Abstract
Rheumatoid arthritis (RA) is a common autoimmune disease that results in significant morbidity. As with other complex disorders, genome-wide association studies (GWASs) have greatly contributed to the current understanding of RA etiology. In this review, we describe the genetic configuration of RA as revealed primarily through GWASs and their meta-analyses. In addition, we discuss the pathologic mechanisms of RA as suggested by the findings of genetic and functional studies of individual RA-associated genes, including HLA-DRB1, PADI4, PTPN22, CCR6 and FCRL3, and the potential use of genetic information for RA treatment in clinical practice.
1. Pathogenesis and genetic factors of rheumatoid arthritis
Rheumatoid arthritis (RA) is one of the most common forms of autoimmune arthritis, with a prevalence of 0.3–1.1% in populations with European ancestry and 0.1–0.5% in those with Asian ancestry.1) RA is characterized by the inflammation of synovial joint tissues and the formation of rheumatoid pannus, which is capable of destroying adjacent cartilage and bone and causing subsequent joint deformity. Hyperplasia of the synovial lining cell layer is a hallmark of RA. Additional characteristic features of RA synovitis include infiltration of immune cells and neo-angiogenesis. Mononuclear cellular infiltrates are typically present in RA deep in the synovial lining layer. These infiltrates are comprised predominantly of macrophages and lymphocytes, which can form aggregates. CD4+, CD45RO+ memory T lymphocytes are common, especially in the areas between aggregates. B lymphocytes and immunoglobulin producing plasma cells are present within and between aggregates and in foci.
Sera from the majority of RA patients contain autoantibodies, such as rheumatoid factor (RF) or anti-citrullinated protein antibodies (ACPAs). The presence of these autoantibodies leads to the classification of RA as an autoimmune disease. Although RF is also found in other autoimmune diseases and immunological conditions, such as chronic infection and inflammation, the presence of ACPAs is highly specific, an indication that autoimmunity to citrullinated proteins is a key factor of RA pathogenesis.2) However, approximately 20% of RA patients lack these autoantibodies, suggesting etiologic heterogeneity of the disease. In clinical practice, the development of biologics that target inflammatory cytokines or molecules on the immune cells has dramatically improved the outcome of RA, although a certain proportion of patients do not respond to these therapies. The limitations of current RA therapies reveal the need for further investigation of disease pathogeneses and identification of new therapeutic targets.
The relative risks of RA in monozygotic twins (λ MZ) and siblings of affected individuals (λ sib) are 12 to 62 and 2 to 17, respectively.3) Consequently, the heritability of RA is estimated to be approximately 60%.4) Therefore, there is strong evidence regarding the existence of genetic factors that increase susceptibility to RA. Indeed, the human leucocyte antigen (HLA) region, the major histocompatibility complex (MHC) in humans, was initially suggested as an RA-associated locus in 1976. The HLA complex allows the immune system to distinguish between self and non-self (foreign) entities. Nonetheless, since HLA-DRB1 was identified as an RA-associated molecule in 1986, several linkage studies have also demonstrated that non-MHC genes are also associated with RA.5),6)
2. HLA class II genes
Serological studies revealed that the frequency of the serotypes of HLA-DR4, one of HLA class II genes, is higher in RA patients compared with controls.7) Other serotypes, such as DR1, are also associated with increased risk for RA, although the increase in risk is moderate compared with that of DR4. The sequencing of HLA-DRB1, which encodes the polymorphic β-chain of the DR molecule, revealed that RA risk HLA alleles differ among ethnic groups. In populations of European ancestry, HLA-DRB1*04:01, *04:04, and *01:01 are the most frequent RA risk alleles, whereas in East Asian populations, HLA-DRB1*04:05 and *01:01 are the most frequent RA risk alleles.1) In addition, several subtypes of the DR4 allele, such as *04:02 and *04:03 appear to confer protection against the disease. These observations led to the widely accepted hypothesis that a conserved amino acid sequence (i.e., QKRAA/QRRAA/RRRAA) spanning residues 70–74 in the third hypervariable region of the β chain (which is referred to as a “shared epitope [SE]”) is associated with RA susceptibility.8)
The importance of ACPAs in RA has become evident over the last decade. A strong association between SE alleles and ACPAs in RA patients has been found in several populations,9)–11) suggesting that DR molecules encoded by SE alleles are actually involved in the presentation of citrullinated peptides to T cells. In fact, a study with human DR4–transgenic mice reported that the presence of citrullinated peptide leads to increased affinity to DR molecules and subsequent activation of CD4 T cells.12) On the other hand, the association between pathology and citrullinated autoantigens is poorly defined because clinical laboratory testing of sera from RA patients is dependent on an artificial cyclic-citrullinated peptide that could react with multiple citrullinated self-proteins. However, highly relevant target molecules such as fibrinogen, α-enolase, vimentin, immunoglobulin binding protein (BiP), and type II collagen, are expressed in the synovial joint tissues.13) Actually, citrullinated vimentin and aggrican-specific CD4 T cells in the peripheral blood HLA-DR4 (*04:01)-positive RA and healthy individuals were observed.
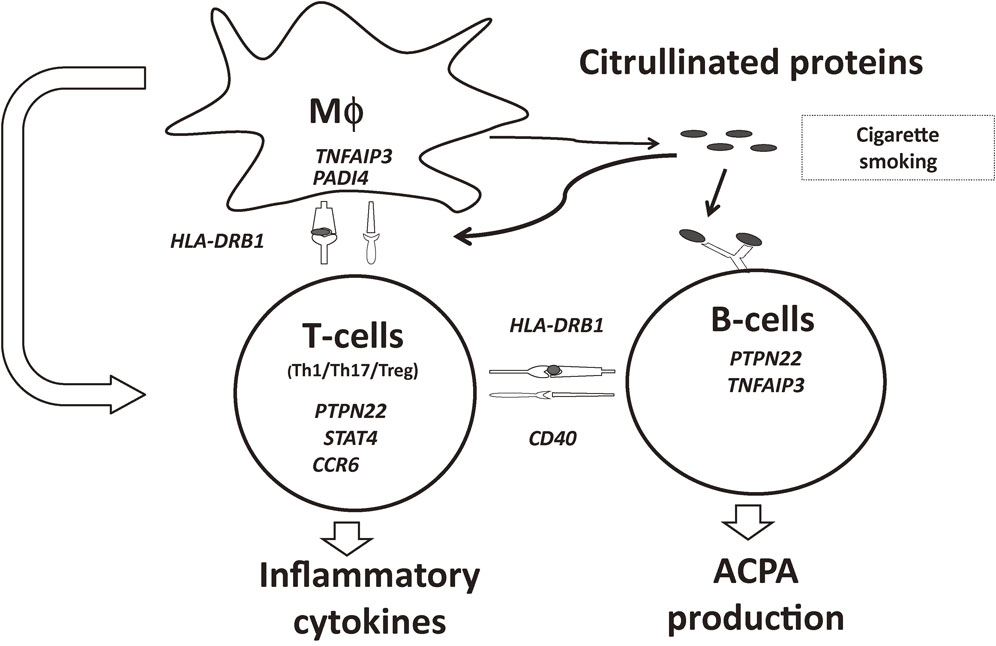
The primary autoantigens could potentially differ among individuals. Patient serum samples can be expected to exhibit spreading of these epitopes, with an increase in the recognition of citrullinated antigens, before the onset of RA.14) Differences in antibody profiles among patients could depend on multiple genetic and environmental factors. Cigarette smoking is an example of an environmental factor that substantially increases the risk of ACPA appearance, and gene-environment interactions between the HLA-DRB1 SE allele and smoking have been reported.15),16) The combination of smoking and genetic factors, including HLA-DRB1, may determine the specificity of ACPAs in RA patients.17)
Significant RA risk of HLA-DRB1 SE alleles has been confirmed in multiple ethnic populations, and the SE hypothesis has been generally accepted for a few decades. However, several attempts have been made to reclassify HLA-DRB1 alleles that can more precisely predict RA risk than the SE hypothesis. Two recent studies clarified that the amino acids at residues HLA-DRb1 11 and 13, rather than the classical SE alleles at residues 70–74, are also independently associated with RA, which may explain the higher risk associated with DR4 (*04:01/*04:04/*04:05) compared with DR1 (*01:01).18)
Classical association of RA with SE alleles has been confirmed in several studies using ACPA-positive RA and controls. However, susceptibility SE alleles have unequal strength on genotypic risk. The group of alleles called DR4(*04SE) seems to be the strongest and DRB1*07, HLA-DRB1*08, HLA-DRB1*11, HLA-DRB1*13, HLA-DRB1*03 are more often low risk than high risk or neutral in European population. The association between HLA-DRB1 and ACPA-negative RA has not been extensively studied due to the high prevalence of APCA-positive RA. HLA-DRB*03:01, which is not the SE allele, is reportedly a risk allele for ACPA-negative RA in European populations, suggesting that the contribution of HLA-DRB1 alleles is distinctly different in ACPA-negative RA.19),20) In fact, a recent study of ACPA-negative patients statistically adjusted for the clinical heterogeneity of ACPA-negative RA identified two independent association signals in HLA-DRB1 and HLA-B gene products: serine 11 (encoded by DRB1*03) and aspartate 9 (encoded by HLA-B*08), providing additional evidence that ACPA-positive and ACPA-negative RA are genetically distinct.21)
3. Pathological insights from GWASs
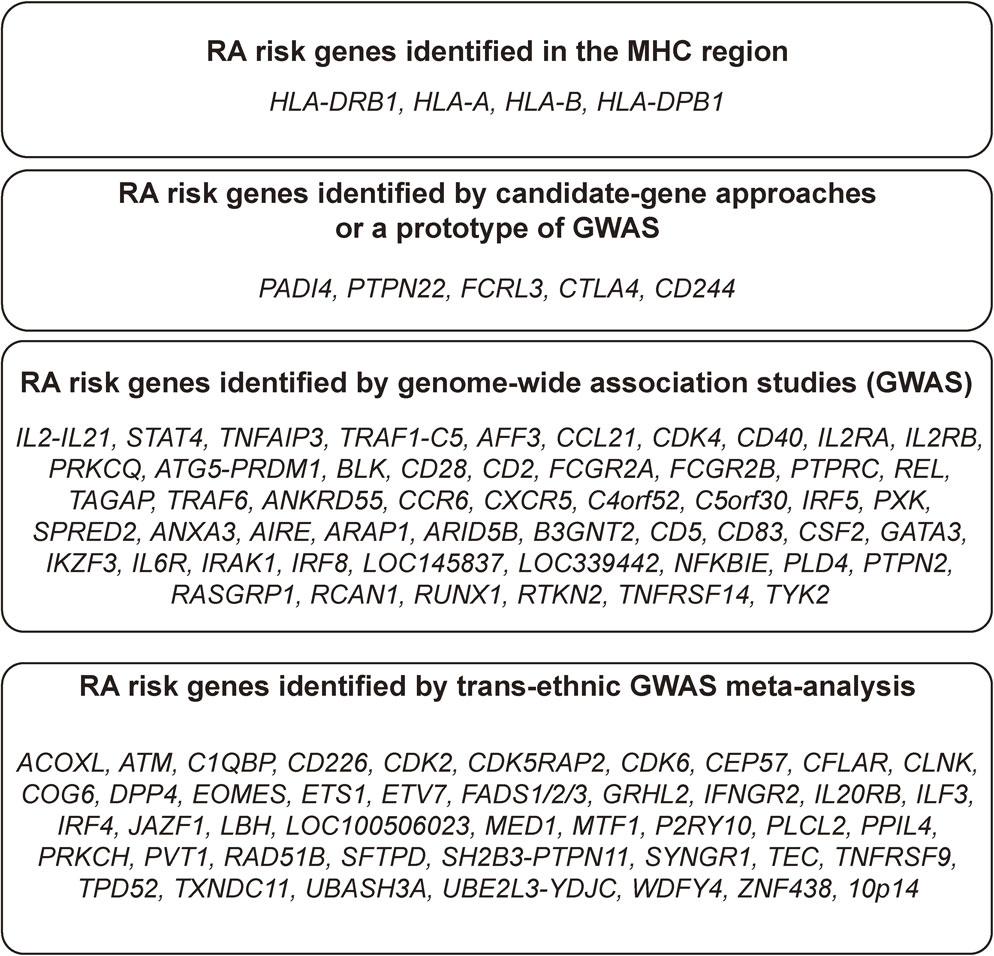
GWASs have been performed for many years following a prototype that was first performed in Japan and covered only gene encoding regions (no intergenic spaces) (Fig. 1).22) In its current form, ∼1 million single-nucleotide polymorphisms (SNPs) are genotyped for affected patients (cases) and controls. GWASs performed using commercially available DNA array technology have led to the identification of RA loci with a larger effect size than in previous linkage-based studies. These individual GWASs identified a number of RA-susceptibility loci, including PADI4,23) PTPN22,24) TNFAIP3,25) TRAF1/C5,26) REL,27) CCR628) and FCRL3.29) However, each of these GWASs lacked sufficient statistical power to detect loci with moderate effect sizes. To overcome this limitation, meta-analyses of GWASs of both European and Asian populations have been conducted. These studies have increased the number of known risk loci.30),31)
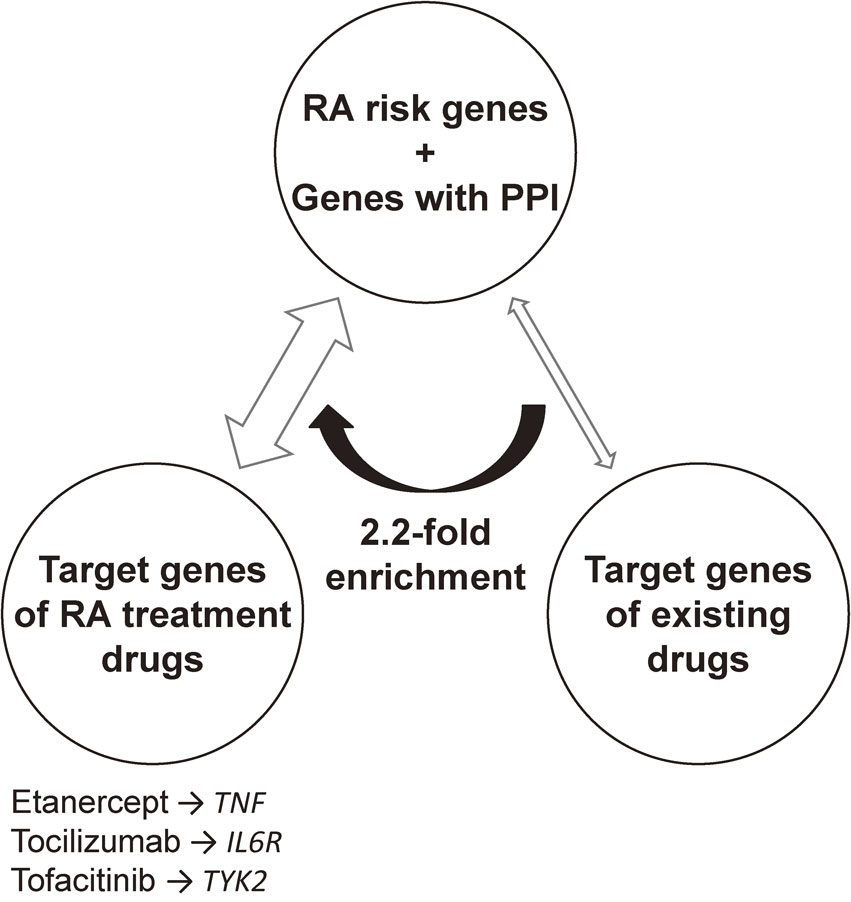
One of the largest meta-analyses of autoimmune disease GWASs was completed in 2014. The collaborative effort produced a multi-ethnic meta-analysis, using more than 100,000 subjects (29,880 RA cases and 73,758 controls) of European or Asian ancestry, which identified 101 RA risk loci.32) A systematic evaluation of the overlap between RA risk genes and the target genes of drugs approved for RA treatment and revealed a significant network of connections (Fig. 2). Empirically, these findings suggest that disease-associated genes are promising resources for drug development. These results also highlight the potential of human genetics not only to identify novel disease susceptibility loci, but to also contribute to the discovery of novel drugs.33)
Although a GWAS can identify disease risk loci, it cannot directly identify the responsible genes or disease-causing variants. The initial findings of GWAS only imply that a disease-causing variant is within the linkage disequilibrium of the identified polymorphism. The disease-causing variants could affect the function of the responsible genes by introducing stop codons or frame-shift mutations, changing the amino acid sequence, affecting alternative splicing, or regulating the level of transcript expression. Of almost 100 risk-associated SNPs in non-HLA RA risk loci, 16% are in linkage disequilibrium with missense SNPs, indicating that the majority of causal variants in the risk loci could affect splicing or the level of gene expression. In fact, 44 out of 100 RA-risk SNPs were found in cis-acting expression quantitative trait loci (cis-eQTL) identified in peripheral blood mononuclear cells.34) This indicates that majority of disease-causing variants in the risk loci affect the expression level of genes in cis. Similar observations have been reported for other autoimmune diseases.
4. RA risk genes and pathogenesis
As described above, meta-analyses of GWASs have identified more than 100 non-HLA RA risk loci. Although the effect of each individual locus is moderate, detailed analyses of an individual locus to identify a disease-causing variant and its effect on the relevant gene are expected to enhance our understanding of the disease (Fig. 3). Here, we discuss the examples of RA risk genes and their role in the pathogenesis of RA.
PADI4.
In 2003, we reported peptidylariginine deiminase type 4 (PADI4) as a susceptibility gene for RA in a Japanese population.23) PADI4 is a member of the PADI gene family that encodes an enzyme which converts arginine residue (peptidylarginine) to citrulline residue (peptidylcitrulline) in a posttranslational modification. PADI4 is highly expressed in bone marrow, macrophages, neutrophils and monocytes.35),36) Peptidylcitrulline is an important molecule in RA, because it is a target antigen of ACPA and only PADs (translated protein from PADI genes) can provide peptidylcitrullines, via modification of protein substrates. Through in vitro assays, we found that transcripts of the risk haplotype of PADI4 are more stable than those of the non-risk haplotype, suggesting that increased expression and function of PAD4 could increase the risk of RA.
The association of PADI4 variants with RA susceptibility has been replicated in Asian populations.37)–39) However, it has not been consistently replicated in European populations,40) although recent meta-analyses suggest PADI4 is also a risk allele in European populations. This suggests ethnic differences in the susceptibility of PADI4. One reason for the lack of definite association of PADI4 with RA in European populations may be related to gene-environmental interaction specific to Asian populations. Cigarette smoking is one of candidates, as smoking rate in Asian males is notably higher than in European males. Direct evidence indicating a link between PADI4 and smoking was presented based on the finding of citrullinated peptides in bronchoalveolar lavage cells and increased expression of PADI enzymes in smokers but not in non-smokers.41) In addition, the gene-environment interaction between PADI4 polymorphisms and smoking has been observed in both European and Asian populations,42),43) in which the effect size (odds ratio) of PADI4 polymorphisms on disease risk has shown to be prominently higher in male smokers. Altogether, the disparity in the contribution of PADI4 to RA between Asians and Europeans might be caused by the difference in the prevalence of smoking in these ethnic groups.
PTPN22.
Of the RA-associated common variants outside the HLA region that have been identified by both conventional association studies and GWAS, a missense variant of the protein tyrosine phosphatase nonreceptor 22 (PTPN22) gene exerts the strongest effect.24),40) This missense variant (PTPN22 R620W) has been reported to be associated with over 20 different autoimmune diseases in European populations, including systemic lupus erythematosus (SLE), type 1 diabetes, and Graves disease.44) On the other hand, this variant is very rare or is not polymorphic in Asian and African populations45),46) and provides another example of genetic heterogeneity among populations. PTPN22 encodes lymphoid tyrosine phosphatase (LYP), which dephosphorylates the phosphotyrosine residues of target proteins in lymphocytes. The disease-associated variant exchanges the arginine residue for tryptophan at position 620. In vitro expression of the R620W risk allele leads to interference of the physical association between LYP and c-Src kinase (CSK), resulting in increased LYP activity.
CCR6.
In 2010, chemokine (C–C motif) receptor 6 (CCR6) was identified as an RA-associated gene in Asian and European populations.28),30) In particular, rs3093024 in the CCR6 gene was the second largest genetic risk allele in a Japanese RA population. A dinucleotide polymorphism (DNP) in the 5′-flanking region influences the binding of nuclear proteins and enhances the transcriptional activity of CCR6. The risk allele exhibits greater enhancing activity and the level of CCR6 transcription is higher in cells with the risk genotype. This variant, CCR6DNP, is also associated with the positive status of IL-17A in the serum of RA patients, suggesting that CCR6DNP influences the activity of Th17 cells. In an animal model, Ccr6+ Th17 cells play a role in the pathogenesis of arthritis.47) Although these data strongly support the hypothesis that Th17 cells are involved in RA, an association between the CCR6 locus and disease development has only been confirmed for Crohn’s and Basedow’s diseases and not for other Th17-related diseases, such as psoriasis and multiple sclerosis, in which a STAT3 variant increases the risk.
FCRL3.
Fc receptor-like 3 (FCRL3) is a member of the FCRL gene family (FCRL1-6) that exhibits a high structural homology to classical Fc receptors. A regulatory SNP in FCRL3 is associated with susceptibility for RA, autoimmune thyroid disease and SLE in a Japanese population.48) This association was replicated with Japanese populations in both an independent RA cohort49) and studies using patients with additional autoimmune disorders such as autoimmune pancreatitis and chronic graft-versus-host disease. As association between FCRL3 and Grave’s disease has also been reproduced in European populations,50) the FCRL3 polymorphism appears to be a common risk factor for Asian and European populations with Grave’s disease. In contrast, inconsistent results have been shown for RA patients of European ancestry. The lack of association in RA patients from several different European populations may reflect a relatively lower contribution of FCRL3 polymorphisms in European populations than in Asian populations, as was observed with the PADI4 polymorphism, and signal that each individual study may have require a greater statistical power to detect an association.
FCRL expression is regulated during B-cell differentiation. In particular, germinal center B-cells express high levels of FCRL3. The cytosolic portion of FCRL3 contains four tyrosine residues that potentially function as an immunoreceptor tyrosine-based activation motif (ITAM) or as an immunoreceptor tyrosine-based inhibitory motif (ITIM). An in vitro study using murine Fc γ RIIB/human FCRL3 chimeric protein revealed that FCRL3 potentially inhibits BCR-mediated signalling through the activity of SH-2 domain-containing phosphatases SHIP, SHP-1, and SHP-2, which are recruited to the ITIMs of FCRL3.
The responsible SNP is located in the promoter region of FCRL3 at −169 bp from the transcription initiation site. This SNP has been shown to alter the binding affinity of NF-κB and regulate gene expression. High levels of FCRL3 expression were observed on B-lymphocytes carrying the risk allele, indicating that a quantitative gain-of-function in FCRL3 leads to disease onset. The potential inhibitory activity of FCRL3 on BCR-signalling, augmented by inhibition of BCR signalling in individuals with the disease susceptible genotype, may lead to autoimmunity.
5. Search for rare variants as the heritability of diseases
GWAS has proven to be a powerful tool in identifying risk loci in common diseases.51) However, it is increasingly obvious that common variants explain only a small proportion of the heritability of these diseases. Thus, the remaining heritability might be explained by the effects of rare variants, which are usually defined as minor allele frequencies <1%. The signals from rare variants are difficult to detect in a conventional GWAS because genetic markers used in this type of study are mainly common variants. However, the development of next-generation sequencing technologies has enabled re-sequencing of the entire genome.52) The contribution of each individual variant to disease development would be potentially higher for a rare missense variant than a common missense variant, fascinating the sequencing of protein-coding regions. In a recent study attempting to determine the roles of rare variants in RA, deep exon sequencing of 25 biological candidate genes from GWAS-identified loci resulted in the identification of an accumulation of missense variants in the IL2RA and IL2RB genes.53) A more comprehensive approach involves whole-exome sequencing, which decodes all protein-coding genes. However, to date, no study has succeeded in identifying RA-associated rare variants using whole-exome sequencing. This might be due to insufficient statistical power, but also could come from the fact that rare variants do not explain a substantial part of heritability of common diseases.
6. Clinical application of genetic information in RA
The use of genetic data is essential because RA is a very heterogeneous disease with variable outcomes that require different treatment regimes. The heterogeneity can be explained, at least in part, by genetic factors. Accordingly, the specific combination of genetic factors in an individual can determine the outcome of the disease. In this regard, GWAS data can be used to predict the disease phenotype of an individual. Phenotype prediction has been largely investigated in RA with respect to disease severity and drug response.
In fact, the nature of progressive joint damage in RA varies among individuals. Patients with more rapid progression of joint damage apparently need more extensive therapy, such as the early use of biologics. Disease severity can be monitored by scoring the degree of joint damage using radiographic imaging. Regression analyses can be performed to test the associations between changes in radiologic scores and variant genotypes. The most extensively investigated gene so far is HLA-DRB1, which appears to have the strongest effect on disease severity.54) Potential associations between various candidate genes and disease severity have also been reported.55)–57) A recent analysis involving a Japanese cohort demonstrated that polymorphisms in PADI4 are associated with radiographic progression of RA.58) Furthermore, a GWAS on the radiological progression rate in autoantibody-positive RA patients recently identified an association in a SNP in the SPAG16 gene, which is expressed in the synovial tissues of RA patients.59) Although these pieces of evidence indicate that genetic variants can influence the severity of RA, individual alleles of these genes could not sufficiently predict disease severity for use in clinical practice.
Another important clinical issue with genetic information is individual drug response. The advances in biologic therapies such as treatment with anti-TNF antibodies have dramatically improved the treatment of RA. Although a substantial proportion of patients (20∼40%) will not respond to a certain biologic therapy, some patients who do not respond to one biologic therapy (e.g., anti-TNF antibodies) may respond to another (e.g., anti-IL-6R antibodies). Accordingly, if the response to a certain biologic agent can be predicted using genetic information, unnecessary cost and potential side effects can be reduced. Data from several GWASs examining the response to anti-TNF antibody therapy have suggested associations between drug response and genes involved in signaling, including the CD84 locus.60),61) However, similar to the prediction of disease severity, individual loci cannot fully predict the drug response of an individual. So far, the results obtained for predictions of disease severity and drug response clearly indicate that single genetic factors are not powerful predictors of clinical phenotype, and that combinatorial approaches using multiple genetic factors and possible environmental information must be established.
Profile

Kazuhiko Yamamoto graduated from the University of Tokyo School of Medicine in 1977.After finishing his training and residency in internal medicine and rheumatology, he conducted basic research in the laboratory of Prof. T. Tada in Tokyo and Prof. G. Haemmerling in Heidelberg. He then started his own research projects and also served as clinical rheumatologist in the University of Tokyo Hospital. He then moved to St. Marianna University School of Medicine as an associate professor and to Medical Institute of Bioregulation, Kyushu University as a professor. He is now a professor and chairman of Department of Allergy and Rheumatology, the University of Tokyo Graduate School of Medicine. He is also serving as a team leader of Laboratory of Autoimmune Diseases, Center for Integrative Medical Sciences, RIKEN. Dr. Yamamoto’s research interests include characterization of newly identified regulatory T cells and genetic analyses of rheumatoid arthritis and other autoimmune diseases.
Profile

Yukinori Okada was born in Shizuoka in 1980. He graduated the University of Tokyo in 2005, and received Ph.D. from the University of Tokyo in 2011. He worked as a research fellow at the Brigham and Women’s Hospital at Harvard Medical School and the Broad institute. From 2013, he is a Tenure-track Junior Associate Professor at Tokyo Medical and Dental University. He has multiple backgrounds as a rheumatologist, a statistician, and a bioinformatician. His research theme is an elucidation of mechanism in which genetic variants of the populations affect human complex diseasess. Through active collaborative partnership among the researchers of the genetics, Dr. Okada enormously conducted genome-wide association studies and identified a number of novel risk genetic loci. Dr. Okada also has experiences for analyzing genetic polymorphisms in the major histocompatibility complex (MHC) region and HLA genes, which have substantial impacts on genetics of human diseases. His recent research interests include development of novel analytic methods of genetics and bioinformatics that integrate Big Data obtained by the latest high-throughput technologies, such as next generation sequencing. Application of his methods contributed to novel drug discovery, disease biomarker identifications, and personalized medicine (Okada Y. et al. Nature 2014, Nature Genetics 2015). He received the President award of the University of Tokyo in 2012, the 49th Baelz award (second) in 2012, and the Young Scientists’ Prize, the Commendation for Science and Technology by the Minister of Education, Culture, Sports, Science and Technology in 2015.
Profile

Akari Suzuki was born in Kitakyushu in 1972. She graduated from Kyushu University in 1994. She received Ph.D. degree in 2001 in Kyushu University under the supervision of Prof. Kenshi Hayashi. From 2001, she moved to RIKEN, where Prof. Kazuhiko Yamamoto was a leader in the Laboratory for Rheumatic Disaseses in SNP Research Center. She principally focusing on the identification of genetic risk factors using case-control association study and has contributed greatly to our understanding of causative genes for rheumatoid arthritis. Peptidylarginine deiminase type4 that she identified in 2003 is known as one of the genetic risk factors of rheumatoid arthritis (RA). She also indicated that deficiency of PADI4 gene in mice suppressed arthritis score of RA model mice. Her recent research theme is enhancing understanding of physiological disease mechanisms by integrating GWAS and RNA-seq gene expression data for functional characterization of disease-causative variants. She was awarded the 49th Baelz award (second) in 2012.
Profile

Yuta Kochi was born in Regina, Canada, and raised in Yokohama. He graduated the University of Tokyo in 1999. After the residency programs in internal medicine and rheumatology at the University of Tokyo Hospital, he received Ph.D. from the University of Tokyo in 2005. Currently, he is a Deputy Team Leader of Laboratory for Autoimmune Diseases at Center for Integrative Medical Sciences, RIKEN. His area of interest is genetics of autoimmune diseases and their pathological mechanisms. He received the Young Scientists’ Prize, the Commendation for Science and Technology by the Minister of Education, Culture, Sports, Science and Technology in 2014. He is also an ancient history enthusiast, enjoying visiting ancient monuments around the world.
References
- 1) Kochi, Y., Suzuki, A., Yamada, R. and Yamamoto, K. (2010) Ethnogenetic heterogeneity of rheumatoid arthritis-implications for pathogenesis. Nat. Rev. Rheumatol. 6, 290–295.
- 2) Lettre, G. and Rioux, J.D. (2008) Autoimmune diseases: insights from genomewide association studies. Hum. Mol. Genet. 17, R116–R121.
- 3) Seldin, M.F., Amos, C.I., Ward, R. and Gregersen, P.K. (1999) The genetics revolution and the assault on rheumatoid arthritis. Arthritis Rheum. 42, 1071–1079.
- 4) MacGregor, A.J., Snieder, H., Rigby, A.S., Koskenvuo, M., Kaprio, J., Aho, K. and Silman, A.J. (2000) Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 43, 30–37.
- 5) Weyand, C.M. and Goronzy, J.J. (2000) Association of MHC and rheumatoid arthritis. HLA polymorphisms in phenotypic variants of rheumatoid arthritis. Arthritis Res. 2, 212–216.
- 6) Fisher, S.A., Lanchbury, J.S. and Lewis, C.M. (2003) Meta-analysis of four rheumatoid arthritis genome-wide linkage studies: confirmation of a susceptibility locus on chromosome 16. Arthritis Rheum. 48, 1200–1206.
- 7) McMichael, A.J., Sasazuki, T., McDevitt, H.O. and Payne, R.O. (1977) Increased frequency of HLA-Cw3 and HLA-Dw4 in rheumatoid arthritis. Arthritis Rheum. 20, 1037–1042.
- 8) Gregersen, P.K., Silver, J. and Winchester, R.J. (1987) The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 30, 1205–1213.
- 9) Auger, I., Sebbag, M., Vincent, C., Balandraud, N., Guis, S., Nogueira, L., Svensson, B., Cantagrel, A., Serre, G. and Roudier, J. (2005) Influence of HLA-DR genes on the production of rheumatoid arthritis-specific autoantibodies to citrullinated fibrinogen. Arthritis Rheum. 52, 3424–3432.
- 10) van Gaalen, F.A., van Aken, J., Huizinga, T.W., Schreuder, G.M., Breedveld, F.C., Zanelli, E., van Venrooij, W.J., Verweij, C.L., Toes, R.E. and de Vries, R.R. (2004) Association between HLA class II genes and autoantibodies to cyclic citrullinated peptides (CCPs) influences the severity of rheumatoid arthritis. Arthritis Rheum. 50, 2113–2121.
- 11) Shimane, K., Kochi, Y., Suzuki, A., Okada, Y., Ishii, T., Horita, T., Saito, K., Okamoto, A., Nishimoto, N., Myouzen, K., Kubo, M., Hirakata, M., Sumida, T., Takasaki, Y., Yamada, R., Nakamura, Y., Kamatani, N. and Yamamoto, K. (2013) An association analysis of HLA-DRB1 with systemic lupus erythematosus and rheumatoid arthritis in a Japanese population: effects of *09:01 allele on disease phenotypes. Rheumatology 52, 1172–1182.
- 12) Hill, J.A., Southwood, S., Sette, A., Jevnikar, A.M., Bell, D.A. and Cairns, E. (2003) Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J. Immunol. 171, 538–541.
- 13) Klareskog, L., Ronnelid, J., Lundberg, K., Padyukov, L. and Alfredsson, L. (2008) Immunity to citrullinated proteins in rheumatoid arthritis. Annu. Rev. Immunol. 26, 651–675.
- 14) van der Woude, D., Rantapaa-Dahlqvist, S., Ioan-Facsinay, A., Onnekink, C., Schwarte, C.M., Verpoort, K.N., Drijfhout, J.W., Huizinga, T.W., Toes, R.E. and Pruijn, G.J. (2010) Epitope spreading of the anti-citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann. Rheum. Dis. 69, 1554–1561.
- 15) Klareskog, L., Stolt, P., Lundberg, K., Kallberg, H., Bengtsson, C., Grunewald, J., Ronnelid, J., Harris, H.E., Ulfgren, A.K., Rantapaa-Dahlqvist, S., Eklund, A., Padyukov, L. and Alfredsson, L. (2006) A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 54, 38–46.
- 16) Linn-Rasker, S.P., van der Helm-van Mil, A.H., van Gaalen, F.A., Kloppenburg, M., de Vries, R.R., le Cessie, S., Breedveld, F.C., Toes, R.E. and Huizinga, T.W. (2006) Smoking is a risk factor for anti-CCP antibodies only in rheumatoid arthritis patients who carry HLA-DRB1 shared epitope alleles. Ann. Rheum. Dis. 65, 366–371.
- 17) Mahdi, H., Fisher, B.A., Kallberg, H., Plant, D., Malmstrom, V., Ronnelid, J., Charles, P., Ding, B., Alfredsson, L., Padyukov, L., Symmons, D.P., Venables, P.J., Klareskog, L. and Lundberg, K. (2009) Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat. Genet. 41, 1319–1324.
- 18) Raychaudhuri, S., Sandor, C., Stahl, E.A., Freudenberg, J., Lee, H.S., Jia, X., Alfredsson, L., Padyukov, L., Klareskog, L., Worthington, J., Siminovitch, K.A., Bae, S.C., Plenge, R.M., Gregersen, P.K. and de Bakker, P.I. (2012) Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 44, 291–296.
- 19) Furuya, T., Hakoda, M., Ichikawa, N., Higami, K., Nanke, Y., Yago, T., Kobashigawa, T., Tokunaga, K., Tsuchiya, N., Kamatani, N. and Kotake, S. (2007) Differential association of HLA-DRB1 alleles in Japanese patients with early rheumatoid arthritis in relationship to autoantibodies to cyclic citrullinated peptide. Clin. Exp. Rheumatol. 25, 219–224.
- 20) Irigoyen, P., Lee, A.T., Wener, M.H., Li, W., Kern, M., Batliwalla, F., Lum, R.F., Massarotti, E., Weisman, M., Bombardier, C., Remmers, E.F., Kastner, D.L., Seldin, M.F., Criswell, L.A. and Gregersen, P.K. (2005) Regulation of anti-cyclic citrullinated peptide antibodies in rheumatoid arthritis: contrasting effects of HLA-DR3 and the shared epitope alleles. Arthritis Rheum. 52, 3813–3818.
- 21) Han, B., Diogo, D., Eyre, S., Kallberg, H., Zhernakova, A., Bowes, J., Padyukov, L., Okada, Y., Gonzalez-Gay, M.A., Rantapaa-Dahlqvist, S., Martin, J., Huizinga, T.W., Plenge, R.M., Worthington, J., Gregersen, P.K., Klareskog, L., de Bakker, P.I. and Raychaudhuri, S. (2014) Fine mapping seronegative and seropositive rheumatoid arthritis to shared and distinct HLA alleles by adjusting for the effects of heterogeneity. Am. J. Hum. Genet. 94, 522–532.
- 22) Ozaki, K., Ihnishi, Y., Iida, A., Sekine, A., Yamada, R., Tsunoda, T., Sato, H., Sato, H., Hori, M., Nakamura, Y. and Tanaka, T. (2002) Functional SNPs in the lymphotoxin-αgene that are associated with susceptibility to myocardial infarction. Nat. Genet. 32, 650–654.
- 23) Suzuki, A., Yamada, R., Chang, X., Tokuhiro, S., Sawada, T., Suzuki, M., Nagasaki, M., Nakayama-Hamada, M., Kawaida, R., Ono, M., Ohtsuki, M., Furukawa, H., Yoshino, S., Yukioka, M., Tohma, S., Matsubara, T., Wakitani, S., Teshima, R., Nishioka, Y., Sekine, A., Iida, A., Takahashi, A., Tsunoda, T., Nakamura, Y. and Yamamoto, K. (2003) Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat. Genet. 34, 395–402.
- 24) Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678.
- 25) Thomson, W., Barton, A., Ke, X., Eyre, S., Hinks, A., Bowes, J., Donn, R., Symmons, D., Hider, S., Bruce, I.N., Wilson, A.G., Marinou, I., Morgan, A., Emery, P., Carter, A., Steer, S., Hocking, L., Reid, D.M., Wordsworth, P., Harrison, P., Strachan, D. and Worthington, J. (2007) Rheumatoid arthritis association at 6q23. Nat. Genet. 39, 1431–1433.
- 26) Plenge, R.M., Seielstad, M., Padyukov, L., Lee, A.T., Remmers, E.F., Ding, B., Liew, A., Khalili, H., Chandrasekaran, A., Davies, L.R., Li, W., Tan, A.K., Bonnard, C., Ong, R.T., Thalamuthu, A., Pettersson, S., Liu, C., Tian, C., Chen, W.V., Carulli, J.P., Beckman, E.M., Altshuler, D., Alfredsson, L., Criswell, L.A., Amos, C.I., Seldin, M.F., Kastner, D.L., Klareskog, L. and Gregersen, P.K. (2007) TRAF1-C5 as a risk locus for rheumatoid arthritis — a genomewide study. N. Engl. J. Med. 357, 1199–1209.
- 27) Gregersen, P.K., Amos, C.I., Lee, A.T., Lu, Y., Remmers, E.F., Kastner, D.L., Seldin, M.F., Criswell, L.A., Plenge, R.M., Holers, V.M., Mikuls, T.R., Sokka, T., Moreland, L.W., Bridges, S.L. Jr., Xie, G., Begovich, A.B. and Siminovitch, K.A. (2009) REL, encoding a member of the NF-kappaB family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat. Genet. 41, 820–823.
- 28) Kochi, Y., Okada, Y., Suzuki, A., Ikari, K., Terao, C., Takahashi, A., Yamazaki, K., Hosono, N., Myouzen, K., Tsunoda, T., Kamatani, N., Furuichi, T., Ikegawa, S., Ohmura, K., Mimori, T., Matsuda, F., Iwamoto, T., Momohara, S., Yamanaka, H., Yamada, R., Kubo, M., Nakamura, Y. and Yamamoto, K. (2010) A regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibility. Nat. Genet. 42, 515–519.
- 29) Kochi, Y., Yamada, R., Suzuki, A., Harley, J.B., Shirasawa, S., Sawada, T., Bae, S.C., Tokuhiro, S., Chang, X., Sekine, A., Takahashi, A., Tsunoda, T., Ohnishi, Y., Kaufman, K.M., Kang, C.P., Kang, C., Otsubo, S., Yumura, W., Mimori, A., Koike, T., Nakamura, Y., Sasazuki, T. and Yamamoto, K. (2005) A functional variant in FCRL3, encoding Fc receptor-like 3, is associated with rehuematoid arthritis and several autoimmunities. Nat. Genet. 37, 478–485.
- 30) Stahl, E.A., Raychaudhuri, S., Remmers, E.F., Xie, G., Eyre, S., Thomson, B.P., Li, Y., Kurreeman, F.A., Zhernakova, A., Hinks, A., Guiducci, C., Chen, R., Alfredsson, L., Amos, C.I., Ardlie, K.G., Barton, A., Bowes, J., Brouwer, E., Burtt, N.P., Catanese, J.J., Coblyn, J., Coenen, M.J., Costenbader, K.H., Criswell, L.A., Crusius, J.B., Cui, J., de Bakker, P.I., De Jager, P.L., Ding, B., Emery, P., Flynn, E., Harrison, P., Hocking, L.J., Huizinga, T.W., Kastner, D.L., Ke, X., Lee, A.T., Liu, X., Martin, P., Morgan, A.W., Padyukov, L., Posthumus, M.D., Radstake, T.R., Reid, D.M., Seielstad, M., Seldin, M.F., Shadick, N.A., Steer, S., Tak, P.P., Thomson, W., van der Helm-van Mil, A.H., van der Horst-Bruinsma, I.E., van der Schoot, C.E., van Riel, P.L., Weinblatt, M.E., Wilson, A.G., Wolbink, G.J., Wordsworth, B.P., Wijmenga, C., Karlson, E.W., Toes, R.E., de Vries, N., Begovich, A.B., Worthington, J., Siminovitch, K.A., Gregersen, P.K., Klareskog, L. and Plenge, R.M. (2010) Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 42, 508–514.
- 31) Okada, Y., Terao, C., Ikari, K., Kochi, Y., Ohmura, K., Suzuki, A., Kawaguchi, T., Stahl, E.A., Kurreeman, F.A., Nishida, N., Ohmiya, H., Myouzen, K., Takahashi, M., Sawada, T., Nishioka, Y., Yukioka, M., Matsubara, T., Wakitani, S., Teshima, R., Tohma, S., Takasugi, K., Shimada, K., Murasawa, A., Honjo, S., Matsuo, K., Tanaka, H., Tajima, K., Suzuki, T., Iwamoto, T., Kawamura, Y., Tanii, H., Okazaki, Y., Sasaki, T., Gregersen, P.K., Padyukov, L., Worthington, J., Siminovitch, K.A., Lathrop, M., Taniguchi, A., Takahashi, A., Tokunaga, K., Kubo, M., Nakamura, Y., Kamatani, N., Mimori, T., Plenge, R.M., Yamanaka, H., Momohara, S., Yamada, R., Matsuda, F. and Yamamoto, K. (2012) Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat. Genet. 44, 511–516.
- 32) Okada, Y., Wu, D., Trynka, G., Raj, T., Terao, C., Ikari, K., Kochi, Y., Ohmura, K., Suzuki, A., Yoshida, S., Graham, R.R., Manoharan, A., Ortmann, W., Bhangale, T., Denny, J.C., Carroll, R.J., Eyler, A.E., Greenberg, J.D., Kremer, J.M., Pappas, D.A., Jiang, L., Yin, J., Ye, L., Su, D.F., Yang, J., Xie, G., Keystone, E., Westra, H.J., Esko, T., Metspalu, A., Zhou, X., Gupta, N., Mirel, D., Stahl, E.A., Diogo, D., Cui, J., Liao, K., Guo, M.H., Myouzen, K., Kawaguchi, T., Coenen, M.J., van Riel, P.L., van de Laar, M.A., Guchelaar, H.J., Huizinga, T.W., Dieude, P., Mariette, X., Bridges, S.L. Jr., Zhernakova, A., Toes, R.E., Tak, P.P., Miceli-Richard, C., Bang, S.Y., Lee, H.S., Martin, J., Gonzalez-Gay, M.A., Rodriguez-Rodriguez, L., Rantapaa-Dahlqvist, S., Arlestig, L., Choi, H.K., Kamatani, Y., Galan, P., Lathrop, M., the RACI consortium, the GARNET consortium, Eyre, S., Bowes, J., Barton, A., de Vries, N., Moreland, L.W., Criswell, L.A., Karlson, E.W., Taniguchi, A., Yamada, R., Kubo, M., Liu, J.S., Bae, S.C., Worthington, J., Padyukov, L., Klareskog, L., Gregersen, P.K., Raychaudhuri, S., Stranger, B.E., De Jager, P.L., Franke, L., Visscher, P.M., Brown, M.A., Yamanaka, H., Mimori, T., Takahashi, A., Xu, H., Behrens, T.W., Siminovitch, K.A., Momohara, S., Matsuda, F., Yamamoto, K. and Plenge, R.M. (2014) Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381.
- 33) Plenge, R.M., Scolnick, E.M. and Altshuler, D. (2013) Validating therapeutic targets through human genetics. Nat. Rev. Drug Discov. 12, 581–594.
- 34) Westra, H.J., Peters, M.J., Esko, T., Yaghootkar, H., Schurmann, C., Kettunen, J., Christiansen, M.W., Fairfax, B.P., Schramm, K., Powell, J.E., Zhernakova, A., Zhernakova, D.V., Veldink, J.H., Van den Berg, L.H., Karjalainen, J., Withoff, S., Uitterlinden, A.G., Hofman, A., Rivadeneira, F., t Hoen, P.A., Reinmaa, E., Fischer, K., Nelis, M., Milani, L., Melzer, D., Ferrucci, L., Singleton, A.B., Hernandez, D.G., Nalls, M.A., Homuth, G., Nauck, M., Radke, D., Volker, U., Perola, M., Salomaa, V., Brody, J., Suchy-Dicey, A., Gharib, S.A., Enquobahrie, D.A., Lumley, T., Montgomery, G.W., Makino, S., Prokisch, H., Herder, C., Roden, M., Grallert, H., Meitinger, T., Strauch, K., Li, Y., Jansen, R.C., Visscher, P.M., Knight, J.C., Psaty, B.M., Ripatti, S., Teumer, A., Frayling, T.M., Metspalu, A., van Meurs, J.B. and Franke, L. (2013) Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243.
- 35) Asaga, H., Nakashima, K., Senshu, T., Ishigami, A. and Yamada, M. (2001) Immunocytochemical localization of peptidylarginine deiminase in human eosinophils and neutrophils. J. Leukoc. Biol. 70, 46–51.
- 36) Chang, X., Yamada, R., Suzuki, A., Sawada, T., Yoshino, S., Tokuhiro, S. and Yamamoto, K. (2005) Localization of peptidylarginine deiminase 4 (PADI4) and citrullinated protein in synovial tissue of rheumatoid arthritis. Rheumatology (Oxford) 44, 40–50.
- 37) Iwamoto, T., Ikari, K., Nakamura, T., Kuwahara, M., Toyama, Y., Tomatsu, T., Momohara, S. and Kamatani, N. (2006) Association between PADI4 and rheumatoid arthritis: a meta-analysis. Rheumatology (Oxford) 45, 804–807.
- 38) Ikari, K., Kuwahara, M., Nakamura, T., Momohara, S., Hara, M., Yamanaka, H., Tomatsu, T. and Kamatani, N. (2005) Association between PADI4 and rheumatoid arthritis: a replication study. Arthritis Rheum. 52, 3054–3057.
- 39) Kang, C.P., Lee, H.S., Ju, H., Cho, H., Kang, C. and Bae, S.C. (2006) A functional haplotype of the PADI4 gene associated with increased rheumatoid arthritis susceptibility in Koreans. Arthritis Rheum. 54, 90–96.
- 40) Begovich, A.B., Carlton, V.E., Honigberg, L.A., Schrodi, S.J., Chokkalingam, A.P., Alexander, H.C., Ardlie, K.G., Huang, Q., Smith, A.M., Spoerke, J.M., Conn, M.T., Chang, M., Chang, S.Y., Saiki, R.K., Catanese, J.J., Leong, D.U., Garcia, V.E., McAllister, L.B., Jeffery, D.A., Lee, A.T., Batliwalla, F., Remmers, E., Criswell, L.A., Seldin, M.F., Kastner, D.L., Amos, C.I., Sninsky, J.J. and Gregersen, P.K. (2004) A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am. J. Hum. Genet. 75, 330–337.
- 41) Klareskog, L., Padyukov, L., Ronnelid, J. and Alfredsson, L. (2006) Genes, environment and immunity in the development of rheumatoid arthritis. Curr. Opin. Immunol. 18, 650–655.
- 42) Mei, L., Li, X., Yang, K., Cui, J., Fang, B., Guo, X. and Rotter, J.I. (2007) Evaluating gene x gene and gene x smoking interaction in rheumatoid arthritis using candidate genes in GAW15. BMC Proc. 1 (Suppl. 1), S17.
- 43) Kochi, Y., Thabet, M.M., Suzuki, A., Okada, Y., Daha, N.A., Toes, R.E., Huizinga, T.W., Myouzen, K., Kubo, M., Yamada, R., Nakamura, Y. and Yamamoto, K. (2011) PADI4 polymorphism predisposes male smokers to rheumatoid arthritis. Ann. Rheum. Dis. 70, 512–515.
- 44) Zheng, J., Ibrahim, S., Petersen, F. and Yu, X. (2012) Meta-analysis reveals an association of PTPN22 C1858T with autoimmune diseases, which depends on the localization of the affected tissue. Genes. Immun. 13, 641–652.
- 45) Mori, M., Yamada, R., Kobayashi, K., Kawaida, R. and Yamamoto, K. (2005) Ethnic differences in allele frequency of autoimmune-disease-associated SNPs. J. Hum. Genet. 50, 264–266.
- 46) Lins, T.C., Vieira, R.G., Grattapaglia, D. and Pereira, R.W. (2010) Allele and haplotype frequency distribution in PTPN22 gene across variable ethnic groups: Implications for genetic association studies for autoimmune diseases. Autoimmunity 43, 308–316.
- 47) Hirota, K., Yoshitomi, H., Hashimoto, M., Maeda, S., Teradaira, S., Sugimoto, N., Yamaguchi, T., Nomura, T., Ito, H., Nakamura, T., Sakaguchi, N. and Sakaguchi, S. (2007) Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J. Exp. Med. 204, 2803–2812.
- 48) Kochi, Y., Yamada, R., Suzuki, A., Harley, J.B., Shirasawa, S., Sawada, T., Bae, S.C., Tokuhiro, S., Chang, X., Sekine, A., Takahashi, A., Tsunoda, T., Ohnishi, Y., Kaufman, K.M., Kang, C.P., Kang, C., Otsubo, S., Yumura, W., Mimori, A., Koike, T., Nakamura, Y., Sasazuki, T. and Yamamoto, K. (2005) A functional variant in FCRL3, encoding Fc receptor-like 3, is associated with rheumatoid arthritis and several autoimmunities. Nat. Genet. 37, 478–485.
- 49) Ikari, K., Momohara, S., Nakamura, T., Hara, M., Yamanaka, H., Tomatsu, T. and Kamatani, N. (2006) Supportive evidence for a genetic association of the FCRL3 promoter polymorphism with rheumatoid arthritis. Ann. Rheum. Dis. 65, 671–673.
- 50) Simmonds, M.J., Heward, J.M., Carr-Smith, J., Foxall, H., Franklyn, J.A. and Gough, S.C. (2006) Contribution of single nucleotide polymorphisms within FCRL3 and MAP3K7IP2 to the pathogenesis of Graves’ disease. J. Clin. Endocrinol. Metab. 91, 1056–1061.
- 51) Wang, W.Y., Barratt, B.J., Claytona, D.G. and Todd, J.A. (2005) Genome-wide association studies: theoretical and practical concerns. Nat. Rev. Genet. 6, 109–118.
- 52) Kiezun, A., Garimella, K., Do, R., Stitziel, N.O., Neale, B.M., McLaren, P.J., Gupta, N., Sklar, P., Sullivan, P.F., Moran, J.L., Hultman, C.M., Lichtenstein, P., Magnusson, P., Lehner, T., Shugart, Y.Y., Price, A.L., de Bakker, P.I., Purcell, S.M. and Sunyaev, S.R. (2012) Exome sequencing and the genetic basis of complex traits. Nat. Genet. 44, 623–630.
- 53) Diogo, D., Kurreeman, F., Stahl, E.A., Liao, K.P., Gupta, N., Greenberg, J.D., Rivas, M.A., Hickey, B., Flannick, J., Thomson, B., Guiducci, C., Ripke, S., Adzhubey, I., Barton, A., Kremer, J.M., Alfredsson, L., Consortium of Rheumatology Researchers of North America, Rheumatoid Arthritis Consortium International, Sunyaev, S., Martin, J., Zhernakova, A., Bowes, J., Eyre, S., Siminovitch, K.A., Gregersen, P.K., Worthington, J., Klareskog, L., Padyukov, L., Raychaudhuri, S. and Plenge, R.M. (2013) Rare, low-frequency, and common variants in the protein-coding sequence of biological candidate genes from GWASs contribute to risk of rheumatoid arthritis. Am. J. Hum. Genet. 92, 15–27.
- 54) MacGregor, A., Ollier, W., Thomson, W., Jawaheer, D. and Silman, A. (1995) HLA-DRB1*0401/0404 genotype and rheumatoid arthritis: increased association in men, young age at onset, and disease severity. J. Rheumatol. 22, 1032–1036.
- 55) Knevel, R., Krabben, A., Wilson, A.G., Brouwer, E., Leijsma, M.K., Lindqvist, E., de Rooy, D.P., Daha, N.A., van der Linden, M.P., Tsonaka, S., Zhernakova, A., Westra, H.J., Franke, L., Houwing-Duistermaat, J.J., Toes, R.E., Huizinga, T.W., Saxne, T. and van der Helm-van Mil, A.H. (2013) A genetic variant in granzyme B is associated with progression of joint destruction in rheumatoid arthritis. Arthritis Rheum. 65, 582–589.
- 56) Mathews, R.J., Robinson, J.I., Battellino, M., Wong, C., Taylor, J.C., Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate (BRAGGSS), Eyre, S., Churchman, S.M., Wilson, A.G., Isaacs, J.D., Hyrich, K., Barton, A., Plant, D., Savic, S., Cook, G.P., Sarzi-Puttini, P., Emery, P., Barrett, J.H., Morgan, A.W. and McDermott, M.F. (2013) Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann. Rheum. Dis. 73, 1202–1210.
- 57) de Rooy, D.P., Zhernakova, A., Tsonaka, R., Willemze, A., Kurreeman, B.A., Trynka, G., van Toorn, L., Toes, R.E., Huizinga, T.W., Houwing-Duistermaat, J.J., Gregersen, P.K. and van der Helm-van Mil, A.H. (2014) A genetic variant in the region of MMP-9 is associated with serum levels and progression of joint damage in rheumatoid arthritis. Ann. Rheum. Dis. 73, 1163–1169.
- 58) Suzuki, T., Ikari, K., Yano, K., Inoue, E., Toyama, Y., Taniguchi, A., Yamanaka, H. and Momohara, S. (2013) PADI4 and HLA-DRB1 are genetic risks for radiographic progression in RA patients, independent of ACPA status: results from the IORRA cohort study. PLoS One 8, e61045.
- 59) Knevel, R., Klein, K., Somer, K., Ospelt, C., Houwing-Duistermaat, J.J., van Nies, J.A., de Rooy, D.P., de Bock, L., Kurreeman, F.A., Schonkeren, J., Stoeken-Rijsbergen, G., Helmer, Q., van der Linden, M.P., Kern, M., Manjarrez-Orduno, N., Rodriguez-Rodriquez, L., Stinissen, P., Huizinga, T.W., Toes, R.E., Gay, S., Gregersen, P.K., Somers, V. and van der Helm-van Mil, A.H. (2013) Identification of a genetic variant for joint damage progression in autoantibody-positive rheumatoid arthritis. Ann. Rheum. Dis. 73, 2038–2046.
- 60) Plant, D., Bowes, J., Potter, C., Hyrich, K.L., Morgan, A.W., Wilson, A.G., Isaacs, J.D., Wellcome Trust Case Control Consortium, British Society for Rheumatology Biologics Register and Barton, A. (2011) Genome-wide association study of genetic predictors of anti-tumor necrosis factor treatment efficacy in rheumatoid arthritis identifies associations with polymorphisms at seven loci. Arthritis Rheum. 63, 645–653.
- 61) Cui, J., Stahl, E.A., Saevarsdottir, S., Miceli, C., Diogo, D., Trynka, G., Raj, T., Mirkov, M.U., Canhao, H., Ikari, K., Terao, C., Okada, Y., Wedren, S., Askling, J., Yamanaka, H., Momohara, S., Taniguchi, A., Ohmura, K., Matsuda, F., Mimori, T., Gupta, N., Kuchroo, M., Morgan, A.W., Isaacs, J.D., Wilson, A.G., Hyrich, K.L., Herenius, M., Doorenspleet, M.E., Tak, P.P., Crusius, J.B., van der Horst-Bruinsma, I.E., Wolbink, G.J., van Riel, P.L., van de Laar, M., Guchelaar, H.J., Shadick, N.A., Allaart, C.F., Huizinga, T.W., Toes, R.E., Kimberly, R.P., Bridges, S.L. Jr., Criswell, L.A., Moreland, L.W., Fonseca, J.E., de Vries, N., Stranger, B.E., De Jager, P.L., Raychaudhuri, S., Weinblatt, M.E., Gregersen, P.K., Mariette, X., Barton, A., Padyukov, L., Coenen, M.J., Karlson, E.W. and Plenge, R.M. (2013) Genome-wide association study and gene expression analysis identifies CD84 as a predictor of response to etanercept therapy in rheumatoid arthritis. PLoS Genet. 9, e1003394.