Reviews
MAP kinase cascades regulating axon regeneration in C. elegans
2015 Volume 91 Issue 3 Pages 63-75
Details
2015 Volume 91 Issue 3 Pages 63-75
Mitogen-activated protein kinase (MAPK) signaling cascades are activated by diverse stimuli such as growth factors, cytokines, neurotransmitters and various types of cellular stress. Our evolving understanding of these signal cascades has been facilitated by genetic analyses and physiological characterization in model organisms such as the nematode Caenorhabditis elegans. Genetic and biochemical studies in C. elegans have shed light on the physiological roles of MAPK cascades in the control of cell fate decision, neuronal function and immunity. Recently it was demonstrated that MAPK signaling is also important for axon regeneration in C. elegans, and the use of C. elegans as a model system has significantly advanced our understanding of the largely conserved molecular mechanisms underlying axon regeneration. This review summarizes our current understanding of the role and regulation of MAPK signaling in C. elegans axon regeneration.
Mitogen-activated protein kinase (MAPK) signaling cascades are evolutionally conserved in eukaryotes from yeast to humans and play key roles in many diverse physiological processes such as development, growth and proliferation, stress responses, and immunity. A MAPK cascade consists of three classes of protein kinases: MAPK, MAPK kinase (MAPKK) and MAPK kinase kinase (MAPKKK).1) MAPK is activated by MAPKK, which itself is activated by MAPKKK. This activation cascade can be downregulated by phosphatases, in particular by members of the MAPK phosphatase (MKP) family, which can remove phosphate groups from activated MAPK.2) The MAPKs are subdivided into three main groups: the extracellular signal-regulated kinases (ERK), the c-Jun N-terminal kinases (JNK) and the p38 kinases.3) The JNK and p38 MAPKs are activated by environmental stresses such as heat, oxidative stress and ionizing radiation and regulate processes such as stress response, apoptosis and inflammation in mammals (Fig. 1).4)–10) The MKK4 and MKK7 MAPKKs have been shown to activate JNK, and the MKK3 and MKK6 MAPKKs serve as the major activators of the p38 MAPK. The individual MAPKKs are themselves phosphorylated and activated by specific MAPKKKs. Different MKPs display different activities toward the ERK, JNK, and p38 MAPKs.
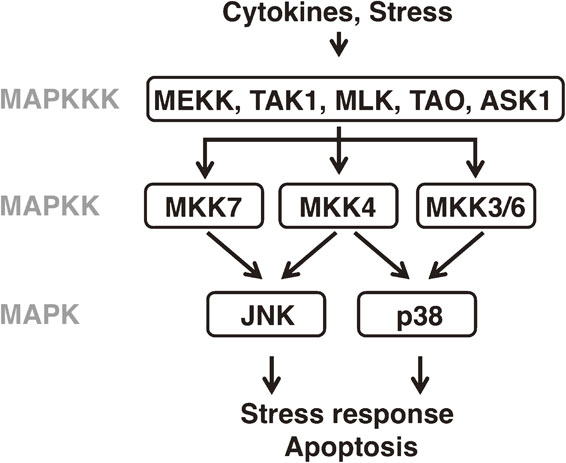
Overview of JNK and p38 MAPK signaling in mammals. The MAPK pathways consist of MAPK, MAPKK and MAPKKK. The JNK and p38 MAPK pathways are activated by cytokines and various types of stress, and regulate numerous processes such as the stress response and apoptosis.
The ease of genetic manipulation has made the nematode Caenorhabditis elegans an excellent model organism for dissecting the molecular mechanisms underlying numerous processes such as programmed cell death, cell migration and differentiation. A number of JNK and p38 MAPK signaling pathways have been described in C. elegans (Fig. 2).11)–17) Genetic studies demonstrate that the C. elegans JNK pathway, consisting of JKK-1 (a MKK7-type MAPKK) and JNK-1 (a JNK-type MAPK), regulates coordinated movement via type D GABAergic (GABA: γ-aminobutyric acid) motor neurons and also plays a role in synaptic vesicle transport. C. elegans possesses another JNK cascade consisting of MLK-1 (a MLK-type MAPKKK), MEK-1 (a MKK7-type MAPKK) and KGB-1 (a JNK-type MAPK), which is involved in the regulation of the response to heavy-metal stress. One of the C. elegans p38 MAPK pathways, consisting of NSY-1 (an ASK-type MAPKKK), SEK-1 (a MKK3/6-type MAPKK) and PMK-1 (a p38-type MAPK), is involved in innate immunity and the oxidative-stress response. The NSY-1 p38 MAPK pathway is also involved in establishing asymmetric cell fate during neuronal development. The second C. elegans p38 MAPK pathway, consisting of DLK-1 (a DLK-type MAPKKK), MKK-4 (a MLK4-type MAPKK) and PMK-3 (a p38-type MAPK), regulates presynaptic development. Another component of the C. elegans JNK and p38 MAPK pathways is the MKP VHP-1, which negatively regulates these MAPK pathways by dephosphorylating KGB-1, PMK-1 and PMK-3.18),19)
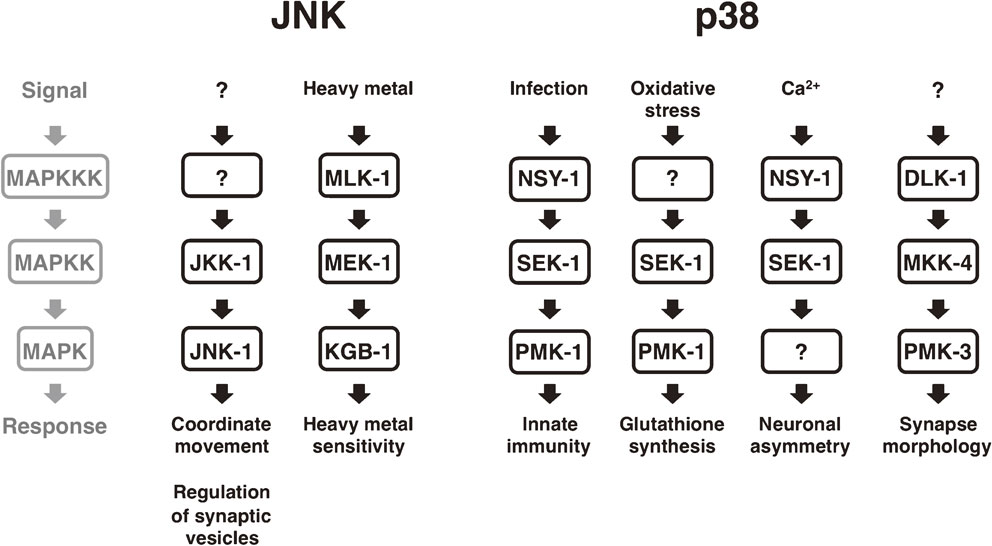
JNK and p38 MAPK signaling pathways in C. elegans. Several distinct JNK and p38 MAPK pathways in C. elegans respond to various stimuli such as heavy metal, infection and oxidative stress, and induce corresponding adaptive responses.
Recent genetic studies have shown that the MLK-1–MEK-1–KGB-1 JNK and DLK-1–MKK-4–PMK-3 p38 MAPK pathways also regulate axon regeneration in C. elegans. Several studies have demonstrated that the homologues of DLK-1 are required for axon regeneration in both Drosophila melanogaster and mice.20)–23) Similarly, JNK-mediated activation of c-Jun is important for axonal outgrowth of neurons in axotomized rat nodose and dorsal root ganglia (DRG) and mouse DRG.24)–26) These discoveries both validate the use of C. elegans as a model system for axon regeneration and suggest that the core machinery that regulates axon regeneration is conserved from worms to mammals.
When an axon is severed, the proximal axonal fragment, which is connected to the cell body, initially retracts and forms a swelling called retraction bulb at its end. If conditions are favorable, the retraction bulb is transformed in a growth cone, a dynamic structure, which extends and guides the regenerating axon towards its target. An axon’s ability to regenerate after damage depends on both its intrinsic capacity and the external environment, and is regulated by a balance of regeneration-promoting and -inhibiting factors (Fig. 3).27) In adult mammals, the regenerative capacity of neurons in the central nervous system (CNS) is limited. In mammals, several myelin-associated factors such as myelin-associated glycoprotein (MAG), oligodendrocyte myelin glycoprotein (OMgp) and Nogo inhibit the regeneration of axons in the CNS.28)–33) On the other hand, injured peripheral neurons activate intrinsic signaling pathways that enable axonal regrowth. When an axon is injured, levels of intracellular calcium (Ca2+) and cyclic adenosine monophosphate (cAMP) are increased.34),35) This increase in cAMP levels activates protein kinase A (PKA), which activates the axon regeneration-promoting transcription factor cAMP response element binding protein (CREB).36)–38) PKA also contributes to the local modification of the cytoskeleton, which is necessary for the formation and maintenance of the growth cone, a specialized structure necessary to initiate regeneration.39) Upon axon severance, a long-range retrograde signal is transported from the site of injury to the cell body where various transcription factors are upregulated and subsequently contribute to increased protein synthesis, cell survival and neurite outgrowth.40) The axonal transport of mRNAs, their stabilization and their local translation provide the structural and regulatory proteins required for axon regeneration in mammals.41) Manipulation of these processes can improve the chances for successful axon regeneration. Nonetheless, our understanding of the axon-regeneration machinery operating in the adult nervous system of mammals remains limited.
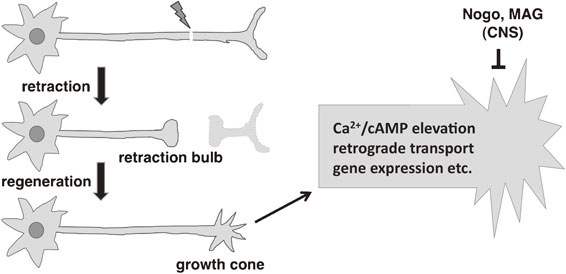
Overview of axon regeneration in mammals. After axon injury, the proximal fragment of the injured axon retracts and forms a retraction bulb. If conditions favor regeneration, the retraction bulb is transformed into a growth cone, which extends in an attempt to rebuild a synaptic connection with its target. The right panel shows an enlarged view of the growth cone where several factors regulate axon regeneration. The regeneration process depends on various events such as increased intracellular concentrations of Ca2+ and cAMP, retrograde transport and gene expression. In the central nervous system (CNS) factors such as Nogo and MAG suppress the regeneration response.
C. elegans is rapidly emerging as a genetic model for probing axon regeneration in the mature nervous system. This animal has a transparent body and it is possible to label specific neurons by cell-specific expression of GFP or other fluorescent proteins. Using a laser device, together with a fluorescent microscope, it is possible to perform laser surgery at the axon level. Moreover, axons of the GABAergic D-type motor neurons of C. elegans can successfully regrow after laser surgery (Fig. 4).42) Soon after the laser surgery approach was developed, it was complemented by the use of β-spectrin mutants whose axons are fragile and spontaneously break, thereby initiating axon regeneration.43) These tools enabled researchers to discover novel regulators of axon regeneration by mutagenizing worms and looking for mutants that were unable to regenerate their axons, or alternatively, that regenerate axons better than the wild-type controls. A similar approach is to downregulate specific genes by feeding the worms bacteria expressing a library of siRNAs and looking for positive or negative effects on axon regeneration ability. These genetic screens have been successful in identifying the p38 and JNK MAPK pathways specifically required for axon regeneration.43),44)
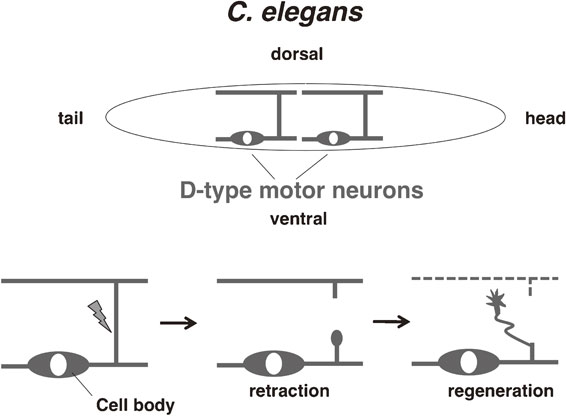
Overview of axon regeneration in C. elegans. The GABA-ergic D-type motor neurons in C. elegans have cell bodies on the ventral side and extend axonal commissures dorsally where they make synaptic connections with muscles. When an axonal commissure is severed with a laser, the proximal axonal fragment initially retracts and after several hours its tip is transformed into a growth cone, which begins to extend dorsally.
An RNAi screen for regulators of axon regeneration identified the MAPKKK DLK-1.43) It was later demonstrated that activation of the p38 MAPK pathway consisting of DLK-1, MKK-4 and PMK-3 is required for axon regeneration of motor and mechanosensory neurons (Fig. 5).45) DLK-1 p38 MAPK signaling promotes axon regeneration by activating the MAPKAP kinase MAK-2, which functions to stabilize the mRNA encoding the bZip transcription factor CEBP-1.45) CEBP-1 in turn promotes axon regeneration, although its targets in this context remain unknown. DLK-1 signaling is also involved in the control of microtubule dynamics during axon regeneration. In mature axons the microtubule cytoskeleton is stably maintained, but after axonal injury it is transformed in a highly dynamic state. This transformation is achieved in two steps: an increase in the number of growing microtubules at the injury site and Δ2 modification of α-tubulin to support persistent microtubule growth.46) DLK-1 signaling is involved in the control of both of these phases. First, it decreases the localization of the microtubule depolymerizing kinesin-13 family member KLP-7 in axons by phosphorylating the N-terminus of KLP-7 and thereby reducing its affinity for microtubules. DLK-1 signaling also upregulates the cytosolic carboxypeptidase CCPP-6, which modifies α-tubulin to the more stable Δ2 form and therefore supports persistent microtubule growth.46) These effects apparently do not depend on CEBP-1. Although the precise steps by which DLK-1 is activated in axon regeneration remain unknown, a Ca2+-dependent mechanism of activation has been recently described.47) In its inactive form DLK-1 is a heteromer consisting of a shorter, inactive isoform DLK-1S that binds to and inhibits the longer, active isoform DLK-1L. An increase in Ca2+ concentration, which occurs for example after axon injury, dissociates DLK-1S from DLK-1L and allows the formation of DLK-1L homodimers, which are the active form. This spatiotemporal mechanism of DLK-1 activation is suitable for its role in axon regeneration.
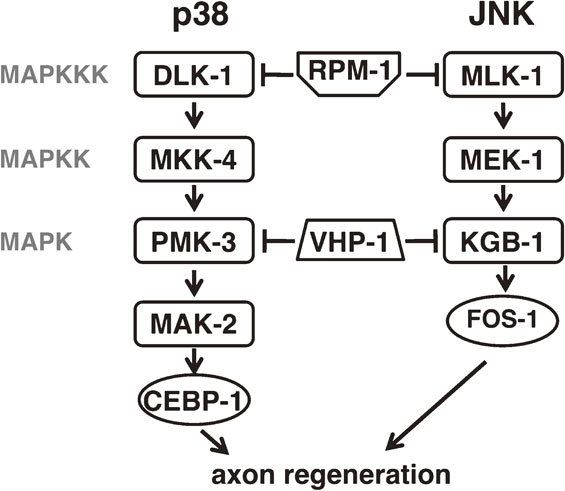
MAPK pathways regulating axon regeneration in C. elegans. The p38 pathway consisting of DLK-1, MKK-4 and PMK-3 and the JNK pathway consisting of MLK-1, MEK-1 and KGB-1 positively regulate axon regeneration. Some factors acting upstream or downstream of these cascades are also indicated.
An RNAi screen for regulators of axon regeneration also identified another MAPKKK, MLK-1, which is involved in the JNK pathway.43) A follow-up study demonstrated that mutation of any of the components of the MLK-1–MEK-1–KGB-1 JNK MAPK pathway significantly impairs axon regeneration (Figs. 5, 6).44) Conversely, overexpression of MLK-1 improves the frequency and timing of growth cone formation, as well as the rate of successful migration of growth cones to their target sites. This effect was observed also in older animals, which usually are very limited in their regeneration ability. One likely target of JNK MAPK signaling is the transcription factor FOS-1.17) A loss-of-function mutation in fos-1 significantly reduces the formation of growth cones.48) FOS-1 is phosphorylated at Thr-304 by the KGB-1 JNK and expression of a fos-1 mutant lacking this site (T304A mutant) fails to rescue the defect in axon regeneration of fos-1 mutants. These observations suggest that phosphorylation of FOS-1 on Thr-304 by KGB-1 is important for axon regeneration, although it is still unknown what genes regulated by FOS-1 function to promote axon regeneration.
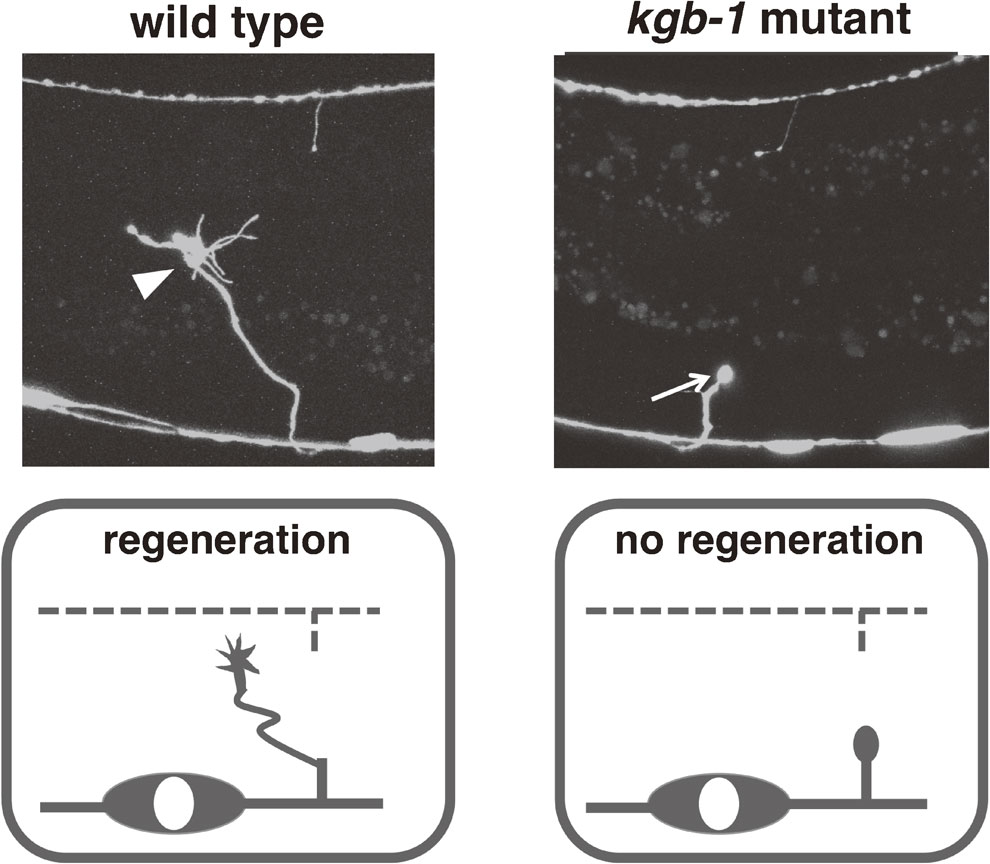
Axon regeneration is strongly suppressed in kgb-1 mutants. Shown are typical pictures of successful (left, wild type) and failed (right, kgb-1 mutant) initiation of axon regeneration. In the wild type, the severed axon has formed a prominent growth cone 24 hrs after the injury (arrowhead). In the kgb-1 mutant, the retraction bulb (arrow) has failed to transform into a growth cone.
Several regulators involved in axon regeneration are common to both the p38 and JNK pathways (Fig. 5). For example, DLK-1 and MLK-1 are both targeted for ubiquitin-mediated degradation by the E3 ubiquitin ligase RPM-1.45) Also, the MAPK phosphatase VHP-1 inactivates both PMK-3 and KGB-1.18),19) As expected, both rpm-1 and vhp-1 mutants display a MAPK-dependent improvement in axon regeneration.44),45) However, despite the similarities in their regulation, the downstream effectors of the p38 and JNK pathways are probably largely distinct.
The JNK MAPK pathway is negatively regulated by the MAPK phosphatase VHP-1 (Fig. 7).18),44) The importance of this negative regulation of MAPK signaling is underscored by the fact that vhp-1 mutants are arrested in their development in the early larval stage and almost never reach adulthood.18) Inactivating mutations in mlk-1, mek-1 or kgb-1 suppress this vhp-1 developmental arrest, indicating that the vhp-1 phenotype is caused by hyperactivation of the JNK MAPK pathway. This phenotype has made possible a genome-wide RNAi screen for regulators of the JNK MAPK pathway (Figs. 7, 8). Briefly, we generated animals heterozygous for vhp-1 in which one allele is mutant and the other allele is normal and linked on the same chromosome to a GFP-expressing marker. The animals were fed bacteria that express RNAi from an RNAi library targeting most of the worm genes. We then looked for animals that reached adulthood but did not express GFP. Lack of GFP would indicate that these animals had become homozygous for the vhp-1 mutant allele, and survival would presumably be due to RNAi downregulation of a gene that contributes to JNK MAPK pathway activity. The genes discovered by this screen were termed svh genes, for suppressors of vhp-1 lethality (Fig. 8). The important role that the JNK MAPK cascade plays in axon regeneration suggests that the svh genes might also function as regulators of axon regeneration. The following sections describe the roles that three svh gene products play in axon regeneration: the growth factor SVH-1, its tyrosine kinase receptor SVH-2, and the endocannabinoid hydrolase SVH-3/FAAH-1.
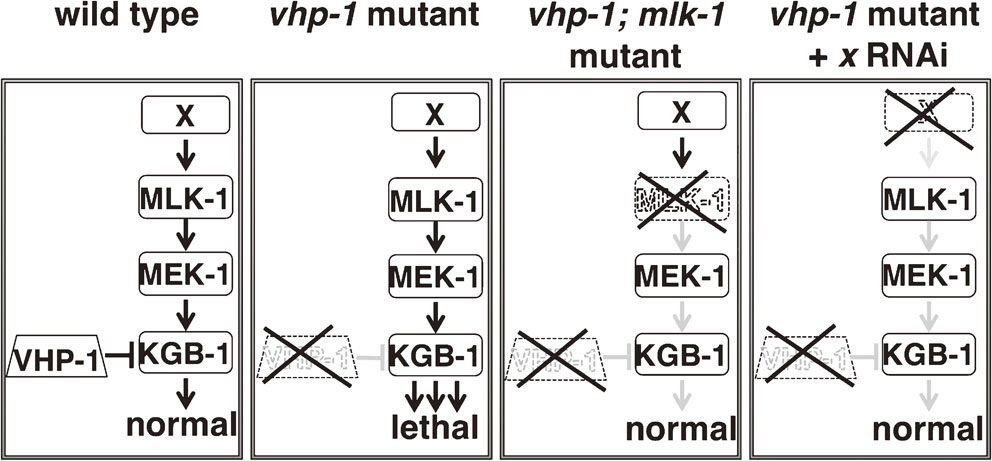
Isolation of vhp-1 suppressors. In wild-type animals, the VHP-1 phosphatase restrains activation of the JNK MAPK cascade to normal levels. In vhp-1 mutants, the hyper-activation of the JNK cascades causes larval arrest and death. Downregulation by RNAi of any of the components of the JNK pathway, for example mlk-1, or a factor that positively regulates the activity of the JNK signaling is sufficient to suppress mutant vhp-1 lethality. The novel genes discovered by this screen were named svh (suppressor of vhp-1 lethality).
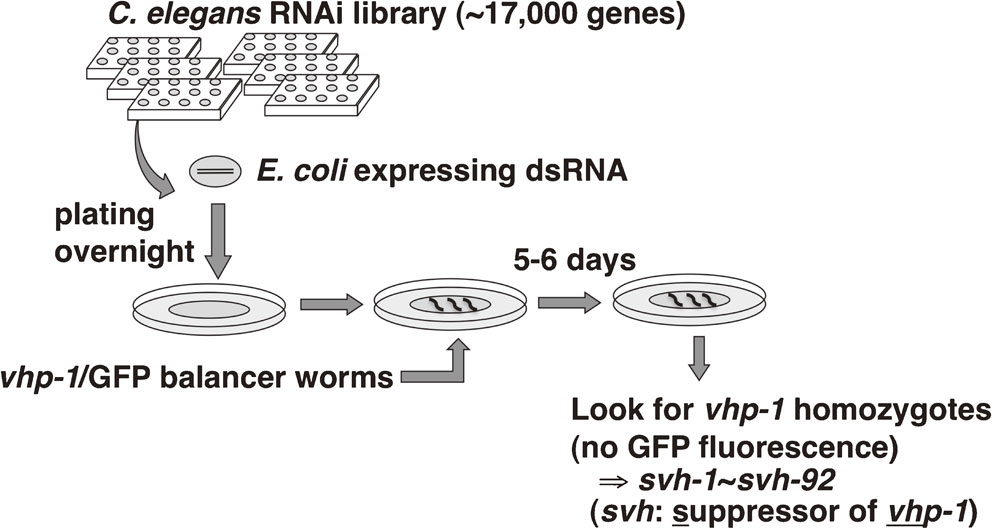
RNAi screen for vhp-1 suppressors. E. coli cultures expressing an RNAi library targeting ∼17,000 genes in C. elegans were plated on individual plates overnight. To each plate were added several vhp-1 heterozygous mutant worms expressing a GFP-tagged balancer linked to the wild-type vhp-1 allele. Ingestion of the dsRNA-expressing bacteria knocks down specific genes in the worm. After several days the plates were screened for homozygous vhp-1 mutants, which have lost the balancer and exhibit no GFP fluorescence and are therefore mutant vhp-1 homozygous. The genes that when knocked down allow vhp-1 homozygous mutants to survive were named svh (suppressor of vhp-1 lethality). In total 92 svh genes were identified.
The first gene identified in the screen for vhp-1 suppressors, svh-1, encodes an extracellular protein possessing a C-type lectin domain, a kringle domain, two LDL receptor-like domains, an N-terminal domain related to plasminogen activation peptide and a serine protease domain (Fig. 9).49) With the exception of the C-type lectin and LDL receptor-like domains, these protein domains are found also in several mammalian proteins: hepatocyte growth factor (HGF), macrophage stimulating protein (MSP) and plasminogen. This suggests that SVH-1 belongs to the HGF/MSP/plasminogen family. The second gene identified, svh-2, encodes a tyrosine kinase receptor, homologous to the HGF receptor Met and the MSP receptor Ron (Fig. 9).49) Both svh-1 and svh-2 mutants are significantly impaired in their axon regeneration ability. Time-lapse movies indicate that these mutants fail to form stable growth cones, which is a required first step for successful axon regeneration.49) Conversely, overexpression of either svh-1 or svh-2 induces the formation of enlarged and highly mobile growth cones, markedly improves regeneration outcome, and partially rescues the decline in axon regeneration ability that progresses with age. These results indicate that SVH-1 and SVH-2 are both necessary for axon regeneration and are able to promote regeneration when overexpressed. Together with this, the sequence homology of SVH-1 to HGF and of SVH-2 to the HGF receptor Met suggests that SVH-1–SVH-2 might function as a ligand–receptor pair in axon regeneration (Fig. 9).
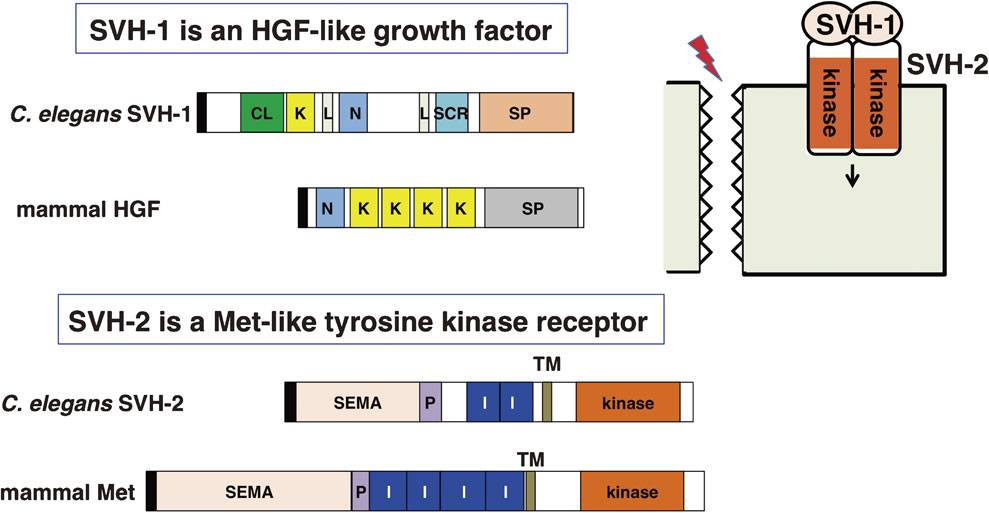
Structures of the HGF-like growth factor SVH-1 and its Met-like tyrosine kinase receptor SVH-2. The figure compares the domain structures of SVH-1, SVH-2 and their mammalian homologues, HGF and Met, respectively. CL: C-type lectin, K: Kringle domain, L: LDL receptor-like domain, N: N-terminal domain, SP: serine protease-like domain, SEMA: sema domain, P: PSI-related domain, I: IPT-like domain, TM: transmembrane domain, kinase: tyrosine kinase domain. Black rectangles indicate signal sequences.
Localization of svh-1 expression can be assessed using a reporter gene consisting of the promoter region of the svh-1 gene and the coding sequence of the fluorescent protein Venus. Analyses with this marker indicate that SVH-1 is constitutively expressed in a pair of sensory neurons (ADL) in the head. Deleting the signal sequence from the svh-1 gene generates a secretion-deficient mutant protein that accumulates in the ADL neurons and is not functional in axon regeneration. Expression of svh-1 from either a pan-neuronal promoter or a pharyngeal muscle-specific promoter is sufficient to rescue the svh-1 mutant defect. Furthermore, SVH-1 is not expressed in motor neurons, even after axon injury. These results indicate that SVH-1 functions in axon regeneration in a cell non-autonomous manner. The fact that SVH-1 is constitutively secreted would suggest that it has functions in addition to the regulation of axon regeneration. SVH-1 contains an intact protease domain whose activity is not necessary for axon regeneration and thus its protease activity has other functions. Indeed, SVH-1 acts as a protease required for larval growth.50) Similar studies looking at the expression pattern of a reporter gene driven by the svh-2 promoter reveal that svh-2 is normally not expressed in motor neurons. However, several hours after axotomy svh-2 expression is induced in the injured motor neurons. It was also found that svh-2 can rescue the svh-2 defect only when expressed in the same neurons that undergo axon regeneration. These results are consistent with the idea that SVH-2 functions to promote axon regeneration in a cell-autonomous manner. The injury-dependent expression of SVH-2 probably serves to ensure that the constitutively secreted SVH-1 can initiate SVH-2 signaling in motor neurons only when an axon regeneration response is necessary (Fig. 10).
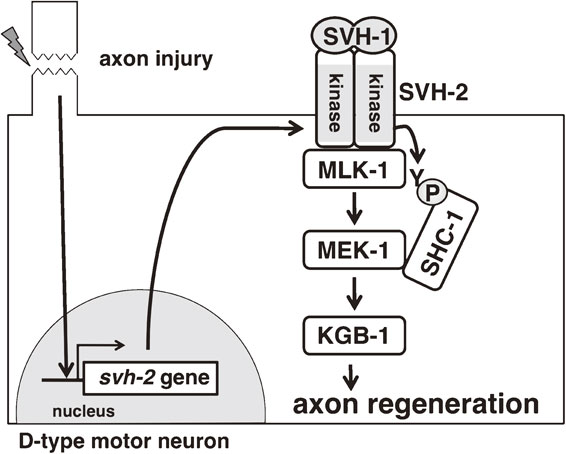
Model for the roles of SVH-1 and SVH-2 in axon regeneration. Axon injury of a D-type motor neuron induces the expression of the tyrosine kinase receptor SVH-2 in the injured neuron. SVH-2 is activated by the HGF-like growth factor SVH-1 and phosphorylates the MAPKKK MLK-1 on a tyrosine residue. The tyrosine-phosphorylated MLK-1 is then recognized by the adaptor protein SHC-1, which also binds the downstream effector of MLK-1, the MAPKK MEK-1. MEK-1 activates KGB-1 JNK, which ultimately promotes the axon regeneration response.
The mammalian homologues of SVH-2, Met and Ron, form a dimer after ligand binding, and trans-phosphorylate their respective kinase activation loops on tyrosine residues.51),52) Several experimental results indicate that SVH-2 functions as a tyrosine kinase in a similar manner. We can create SVH-2 dimers by fusing a dimerization leucine zipper motif (Tpr, translocated promoter region) to the kinase domain and C terminus of SVH-2, thus mimicking ligand-induced dimer formation. When expressed in COS-7 cells and subjected to Western blotting, this hybrid protein Tpr::SVH-2C is recognized by an anti-phosphotyrosine antibody, indicating that it is phosphorylated on tyrosine residues. A catalytically inactive mutant form of Tpr::SVH-2C is not recognized by the anti-phosphotyrosine antibody, suggesting that Tpr::SVH-2C autophosphorylates at tyrosine residues. When expressed in C. elegans, the catalytically inactive form of svh-2 is unable to rescue the axon regeneration defect of svh-2 mutants. This demonstrates that this tyrosine kinase activity is essential for SVH-2 function in axon regeneration.49)
SVH-2 was identified in a screen designed to discover regulators of the JNK MAPK pathway. Western blotting analysis using COS-7 cells expressing MLK-1 MAPKKK and SVH-2 showed that MLK-1 and SVH-2 can physically interact. Additionally, MLK-1 was found to be tyrosine-phosphorylated in the presence of the wild-type, but not a kinase-dead mutant form of SVH-2, suggesting that SVH-2 directly phosphorylates MLK-1. The most likely site in MLK-1 phosphorylated by SVH-2 is the tyrosine in the sequence NPXY, which creates a docking site for the phospho-tyrosine binding (PTB) domain of SHC-1. SHC-1 is an adaptor protein that connects MLK-1 and its downstream MAPKK MEK-1. Western blotting analysis indicates that SHC-1 binds to MLK-1 only when a kinase-active form of SVH-2 is co-expressed, suggesting that SVH-2 phosphorylation is necessary for SHC-1 binding to MLK-1 (Fig. 10).53)
Taken together, these results support a model in which SVH-1 is constitutively expressed and secreted, while its tyrosine kinase receptor SVH-2 is conditionally expressed in neurons in response to axon injury, whereupon it can be activated by SVH-1. SVH-2 activation induces MLK-1 phosphorylation, leading to enhanced JNK MAPK signaling, which promotes the axon regeneration response (Fig. 10).49)
A third gene, svh-3, was also identified in the screen for regulators of the JNK MAPK pathway. svh-3 is identical to faah-1, which encodes the worm homologue of the fatty acid amide hydrolase, FAAH (Fig. 11).53) FAAH is an enzyme that hydrolyzes and thereby inactivates several types of endocannabinoids, derivatives of polyunsaturated fatty acids such as arachidonic acid. Endocannabinoids regulate various processes such as synaptic plasticity, pain sensation and memory formation and are also found in worms, where they were discovered to mediate the effects of diet on lifespan.54),55) Mutation of faah-1 significantly reduces axon regeneration. When faah-1 is knocked down by siRNA, the concentrations of various endocannabinoids increase several-fold, indicating that FAAH-1 is required for maintenance of appropriate low levels of endocannabinoids.55) This suggested that the defect in axon regeneration observed in faah-1 mutants might be due to the abnormal accumulation of endocannabinoids. Indeed, incubation of wild-type worms with medium containing one of the FAAH-1 substrates arachidonoyl ethanolamide (AEA) strongly inhibits axon regeneration. Several other endocannabinoids, such as eicosapentaenoyl ethanolamide (EPEA), have no effect on axon regeneration, indicating that AEA specifically suppresses axon regeneration.
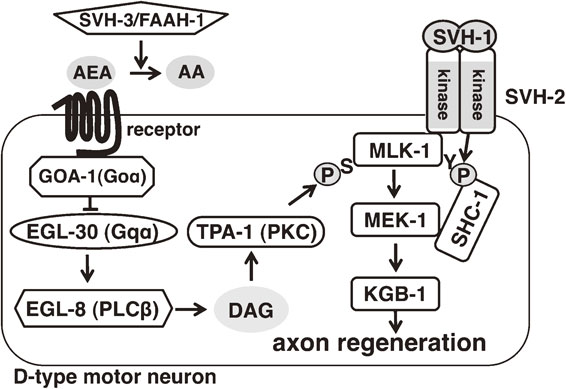
SVH factors regulating axon regeneration in C. elegans. SVH-2 regulates the tyrosine phosphorylation state of MLK-1, thereby regulating MLK-1 interaction with the adaptor protein SHC-1 and transmission of signals to MEK-1. SVH-3/FAAH-1 controls the levels of the endocannabinoid arachidonoyl ethanolamide (AEA) by hydrolyzing it to arachidonic acid (AA). AEA signals through a yet-unidentified receptor to activate GOA-1 (Goα homologue). GOA-1 antagonizes a signaling cascade consisting of EGL-30 (Gqα homologue), EGL-8 (PLCβ homologue) and TPA-1 (protein kinase C homologue). TPA-1 induces MLK-1 activity by phosphorylation on Ser-355 in its activation loop.
In mammals the endocannabinoids bind to and activate several transmembrane receptors such as CB1 and CB2. The activated CB1/CB2 receptors in turn activate G proteins, which convey signals downstream. If a similar endocannabinoid signaling mechanism exists in worms, then loss-of-function mutations in any of its components should confer resistance to AEA. It is difficult to test this hypothesis on the receptor level because CB1 and CB2 have no apparent worm homologues, but there is a well-conserved homologue for mammalian Goα, GOA-1.56),57) Indeed, loss-of-function goa-1 mutants are resistant to the effect of AEA on axon regeneration. Conversely, worms expressing a constitutively active mutant form of goa-1 have a reduced axon regeneration rate even in the absence of AEA. These results suggest that GOA-1 is the downstream effector of AEA. Further analysis of this downstream signaling pathway supports a model in which GOA-1 antagonizes the Gqα protein EGL-30, which in turn activates the phospholipase EGL-8. EGL-8 generates diacyl-glycerol (DAG), an activator of the protein kinase C (PKC) homologue TPA-1. As expected from this model, mutations in egl-30, egl-8 or tpa-1 result in decreased axon regeneration. The most likely target of TPA-1 in the context of axon regeneration is MLK-1 MAPKKK. Within the activation loop of MLK-1 is a consensus site for PKC phosphorylation (sequence RFS), and the serine within this sequence, Ser-355, is phosphorylated by TPA-1. The link between TPA-1 and MLK-1 is further established by the observation that expression of a phospho-mimetic form of MLK-1(S355E) can suppress the regeneration defect of tpa-1 mutants. These results confirm that MLK-1 phosphorylation on Ser-355 is a crucial step for its activation and activity in axon regeneration. In summary, increased signaling from the endocannabinoid pathway leads to suppression of the EGL-30–EGL-8–TPA-1 signaling cascade, which suppresses activation of the MLK-1 JNK MAPK cascade and thereby decreases axon regeneration (Fig. 11).
Results from these studies on SVH-1, SVH-2 and SVH-3/FAAH-1 indicate that the activity of MLK-1 MAPKKK in promoting axon regeneration is regulated by at least two independent mechanisms: it is serine phosphorylated by TPA, which induces its kinase activity, and it is tyrosine phosphorylated by SVH-2, which promotes its interaction with the downstream target MEK-1 MAPKK via the adaptor protein SHC-1 (Fig. 11). Thus, MAPK signaling is regulated at multiple steps, which presumably provides spatiotemporal specificity and ensures responsiveness to the appropriate environmental cues. Negative regulation by endocannabinoids might further restrict the timing of the axon regeneration response to times when conditions are favorable. The synthesis of endocannabinoids increases following several types of injury, such as spinal cord injury and nerve inflammation, and these lipids might therefore function as injury signals.58),59) The regenerating axon constantly depends on contact with extracellular matrix components both for mechanical support and for guidance towards its target and a significant injury might destroy most of the substrate that the growing cone depends on for proper extension and guidance. In these circumstances, the endocannabinoids might provide an injury signal to restrict the axon regeneration response until the surrounding tissue has recovered sufficiently to be able to support the regenerating axon.
It is still unclear how exactly MAPK signaling regulates axon regeneration but at least some of the regulation involves known transcription factors. The downstream effector of DLK-1 p38 MAPK signaling in axon regeneration is the MAPK-activated kinase 2 MAK-2, which regulates stability of the cebp-1 mRNA encoding a bZip family transcription factor.45) In addition, one target of JNK MAPK signaling is the transcription factor FOS-1.17),48) However, it is still unknown what genes are regulated by CEBP-1 and FOS-1 to promote axon regeneration.
MAPK signaling might regulate also axon regeneration via its action on the cytoskeleton and associated regulatory factors. Cytoskeleton reorganization is a major step in the transformation of the inert retraction bulb that forms after axotomy into the highly dynamic growth cone.39) The formation and maintenance of this complex structure requires precise local control of its building blocks and regulators. MAPK signaling has been implicated in numerous aspects of cytoskeleton regulation. JNK exerts its effects on the cytoskeleton by phosphorylating numerous microtubule-associated proteins (MAPs) such as MAP2, MAP1B, tau and the stathmin family microtubule-destabilizing protein SCG10.60)–63) Recently it was demonstrated that one major role of DLK-1 signaling in axon regeneration is the control of microtubule dynamics via the kinesin-13 family member KLP-7 and the microtubule-modifying enzyme cytosolic carboxypeptidase CCPP-6.46) The JNK substrate SCG10 may also be a target of MAPK signaling during axon regeneration, as it is upregulated in rat motor and dorsal root ganglion (DRG) neurons following sciatic nerve crush.64),65)
Many of the MAPK-associated axon regeneration factors discovered in C. elegans have not yet been verified in other animal systems. However, the conserved central role of MAPK signaling in axon regeneration suggests that at least some of these factors may be relevant for axon regeneration in other animals.

Kunihiro Matsumoto was born in Nagasaki in 1951, and graduated Faculty of Engineering, Osaka University in 1974. At the second year of his Ph.D. course, he started his research career with studies on signal transduction in yeast as Assistant Professor at Tottori University in 1977. He received his Ph.D. from Osaka University in 1982. He then moved to DNAX Research Institute in California as Senior Scientist in 1985. He continued his studies on signal transduction, especially MAP kinase pathways, in yeast. In 1990, he returned to Japan as Professor at Nagoya University, where he switched his research area to MAP kinase pathways in mammalian cells and the nematode Caenorhabditis elegans. For his great achievements on molecular genetics, he was honored to receive Japanese Genetic Society Young Investigator Award in 1985, Nissan Science Foundation Award in 2001, Kihara Memorial Foundation Award in 2001, Inoue Prize for Science in 2002, Mochida Memorial Foundation Award in 2012, National Prize of Purple Ribbon Medal (Japan) in 2012 and Chunichi Cultural Award in 2013. He becomes the Dean of Graduate School of Science at Nagoya University in 2015.