pathways in various plant species. From this integrated analysis, we isolated genes encoding enzymes involved in glucosinolate biosynthesis,72),73) MYB transcription factors74) controlling aliphatic glucosinolate production, and other novel regulatory factors.75) Furthermore, we explored practical applications, such as conferring tolerance to sulfur-containing environmental pollutants76) and to heavy metals77),78) using the identified genes in transgenic plants.
Based on these fundamental molecular and genomic studies in A. thaliana, the whole sulfur assimilation pathway, its enzymes with the encoding genes, and the multiple regulatory mechanisms have been clarified (Fig. 5), as summarized in our reviews.35),36),79)–81) The contribution of our group to the improvement of this understanding is immense, as evidenced by the substantial number of highly cited publications.
3.2. Diversified flavonoid metabolism in A. thaliana.
Flavonoids along with glucosinolates are representative specialized metabolites of A. thaliana. Flavonoids in A. thaliana include anthocyanins, flavonols, and proanthocyanidins.23),82) We elucidated the diversified flavonoid metabolism through a phytochemical genomics approach, together with comprehensive flavonoid profiling in A. thaliana (Fig. 6A). First, a comprehensive catalog of candidate genes involved in anthocyanin production was created utilizing the transcriptomics and metabolomics of a gain-of-function mutant that overexpresses a single MYB transcription factor and thus accumulates high levels of anthocyanins.83) Through metabolite analysis of knockout mutants of these candidate genes, their functions have been identified in a straightforward manner using a reverse genetics approach.83)
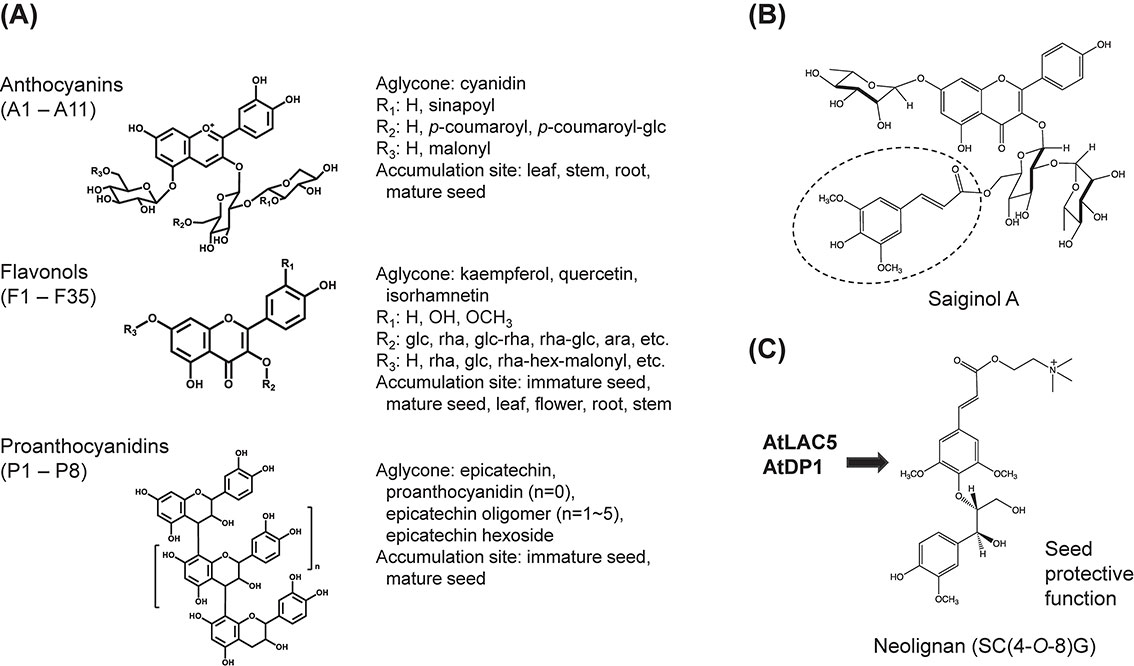
This method was extended to analyze gene co-expression relationships using publicly available A. thaliana transcriptome data.84) This generalized approach includes extracting expression modules of flavonoid biosynthesis genes, predicting the functions of those genes by considering the known genes in them, based on the “guilt-by-association” concept, and finally determining gene function through extensive metabolite analysis of their knockout mutants.4),85)–93) Through these comprehensive approaches, we identified the biosynthetic genes for the basic structural framework of flavonoids and also almost all of the modifying enzyme genes, such as glycosyltransferases and acyltransferases, that contribute to their structural diversity in A. thaliana by the mid 2010s.82)
We further explored the genomic and metabolomic diversity of the natural variants of A. thaliana. Through extensive analysis of metabolomic variations in 64 ecotypes of A. thaliana, we isolated a class of new phenyl acylated-flavonols (saiginols) as specialized ultraviolet (UV)-protective metabolites from certain ecotypes (Fig. 6B).94) Further comparative genomic investigations led to the identification of key biosynthetic genes that are presumed to have recently evolved under natural UV selection.94) Extending from A. thaliana to diverse plants, a study on the general regulation of flavonoid biosynthesis resulted in the discovery of a conserved strategy for chalcone isomerase-like proteins to rectify the product specificity of promiscuous chalcone synthase, facilitating the efficient influx of substrates into the pathway.95)
The first molecular identification of enzymes and genes involved in the biosynthesis of seed-coat protective neolignans, phenylpropanoid-derived products similar to flavonoids, was performed in A. thaliana.96) Through extensive metabolomic analysis during the development of A. thaliana, for which transcriptome information was available, the structural features of 167 metabolites among 1,589 metabolite signals were elucidated.30),31) The association relationships between gene expression and metabolite accumulation suggested a close correlation between an unknown neolignane-type compound and one of the dirigent protein genes. Further detailed molecular investigations combined with a solid chemical study identified a dirigent protein and a laccase responsible for the stereoselective dimerization of coniferyl alcohol to produce neolignans, which exhibit a seed-coat protective function96) (Fig. 6C).
These achievements represent a historic breakthrough as the first clarification of the genomic diversity underlying the structural diversity of a group of plant specialized metabolites in higher plants.11),82) This could provide valuable insights into the origin and evolution of plant flavonoid metabolism.97) Furthermore, the identified genes with mutants or transgenic plants have been applied in biotechnology to generate plants with enhanced useful traits, such as increased antioxidant activity.94),98),99) In particular, a landmark study99) overexpressing two transcription factors involved in regulating flavonoid biosynthesis in transgenic A. thaliana provided the first experimental evidence that the accumulation of flavonoid molecules confers tolerance to oxidative stress and drought resistance.100) This paper has been cited approximately 1,000 times since 2014.
3.3. Stress-related lipid metabolism in A. thaliana.
Because the chemical properties of lipids are distinct from those of other plant metabolites, an analytical platform for the lipidome has been separately established.18) Using this lipidomics platform, novel genes and metabolites involved in stress-related lipid metabolism were identified in A. thaliana. In relation to sulfur metabolism, a novel glycosyltransferase (UGP3) specifically involved in the first step of sulfolipid biosynthesis in chloroplasts has been identified101) (Fig. 7A). Furthermore, a new class of plant lipids, glucuronosyldiacylglycerol (GlcADG), containing glucuronic acid has been isolated from A. thaliana subjected to nutrient stress caused by phosphorus depletion102) (Fig. 7A). This novel lipid alleviates phosphorus deficiency in A. thaliana and crop plants102)–105) (Fig. 7B). We elucidated the biosynthetic pathway and identified the enzyme (SQD2) involved in this pathway102)–104) (Fig. 7A).

For the metabolomic response to other environmental stresses, such as high temperature, a detailed investigation of the lipidomics and transcriptomics of A. thaliana revealed an extensive lipid remodeling under heat stress.106) Based on this lipidomic study, we identified a novel heat-inducible lipase (HIL1) with an essential role in mitigating heat stress in chloroplasts.107),108) This lipase is responsible for liberating α-linolenic acid from chloroplastic glycolipids resulting in a heat-induced lipid remodeling (Fig. 8).
These findings in lipid metabolism reveal new molecular mechanisms of tolerance to environmental stresses, such as phosphorus depletion and high temperatures, which are critical for plant survival under harsh environmental conditions.103),108)
3.4. Other miscellaneous metabolism in A. thaliana.
The metabolomic analysis of A. thaliana under abiotic stresses, such as sulfur70) and phosphorus102) nutrient deficiencies, as described previously, nitrogen depletion,70),109),110) UV radiation,94),111) drought,112) cold,113) and heat,106),107) and in metabolic gene mutants,114)–118) led to the successfully identification of novel genes, metabolites, and metabolic networks.100),119) These environmental stresses induce the production of certain groups of metabolites, e.g., amino acids, sugars, phenylpropanoids, and flavonoids, which play roles in stress tolerance. Additionally, metabolomic analysis of circadian clock gene mutants revealed a close interaction between the biological clock, mitochondrial metabolism,120),121) and the role of the circadian clock in the cold stress response.122)
4. Application to crops and medicinal plants
Basic plant science research can be most efficiently advanced using A. thaliana as a model plant. However, research aimed at agricultural and pharmaceutical applications must be conducted on important crops and medicinal plants. Therefore, the principles and techniques for phytochemical genomics developed using A. thaliana have been applied to important crops and medicinal plants, enabling the identification of the functions of many genes and the further extension of their application to biotechnology.
To identify gene functions using reverse genetics approaches, plants must be generated in which target genes are disrupted or overexpressed. In addition, for biotechnological applications, such manipulating the production of bioactive metabolites, it is essential to establish techniques for creating transgenic (genetically modified) plants of non-model species, such as crops and medicinal plants. Since the 1990s, we have successfully developed methods for creating transgenic medicinal plants using the Agrobacterium-Ti or Ri plasmid systems for the first time.123) This research involved the introduction and expression of model genes or herbicide resistance genes in representative medicinal plants such as Glycyrrhiza (licorice),124) Digitalis (foxglove),125) Atropa belladonna (belladonna),126) Scoparia dulcis,127) Perilla frutescens (perilla),128) and Ophiorrhiza pumila.129) These studies have laid an important foundation for the development of functional genomic and metabolic engineering of medicinal plants.
4.1. Flavonoid and anthocyanin biosynthesis.
Phytochemical genomics of flavonoid and anthocyanin biosynthesis have been performed extensively in A. thaliana, as described in the previous sections. However, substantial investigations have also been performed on medicinal plants and crops, as flavonoids and anthocyanins are often the main bioactive products in these plants and exhibit useful properties.130)
The leaves and branch tips of P. frutescens var. crispa (perilla, Shiso in Japanese) are widely used in traditional Chinese medicine, folk remedies, and as a food ingredient under the name soyou.131) The main components include polyphenols, such as anthocyanins and rosmarinic acid, and essential oils primarily composing of monoterpenes.
P. frutescens has two known leaf color chemotypes owing to the accumulation of anthocyanin pigments: red shiso, which produces anthocyanins, and green shiso, which does not. Using these two varieties, the comprehensive isolation of genes encoding anthocyanin biosynthetic enzymes132)–135) and transcriptional regulators136)–139) was performed, followed by functional characterization of these genes.140),141) In particular, the analysis of mRNA differential display,142) which identified cDNA fragments exhibiting differential expression between red and green perilla,143),144) resulted in the isolation of a novel anthocyanin 5-O-glycosyltransferase gene for the first time.134),145) This first molecular cloning of a new gene led to the subsequent isolation of similar genes from plant species other than perilla.146),147) Moreover, an anthocyanidin synthase gene, which is involved in anthocyanin coloration, was isolated from perilla, and its enzymatic activity was successfully detected in vitro for the first time.133),148) The biochemical mechanisms underlying this coloration stage were further elucidated in detail using computational chemistry approaches.149)–152) These molecular biology studies on anthocyanin biosynthesis have become the basis for an in-depth understanding of the anthocyanin chemotypes of P. frutescens, particularly by combining metabolomic analyses of red and green perilla153) and deep transcriptome analyses using next-generation sequencing technologies.154)
Because flavonoids are representative plant specialized products that are widely distributed in the plant kingdom, we extended the molecular study to ornamental plants and crops besides A. thaliana and P. frutescens, which are model plant systems. These include the molecular characterization of biosynthetic genes from Petunia hybrida,146),149),155) Torenia fournieri,148) snapdragon, and maize149); metabolomic and genetic analysis of specialized metabolites, including flavonoids of rice156),157); and cloning of genes for modification of flavonoids from soybean.158)–160)
4.2. Quinolizidine and other alkaloid biosynthesis.
Quinolizidine alkaloids with a quinolizidine ring are one of the most widely distributed plant alkaloid groups. This alkaloid group, also known as lupine alkaloids, often accumulate in Lupinus plants and comprise over 200 structurally related alkaloids in nature.161),162) The chemical study of the quinolizidine alkaloids of the Leguminosae family, represented by the main component matrine in the traditional KAMPO medicine Kushen (Sophora root), has a long history of natural plant product research in pharmaceutical chemistry in Japan since the 19th century.163) While continuing plant physiological and chemical taxonomy research on the quinolizidine alkaloids of the Leguminosae family, we have also initiated biotechnological and molecular studies, such as alkaloid production using plant tissue cultures164),165) and the purification of the enzymes involved in the biosynthetic pathway.166),167) Finally, for the first time, we succeeded in purifying168) and molecularly cloning169) an alkaloid acyltransferase (13α-hydroxymultiflorine/13α-hydroxylupanine acyltransferase) involved in the biosynthesis of acylated quinolizidine alkaloids in Lupinus species (Fig. 9).

In addition to these biochemical approaches, we took molecular biological approaches using alkaloid-containing “bitter” and alkaloid-free “sweet” chemotypes of Lupinus angustifolia. We analyzed alkaloid patterns170) and genetic and biochemical markers,171),172) followed by differential gene expression analyses. Through differential gene expression analysis between the “bitter” and “sweet” types, we identified several gene fragments potentially associated with alkaloid production. These fragment sequences allowed us to annotate the functions of the genes and isolate full-length cDNAs. Consequently, we successfully cloned, for the first time, the cDNA of lysine decarboxylase (lysine/ornithine decarboxylase) from L. angustifolius, which catalyzes the decarboxylation of lysine, resulting in the production of cadaverine, the first committed step in quinolizidine alkaloid biosynthesis173) (Fig. 9). Furthermore, we cloned the lysine decarboxylase gene from other quinolizidine alkaloid-producing plants, such as Sophora flavescens, Echinosophora koreensis, Thermopsis chinensis, and Baptisia australis. These enzymes are considered mutated versions of ornithine decarboxylases, which are involved in the production of polyamines during primary metabolism. A unique mutation in a single amino acid residue of the enzyme is essential for the lysine decarboxylation activity. This mutation determines the size of the substrate-binding cavity in the active center of ornithine/lysine decarboxylases.173)–175) This critical amino acid mutation was conserved in lysine decarboxylases from distantly related plants that produce lysine/cadaverine-derived alkaloids, such as Lycopodium clavatum and Huperzia serrata.176) Evolutionary analyses further demonstrated that this mutation is under positive selection pressure, proving the convergent evolution of alkaloid biosynthetic genes in distantly related families, such as Leguminosae and Lycopodiaceae.176),177) While screening for differentially expressed genes between “bitter” and “sweet” chemotypes of L. angustifolius, we isolated an additional acyltransferase-like gene whose function was uncertain at that time.178) However, it has recently been reported that this enzyme is involved in the hidden acetylation step of a biosynthetic intermediate, a key step in quinolizidine alkaloid biosynthesis.179)
For biotechnological applications using the isolated lysine decarboxylase gene, we engineered lysine-derived alkaloid biosynthesis in tobacco hairy roots, such as anabasine production.180) Metabolomic analysis revealed that transgenic A. thaliana plants ectopically expressing lysine decarboxylase gene from L. angustifolius produced several cadaverine-derived specific compounds such as 5-aminopentanal, 5-aminopentanoate, and δ-valerolactam181) (Fig. 9). This study indicated that the promiscuous activity of such enzymes may have contributed toward the diversification of pathways, and consequently, to an expansion of chemodiversity in plant evolution.181),182)
In addition to quinolizidine alkaloids, we investigated the production of representative alkaloids in Solanaceae plants, such as tobacco and potato (described later), at the genome level. Genome sequences of several alleles of low-nicotine varieties of Nicotiana tabacum (tobacco) have been characterized to reveal the critical role of ethylene response factor (ERF) transcription factors in regulating nicotine biosynthesis.183) The CRISPR/Cas9-mediated knockout of these ERF transcription factors has resulted in the successful breeding of elite tobacco lines with ultra-low levels of nicotine over naturally occurring variants.183) These ultra-low nicotine lines are promising hosts for future biotechnology using tobacco.184)
4.3. Camptothecin biosynthesis.
Through functional genomic studies, several new discoveries have been made regarding the biosynthetic mechanism of the quinoline alkaloid camptothecin (CPT), which is derived via the monoterpenoid indole pathway and is a clinically used anticancer plant product.185),186) First, hairy root cultures of Ophiorrhiza pumila (Rubiaceae)129) and related species O. liukiuensis and O. kuroiwai187) were established using the Agrobacterium-Ri plasmid system. Hairy root cultures exhibited stable camptothecin production. From these hairy root cells, several enzyme genes involved in camptothecin biosynthesis have been isolated.188),189) Moreover, we established production systems using bioreactors190) and regeneration systems for O. pumila plants from hairy root cells.191) Isotope tracer experiments using 13C-labeled glucose assisted by an in silico tracer study confirmed that camptothecin is biosynthesized via a combined pathway involving secoiridoides derived from the non-mevalonate methylerythritol phosphate (MEP) pathway and tryptamine derived from the shikimate pathway192) (Fig. 10A).

In addition, by hormonally treating of hairy roots, we established a cell suspension culture that exhibited no camptothecin production. Targeted analysis of camptothecin-related metabolites193) and metabolomic and transcriptomic analyses194) were conducted on camptothecin-producing (hairy root) and non-producing (suspension culture) cells, two of which are genetically identical. In general, differential gene expression analysis between cells that produce specialized metabolites and those that do not is a powerful strategy for identifying biosynthesis-related genes. In the case of O. pumila, PCR-select subtraction and RNA-Seq analysis using next-generation sequencing194) were particularly effective, leading to the identification of two novel transcription factor genes195),196) involved in camptothecin biosynthesis. We also found that the hairy roots of O. pumila produce monoterpenoid indole alkaloids, such as camptothecin, anthraquinones, and chlorogenic acids, as specialized metabolites.193) Candidate genes involved in the biosynthesis of these compounds have also been identified.194) Furthermore, evolutionary insights into the pathways and genes involved in the biosynthesis of camptothecin-related indole alkaloids have been recently obtained through chromosome-level genome assembly of O. pumila.197)
To characterize unknown metabolites and genes of interest narrowed down through integrated omics analyses, RNA interference-based knockdown of specific genes is a feasible approach. Using the hairy roots of O. pumila, in which the tryptophan decarboxylase gene involved in the initial step of camptothecin biosynthesis was knocked down via RNA interference, unknown metabolite peaks of presumed biosynthetic intermediates were detected through metabolome analysis using ultra-high-resolution LC-FT-ICR-MS.198) To identify the function of the gene, we determined the in vivo function of the MYB transcription factor,195) which negatively regulates specialized metabolite biosynthesis, and the ERF transcription factor,196) which positively regulates the monoterpenoid indole alkaloid pathway, using RNA interference in recombinant hairy roots.
4.4. Mechanism of self-resistance against camptothecin in its producing plants.
Camptothecin acts on DNA topoisomerase I, which is present in the nucleus of all eukaryotic cells, inhibiting its activity and consequently halting cell division, exerting anticancer effects. Given this intrinsic activity, plant cells that produce camptothecin must possess self-resistance mechanisms to protect themselves from toxicity. Several mechanisms for self-resistance against such toxic compounds in plants are known,199) including the sequestration of the target molecule through specific transport and storage mechanisms. Although the intracellular and extracellular transport and sequestration of camptothecin have been investigated,200) no clear evidence has been obtained for self-resistance by such sequestration mechanisms. Thus, the mode of self-resistance is unlikely to be due to the specific transport and sequestration of camptothecin in plants. However, we identified a critical point mutation in DNA topoisomerase I, the target protein of toxicity, in camptothecin-producing plants as a unique mechanism conferring self-resistance to camptothecin.199),201)
This mutation involves a single amino acid change from asparagine to serine adjacent to the active center of topoisomerase I, which forms an intermediary covalent bond with the nicked DNA during the enzymatic reaction.201) Remarkably, the same mutation has also been found in human leukemia cells that acquired resistance to camptothecin.202),203) Detailed studies have revealed that this highly conserved asparagine-to-serine mutation maintains the enzymatic activity of DNA topoisomerase I while preventing the formation of hydrogen bonds between camptothecin and topoisomerase I during the intermediary reaction.204) As a result, the camptothecin-enzyme-DNA ternary complex cannot form, granting resistance to camptothecin but maintaining its topoisomerase activity. This mutation was the only one that conferred resistance while preserving the enzyme function.
Interestingly, this mutation was consistently found in all camptothecin-producing plant species, including those within the Rubiaceae family and Ophiorrhiza genus, but was absent in camptothecin-non-producing species, even among closely related species, showing a strict correlation with camptothecin productivity201),205) (Fig. 10B). Based on these findings, a new hypothesis regarding the genetic evolution of plant specialized metabolism and acquisition of self-resistance was proposed,203),206) suggesting that the acquisition of camptothecin productivity and self-resistance mechanisms co-evolved closely. Further theoretical studies using computational chemistry have explored the structure of DNA topoisomerase I and camptothecin toxicity, including other possible mutations with minor contributions.207) These findings contribute to the fundamental molecular evolutionary understanding of plant specialized metabolism and extend the potential for the future development of anticancer drugs and the elucidation of drug resistance mechanisms.208)
4.5. Glycyrrhizin biosynthesis and genomics of licorice plants.
Licorice is one of the most important crude drugs and raw materials for natural sweeteners prepared from the dried roots and stolons of Glycyrrhiza uralensis or related plant species. Licorice is used in more than 70% of traditional Chinese and Japanese prescriptions. Various pharmacological activities, such as anti-inflammatory effects and the improvement of liver function, have been reported for its main chemical constituent, glycyrrhizin, which has a sweet taste. The early stages of our research on licorice involved DNA extraction, the identification of licorice crude drugs (dry roots and stolons),209) and the introduction and expression of foreign genes into the hairy roots of G. uralensis,124) opening up long-lasting biotechnology research on licorice.210)
Genomic research on licorice and glycyrrhizin biosynthesis has been promoted by the establishment of an unofficial research consortium at the national level in Japan, leading to productive collaboration with many colleagues. A full-length cDNA library of G. uralensis, a representative licorice source plant, was constructed and an expressed sequence tag database of terminal read sequence data for each cDNA clone was established.211) Based on these data, several genes annotated as cytochrome P450 were extracted. Among these, clones whose expression patterns correlated well with glycyrrhizin accumulation were identified as candidate genes involved in glycyrrhizin biosynthesis, and their function was demonstrated by expression in a heterologous system, such as in yeast. As a result, two cytochrome P450 genes involved in the biosynthesis of glycyrrhetinic acid—the aglycone of glycyrrhizin—from β-amyrin, a common triterpenoid intermediate, were identified212),213) (Fig. 11). Subsequently, glycyrrhetinic acid production was achieved by expressing these newly-identified P450 genes together with β-amyrin synthase gene in recombinant yeast.213) The enzyme, CYP88D, involved in the oxidation at position 11 of β-amyrin was found to be the committing enzyme in glycyrrhizin biosynthesis.212) A comparison of the functions of CYP88D genes isolated from glycyrrhizin-producing and non-glycyrrhizin-producing species of the Glycyrrhiza genus revealed that the product specificity of these enzymes determines the ability of glycyrrhizin production.214) Furthermore, two glycosyltransferase enzymes that add two glucuronic acid moieties to glycyrrhetinic acid were identified by the collaborators,215),216) and the heterologous expression of these glycosyltransferase genes together with the previously identified P450 genes enabled the de novo biosynthesis of glycyrrhizin in recombinant yeast216) (Fig. 11).

In genome-wide research, RNA-Seq analysis of multiple tissues of G. uralensis varieties with differing glycyrrhizin content was conducted to establish a genome-related resource of licorice.217) Subsequently, a draft genome sequence (approximately 379 Mb) of an elite line of G. uralensis with high glycyrrhizin content was completed.218) This draft genome assembly revealed that genes encoding three enzymes involved in the biosynthesis of isoflavonoids, one of the pharmacologically active ingredients of licorice other than glycyrrhizin, are closely localized as a gene cluster in the genome.218) Furthermore, using cutting-edge DNA sequencing technology, we completed high-quality chromosome-scale genome assembly of G. uralensis219) (Fig. 12). Based on this nearly complete genome assembly, we discovered that the key genes involved in the biosynthesis of glycyrrhizin and other related triterpenoids are localized in a few narrow clusters on the chromosome. This represents the first high-quality chromosome-scale genome sequencing of a medicinal plant used in traditional Chinese/Japanese medicine, providing a foundation for future research on licorice and biotechnological applications, such as efficient genome-based breeding or the production of pharmaceutical components employing biotechnology.

Potatoes in the Solanaceae family contain toxic steroidal glycoalkaloids (SGAs), such as α-solanine and α-chaconine. Although cholesterol biosynthesis in plants remained largely unclear, these compounds were believed to be synthesized via cholesterol. To elucidate the molecular mechanism of SGA production, we identified a cDNA clone in the potato cDNA database with a high similarity to the human enzyme 24-dehydrocholesterol reductase, a key enzyme in cholesterol biosynthesis in animals. Functional analysis of this cDNA using recombinant yeast revealed that the encoded protein, sterol side chain reductase 2 (SSR2), catalyzes the reduction of the 24–25 double bond, converting cycloartenol to cycloartanol or desmosterol to cholesterol220) (Fig. 13). Although SSR2 is a homolog of sterol side chain reductase 1 (SSR1), which is involved in the biosynthesis of the plant hormone brassinolide, SSR2 acts as a key enzyme, specifically branching from cycloartenol to steroidal glycoalkaloid biosynthesis within phytosterol metabolism. Consequently, RNA interference silencing or gene knockout using genome editing technology can significantly reduce the levels of toxic compounds in potatoes.220) Recently, using Agrobacterium-mediated transient genome editing via transcription activator-like effector nuclease (TALEN), mutant potato lines lacking SSR2 with a reduced level of toxins were efficiently developed.221) These mutant lines are currently undergoing field trials aimed at practical cultivation of toxin-less potatoes. The all enzymes involved in the biosynthesis of SGAs have recently been completely identified.222),223) This research advances our understanding of steroidal glycoalkaloid biosynthesis in Solanum species, such as potato and tomato,224) and will aid the societal implementation of non-toxic potatoes using cutting-edge biotechnologies.

In some Solanaceae plants, such as Physalis species and Withania somnifera (Indian Ayurvedic ashwagandha), steroidal lactones, known as withanolides, derived from 24-methylenecholesterol, uniquely accumulate (Fig. 13). Through transcriptomic and metabolomic analyses of Physalis alkekengi, P. peruviana,225) and W. somnifera, a third homologous gene of SSR1 and SSR2, sterol Δ24-isomerase (24ISO) gene, was identified in addition to the conventional SSR1 and SSR2. Functional analysis of recombinant proteins in yeast and Nicotiana benthamiana and virus-induced gene silencing in W. somnifera demonstrated that 24ISO catalyzes the conversion of 24-methylenecholesterol to 24-methyldesmosterol,226) which is the committing step in withanolide biosynthesis (Fig. 13). Evolutional speculation suggested that SSR1 duplication in the evolutionary process first led to the emergence of SSR2, which resulted in increased cholesterol production in solanaceous plants, whereas a second duplication of SSR2 produced 24ISO which became specifically committed to withanolide biosynthesis in Physalis and Withania plants (Fig. 14).226)

An ERF transcription factor that regulates nicotine biosynthesis in tobacco183) as described previously (Section 4.2), similarly coordinates the expression of various genes involved in specialized metabolism across other plant species. In petunia, transcriptomic and metabolomic analyses have indicated that ectopic overexpression of the petunia ERF regulator, PhERF1, induces the expression of over 100 phytosterol biosynthesis-related genes, including unknown enzyme genes.227) Simultaneously, a significant accumulation of petunia-specific steroid compounds (petuniolides and petuniasterones) was observed in a short time of cultivation.227) This study highlights the potential of evolutionarily conserved transcriptional regulators in enhancing the production of specialized metabolites in plants through downstream transcriptional reprogramming.228)
4.7. Sulfur metabolism and omics of health-promoting sulfur-containing metabolites.
Research on the biosynthesis of sulfur-containing compounds has been based on studies on A. thaliana, as mentioned previously (Section 3.1). However prior to genomic studies of A. thaliana, the first successful cloning of a cysteine synthase cDNA from spinach marked the beginning of the development of this field.38) The cloning method at that time involved determining the partial amino acid sequences of purified enzymes and synthesizing a degenerate DNA probe based on these peptide sequences to screen a cDNA library. Subsequent screening involved genetic complementation with Escherichia coli mutants. Through these techniques, several cDNAs of related metabolic enzymes were isolated and characterized: these include additional cysteine synthases with different subcellular localizations in spinach, β-cyanoalanine synthase,39),42),43),229),230) the enzyme involved in the biosynthesis of β-substituted alanines from watermelon,231),232) and serine acetyltransferase.45),233)–235) This early pioneering molecular biology research laid the groundwork for later functional genomics in A. thaliana on sulfur assimilation and cysteine biosynthesis as described previously (Section 3.1).
Sulfur-containing specialized plant metabolites often exhibit beneficial health-promoting activities in humans, including anticancer, antihypertensive, and cholesterol-reducing activities. Regarding such specific sulfur-containing phytochemicals, molecular biological insights have been gained regarding the biosynthesis and metabolism of alkyl (alkenyl) cysteine sulfoxides, e.g., alliin, which accumulate in Allium plants, such as garlic, onion, and chives, as pungent compounds with characteristic biological activities. Early in this study, alliinase cDNA, which participates in the breakdown of alliin to produce its odorous compounds, was cloned and characterized,236) and the localization of alliinase was further investigated.237) This study further extended to the first cloning of a plant cystine lyase catalyzing the C–S bond cleavage reaction.238) For the biosynthesis of alliin in Allium plants, the cDNAs of γ-glutamyl transpeptidase, involved in the de-glutamylation step of the biosynthetic intermediate,239) and a flavin-containing monooxygenase catalyzing the reaction of sulfoxide formation at the terminal step in alliin biosynthesis,240) were successfully identified from garlic (Fig. 15A). These studies have paved the way for a better molecular understanding of the biosynthesis and metabolism of alkyl (alkenyl) cysteine sulfoxides, a unique group of plant specialized metabolites containing sulfur atom.241)

As a sub-technology of metabolomics, a comprehensive analytical method targeted to sulfur-containing compounds, S-omics, was developed using ultra-high-resolution LC-FT-ICR-MS.19),242),243) This technology has been applied for the comprehensive profiling of sulfur compounds in Allium plants.244) Using this method, a novel sulfur-containing metabolite with potential blood pressure-lowering effects, asparaptine A, was isolated from Asparagus officinalis (asparagus)245) (Fig. 15B). Furthermore, several further analogs of asparaptine, biosynthetic mechanisms, and blood pressure-lowering activity in hypertensive mice have been revealed246) together with the spatial localization of the compound in A. officinalis.247)
By extending the fundamental study on the regulation of glucosinolate biosynthesis in Arabidopsis,74) homologous regulatory factors for glucosinolate biosynthesis were also identified and functionally analyzed in kale, Brassica oleracea, an important crop belonging to the Brassicaceae family, similar to A. thaliana.248) Sulfur-containing plant compounds often exhibit unique biological activities together with characteristic biosynthetic mechanisms and enzymes. Thus, sulfur-containing phytochemicals are fascinating from the perspective of environmental adaptation through these metabolites in producing plants and the evolution of biosynthesis of these specific metabolites.
4.8. Metabolomics and phytochemical genomics in other major crops and medicinal plants.
Phytochemical genomics based on metabolomics should be extended beyond model plants to include major crops such as rice and diverse medicinal plants. In rice, a systematic metabolomic study has been conducted using rich bioresources (expression panels of full-length cDNAs, core collections, recombinant inbred lines, and specific gene-knockout mutants). This has led to the identification of novel rice genes that control nitrogen and specialized metabolism,15),24),249)–255) metabolites and genes related to rice traits,110),251),256),257) the roles of amino acid biosynthesis genes and metabolic networks,258)–262) and the molecular elucidation of morphogenesis and environmental stress responses.263)–269) Genome-wide metabolomic studies have further identified quantitative trait loci (QTLs) responsible for the accumulation of specific metabolites,156),257) extended to metabolome-genome-wide association studies (GWAS),157) and gained insights into the metabolic features of rice breeding and useful gene loci.270)–272) Additionally, metabolomic analyses have been used to assess the substantial equivalence between genetically modified tomatoes273) and soybeans.274) These studies have addressed the practical and social issues of genetically modified organisms from objective scientific viewpoints.
For representative medicinal plants containing intriguing specialized metabolites, deep transcriptome and genome analyses have been conducted together with systematic metabolomic analyses as the foundation for phytochemical genomics studies. Transcriptomics using RNA-Seq was initially completed for the previously mentioned Perilla frutescens (perilla),154) Ophiorrhiza pumila,194) Glycyrrhiza uralensis (licorice),217) followed by Pueraria lobata (kudzu),275) Sophora flavescens (kushen),276) Panax japonicus (Japanese ginseng),277) Swertia japonica,278) Physalis sp. (ground cherry),225) Lonicera japonica (honeysuckle),279) Aconitum sp. (aconite),280),281) Lithospermum officinale,282) Forsythia sp.,283) Gleditsia sinensis,284) Pueraria candollei var. mirifica,285) Cornus officinalis,286) Mallotus japonicus,287) and Magnolia obovata.288) These transcriptomic analyses have been conducted on multiple plant parts, including medicinal parts, and have integrated RNA-Seq and metabolome data from closely related species with different chemical constituents to narrow down the candidate genes involved in the biosynthesis, metabolism, and transport of specific bioactive compounds. Additionally, as described previously, high-quality chromosome-scale genome decoding of O. pumila197) and G. uralensis219) genomes was achieved soon after the completion of the draft genome assembly.218) Currently, pan-genome analyses of multiple varieties of G. uralensis and comparative genomics of licorice species with and without glycyrrhizin, including G. glabra, are underway. We are confident that these genome-related studies on medicinal plants will form a crucial research foundation for future resources.289)–291)



