Abstract
Originating from cancer research in mammalian cultured cells, the entirely new field of the unfolded protein response (UPR) was born in 1988. The UPR is a transcriptional induction program coupled with intracellular signaling from the endoplasmic reticulum (ER) to the nucleus to maintain the homeostasis of the ER, an organelle which controls the quality of proteins destined for the secretory pathway. Extremely competitive analyses using the budding yeast Saccharomyces cerevisiae revealed that although signaling from both the ER and cell surface is initiated by activation of a transmembrane protein kinase, the mechanism downstream of ER-resident Ire1p, a sensor molecule of the UPR, is unique. Thus, unconventional spliceosome-independent mRNA splicing is utilized to produce the highly active transcription factor Hac1p. This is the autobiographical story of how a young and not yet independent scientist competed with a very famous full professor in the early days of UPR research, which ultimately lead to their sharing Lasker Basic Medical Research Award in 2014.
Introduction
Every protein synthesized according to the “central dogma” emerges in the cell as a linear polypeptide, and must be folded into its specific tertiary and quaternary structures to fulfill its function as assigned by genetic code. “Anfinsen’s dogma” says that a protein’s structure is primarily determined by its amino acid sequence, but such spontaneous protein folding is very difficult to achieve inside the cell, where the extremely high protein concentrations can easily lead to protein misfolding or even aggregation. Therefore, essentially all living cells are equipped with special types of protein termed “molecular chaperones”, which assist protein folding in the cell. The name is apt: in western countries, “chaperone” was the name given to a person who used to accompany young unmarried woman to social events to ensure proper behavior. Thus, molecular chaperones transiently associate with newly synthesized polypeptides, suppress inappropriate interactions between sticky hydrophobic amino acids, help them fold according to the Anfinsen’s dogma, and then dissociate from the now correctly folded proteins, allowing them to function.1)
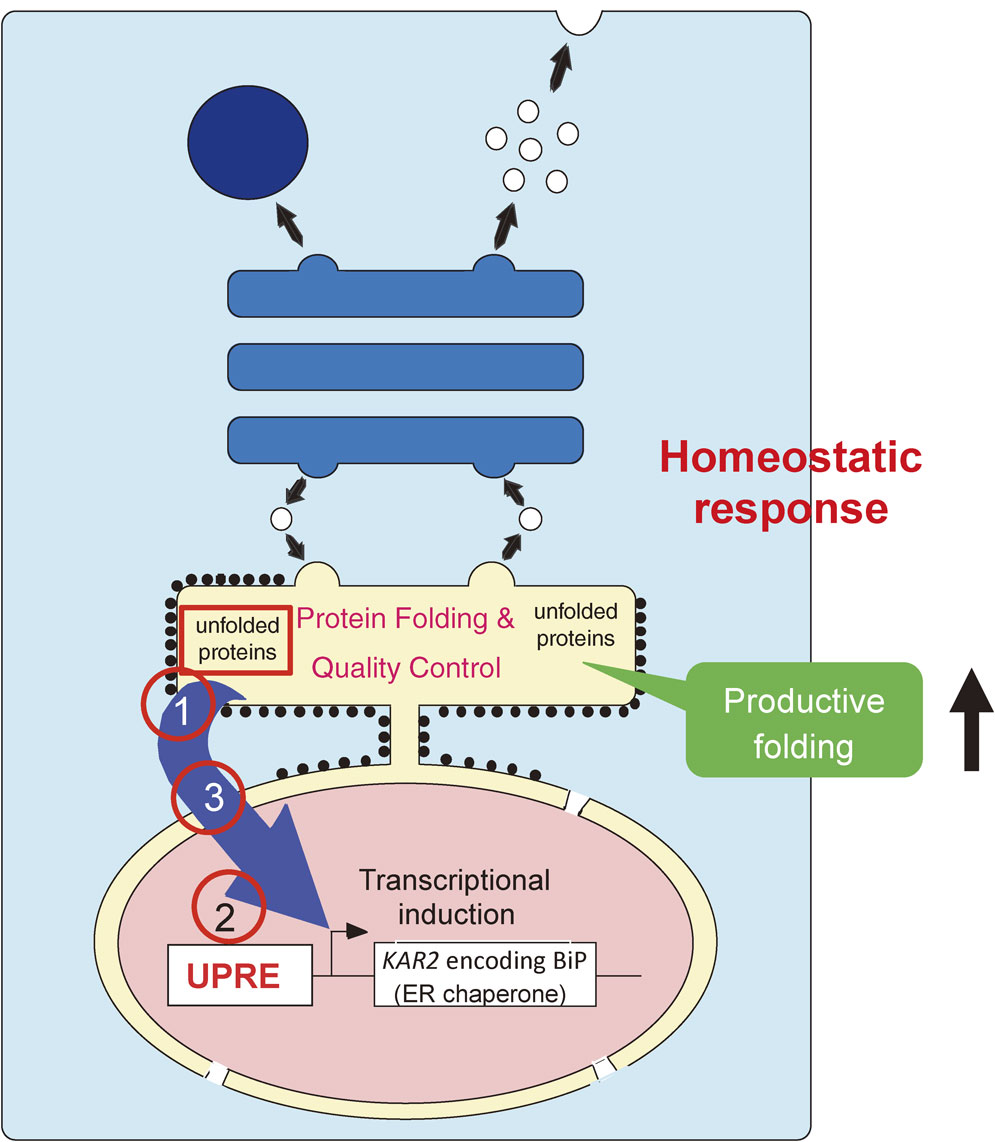
Secretory and transmembrane proteins accounting for one-third of total cellular proteins are synthesized on ribosomes bound to the endoplasmic reticulum (ER) membrane, translocated into the lumen of the ER, and then folded with the assistance of molecular chaperones and folding enzymes (protein disulfide isomerase etc.) abundantly expressed in the ER (ER chaperones hereafter). Only correctly folded molecules are permitted to leave the ER toward their final destinations.2) Molecules unable to assume the correct three-dimensional structure even after the assistance of ER chaperones are retained in the ER and eventually degraded. This degradation of proteins unfolded (not matured) or misfolded in the ER occurs in the cytoplasm, implying that they are somehow recognized in the ER, then retrotranslocated back to the cytoplasm, ubiquitinated and finally degraded by the proteasome, a series of events collectively termed ER-associated degradation.3) Thus, the quality of proteins in the ER is ensured by two distinct mechanisms with opposite directions, namely ER chaperone-mediated productive folding and ER-associated degradation.
Under a variety of physiological and pathological conditions, however, the protein quality control system is compromised, resulting in the accumulation of unfolded or misfolded proteins in the ER. This “ER stress” is problematic for two reasons. First, as only correctly folded molecules are allowed to reach their final destination, prolongation of ER stress eventually causes the cell to suffer a shortage of necessary proteins. Second, as unfolded or misfolded proteins expose sticky hydrophobic amino acids which are normally confined inside the molecule, they exhibit proteotoxicity. In the face of these dangers, essentially all eukaryotic cells have developed ways to cope with ER stress promptly and adequately. This remarkable conservation highlights the extreme importance of protein folding.4) This review summarizes the discovery of the cellular homeostatic response in eukaryotes, termed the ER stress response or the unfolded protein response (UPR), and its mechanism in the budding yeast Saccharomyces cerevisiae.
Discovery of the UPR
The prototype of the UPR was discovered in the mid 1970’s. Scientists found that the synthesis of two cellular proteins of 78 kDa and 94 kDa was markedly induced when mammalian cultured cells were transformed with Rous sarcoma virus.5),6) Contrary to expectations, however, this induction was not directly associated with malignant transformation. Rather, raid growth of transformed cells depleted glucose levels in the culture medium, and it was this depletion that triggered the induction of the proteins. For untransformed cells, in other words, the deprivation of glucose in the culture medium also caused the induction of the two proteins. Based on these findings, the two proteins were designated glucose-regulated proteins (GRP78 and GRP94) in 1977, as their expression levels are regulated by glucose in medium.7) Many scientists might then have lost interest in GRPs owing to their lack of any association with malignancy, and moved onto the new field of “tyrosine phosphorylation” following the discovery in 1980 that the transforming gene product of Rous sarcoma virus phosphorylated tyrosine instead of serine or threonine.8) Nonetheless, a few scientists continued working on GRPs with two primary goals to identify their function and their mechanism of induction.
In the mid 1980’s, GRP78 was found to be identical to immunoglobulin heavy chain binding protein (BiP). BiP was initially discovered as an intracellular protein which binds to immunoglobulin heavy chain in pre-B cells synthesizing heavy chain only, and not in plasma cells synthesizing both heavy and light chains.9) Furthermore, BiP/GRP78 was found to be a member of the heat shock protein 70 family, whose emerging role at that time was as a molecular chaperone. As BiP/GRP78 contains a signal peptide sequence at its N-terminus and an ER-retrieval signal at its C-terminus, it was considered to function as an ER chaperone.10)
Despite extensive work over the decade from 1977, it was not until 1988 when the real trigger for the induction of GRPs was pinpointed. Mary-Jane Gething and Joseph Sambrook at the University of Texas Southwestern Medical Center at Dallas revealed that the accumulation of misfolded proteins in the ER (ER stress) triggered the induction of GRPs in mammalian cells, culminating in the induction of ER chaperones which attempt to refold misfolded proteins accumulated in the ER11) (Fig. 1). Glucose depletion turned out to cause ER stress by inhibiting protein N-glycosylation in the ER. They designated this homeostatic response the UPR, in the sense that GRPs deal with not only misfolded proteins but also unfolded proteins, such as lonely immunoglobulin heavy chain, which is not misfolded but rather unfolded (not maturated) in the absence of light chain. To pursue the induction mechanism, they then switched analytical system from mammalian cells to the budding yeast Saccharomyces cerevisiae, with the hope that induction mechanism might be simpler in yeast, and because the power of yeast genetics was quite attractive. In retrospective, this decision turned out to be perfect. In 1989, they and Mark Rose independently reported that yeast has the UPR, namely a transcriptional induction program coupled with intracellular signaling from the ER to the nucleus.12),13) Nonetheless, molecular mediators of this response had yet to be identified.
Molecular cloning of the IRE1 gene encoding ER stress sensor Ire1p
In April, 1989, I became a post-doc in Mary-Jane and Joe’s laboratories and took over the yeast UPR project. First, I characterized the promoter region of the yeast KAR2 gene encoding yeast BiP, the only known target of the yeast UPR at that time. By constructing and analyzing 5′ and internal deletion mutants of the yeast KAR2 promoter, I succeeded in identifying a cis-acting element responsible for transcriptional induction of the yeast KAR2 gene in response to ER stress as a 22 bp UPR element (UPRE) in 1992.14),15) Later, in 1998, Hiderou Yoshida and I found a cis-acting element in mammals I designated the ER stress response element (CCAAT-N9-CCACG), which was triplicated with minor modifications in its consensus sequence (CCAAT-N9-CCACG → CCAAT-N9-CCAac → CgAAT-N9-CCAgc) in the BiP promoter. This redundancy caused the ERSE to remain unidentified for two decades.16) As Mary-Jane and Joe had anticipated, the yeast system was much simpler than the mammalian system.
Since I started my research career as a biochemist, I wanted my next project to be something new, rather than simply purifying a trans-acting factor capable of binding to UPRE biochemically. So I decided to genetically screen yeast to identify molecules involved in the yeast UPR (Fig. 2). I constructed a reporter gene in which the UPRE-containing KAR2 promoter was placed upstream of an Escherichia coli β-galactosidase gene. Colonies carrying this reporter gene which formed on agar plates containing X-Gal (no color) were white as usual but were expected to turn blue upon ER stress: the induced expression of not only yeast BiP but also β-galactosidase results in the hydrolysis of X-Gal, and X is blue. I utilized a temperature-sensitive sec53 mutant as parent strain; sec53 mutant cells are normal at the low temperature 23 °C, but became ER-stressed at 30 °C, owing to a defect in phosphomannomutase activity which, in the same way as glucose depletion, blocks N-glycosylation of newly synthesized proteins in the ER, resulting in the accumulation of misfolded proteins in the ER.13) I could therefore induce ER stress in the reporter strain (sec53 cells carrying the KAR2 promoter-β-galactosidase reporter gene in a plasmid vector) by simply increasing the incubation temperature. I randomly mutagenized the genome of the reporter strain with ethylmethanesulfonate, a chemical mutagen. If any gene involved in the UPR was mutated in a cell, colonies formed at 23 °C were white, and remained so even after temperature was upshifted to 30 °C, because neither yeast BiP nor β-galactosidase was induced. I conducted this simple blue-white screening for 100,000 independent colonies (200 colonies/plate × 100 plates × 5 times). I obtained 100 colonies which did not turn blue very well on plate at 30 °C. By measuring β-galactosidase activity in these cells on culture in liquid medium at 23 °C or 30 °C, I was able to select three mutant cells defective in the UPR.17) This took half a year.
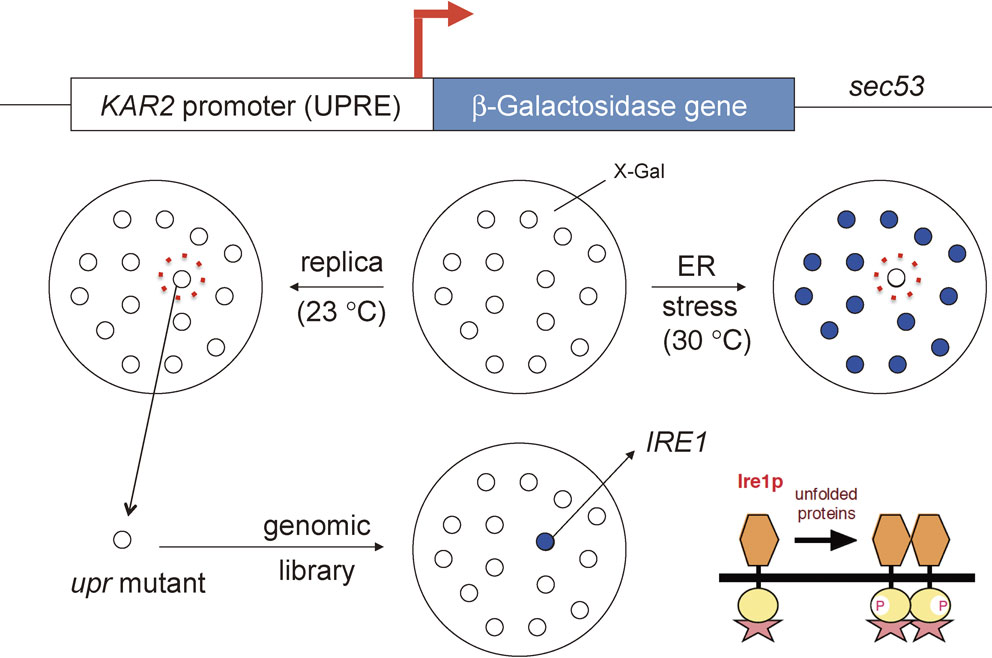
From its signaling nature, the UPR requires at least three mechanisms for completion (Fig. 1; see the dark blue arrow from the ER to the nucleus). The first is the mechanism by which ER stress is sensed in the lumen of the ER and transmitted across the ER membrane; the second achieves the activation of transcription of the KAR2 gene in the nucleus; and the third connects the event in the ER (ER stress sensing) with that in the nucleus (transcriptional activation). I tried to identify the defective gene(s) in my three upr mutants by transformation with a yeast genomic library; if the defective gene was complemented, the white colony should have turned blue again after a temperature upshift to 30 °C. For this I needed more than 10,000 independent transformants, and needed to introduce a different vector clone from a genomic library into each, to cover the entire genome. However, improving transformation efficiency proved quite a difficult task. I struggled with this for half a year. In December, 1991, by incorporating the reporter gene into the genome of upr mutants, I finally succeeded in obtaining a single vector clone, which turned out to complement all three upr mutants — 1 gene from 100,000 colonies!
Sequencing analysis predicted that the gene in the vector clone encoded a type I transmembrane protein with a protein kinase domain located at the cytoplasmic side, suggesting that the protein functions as an ER stress sensor (Fig. 2). It was a new gene when I sequenced it (it took three months) but was found to be identical to the IRE1 gene encoding inositol-requiring enzyme 1 (Ire1p), which was identified by Jun-ichi Nikawa in 1992 as a gene capable of complementing a yeast mutant requiring exogenous inositol for its growth.18) Ire1p is the only transmembrane protein kinase among approximately 6,000 yeast gene products. I demonstrated that its protein kinase activity is essential for UPR signaling by mutating amino acids indispensable for enzymatic activity. I also raised two polyclonal antibodies against the N-terminal (luminal) region of Ire1p and was about to finish demonstrating its type I transmembrane nature. And then came the day, probably in April, 1993, that Mary-Jane came back from San Francisco with terrible news. Peter Walter, a famous young professor at the University of California, San Francisco, had conducted similar yeast genetic screening and obtained the same IRE1 gene, and his paper was in press in ‘Cell’! We all were deeply depressed but Mary-Jane and Joe told me that “you have only a few experiments to go, so do not give up, do all that we planned to do, and then let’s submit to Cell”. Peter’s paper was published in the June 18 issue.19) Although a very exceptional case, my paper, with a significant characterization of Ire1p, was accepted and published in the August 27 issue.17) The publication of these two ‘Cell’ papers in 1993 brought world-wide recognition and opened up an entirely new field, “the UPR”.
Molecular cloning of the HAC1 gene encoding yeast UPR-specific transcription factor Hac1p
After publication of the IRE1 paper, I came back to Kyoto in September, 1993, and obtained a postdoctoral position at HSP Research Institute. The institute was directed by Takashi Yura, who had just retired from the Virus Research Institute, Kyoto University. He was a pioneer of the heat shock protein research field in Japan. Takashi served as chairman when I reported my IRE1 story at a meeting held at the University of Occupational and Environmental Health, Kyushu, Japan in November, 1992.20) He liked my work and accepted me into his Institute; it was a joint program between the Japanese government and four Japanese pharmaceutical companies, which existed only for 7 years by contract (April, 1993 to March, 2000).
My next target was the yeast UPR-specific transcription factor. Yeast researchers often perform multicopy suppressor screening after isolation of yeast mutant cells. I also intended to isolate genes which suppressed the UPR signaling defect in my ire1 mutant cells when overexpressed from a multicopy vector, but Takashi rejected this plan. If a protein kinase cascade occurred downstream of Ire1p, such screening would likely identify genes existing between IRE1 and my transcription factor gene, such as many protein kinase genes. This would prevent the identification and characterization of my transcription factor within my limited 7 years. Takashi continued that as the HSP Research Institute focuses on transcription, I should come up with a method which would allow me to obtain my transcription factor directly. It was a difficult task. Given my biochemistry background, perhaps Takashi was thinking of biochemical purification of the factor, but I did not like this approach because protein purification is laborious and the determination of amino acid sequence was particularly difficult at that time. Rather, I wanted to obtain the factor gene by a molecular biology method.
During this period, in parallel, I extensively characterized the 22 bp UPRE by constructing and analyzing 22 single point mutants. This revealed that the sequence CAGCGTG is essential for UPRE activity. CANNTG is called the E box, to which basic region-containing transcription factors bind; and a characteristic of the yeast UPRE is that its E box contains a nucleotide spacer C, which is also important for UPRE activity.21) I showed later that this “E box containing one nucleotide spacer” structure is conserved in the promoter regions of other ER chaperone genes.22)
As this kind of cis-acting element analysis immediately gave me a UPRE mutant with much weaker transcriptional activity than wild-type UPRE, I thought of and indeed conducted multicopy suppressor screening with this UPRE mutant, because I thought that only overexpression of the trans-acting factor would suppress the weak activity of the cis-acting element. After initial screening I was excited to obtain a single clone encoding a basic-region-containing protein (candidate A). However, I was immediately disappointed because knockout of this gene had no effect on the UPR, unlike knockout of IRE1. Nonetheless, I could not give up. Candidate A seemed too good as it is a basic region-containing transcription factor. I sought an excuse to explain why knockout of candidate A did not show the phenotype I expected. I came up with the idea that because yeast cells have only Ire1p as an ER stress sensor, knockout of IRE1 abolished the UPR signaling. However, because basic region-containing transcription factors usually work as a dimer, two yeast UPR-specific transcription factors might be present, A and B. So, knockout of candidate A alone would not suffice to shut down UPR signaling. But if I obtained B as the dimerization partner of candidate A, and knocked out both A and B, this double knockout would shut down UPR signaling completely.
Scientists often employ two-hybrid screening to identify the partner protein of a protein they already have (Fig. 3). I wondered whether I should also conduct two-hybrid screening to identify candidate B as the partner protein of candidate A, which I already had. In two-hybrid screening, the gene encoding candidate A is fused to the gene encoding the GAL4 DNA-binding domain (one hybrid), and the cDNA library contains various genes (including the gene encoding candidate B) fused to the gene encoding the GAL4 activation domain (another hybrid). If candidate A associates with candidate B in a yeast strain employed for screening, this two-hybrid has all the function of an active transcription factor that enhances transcription of the reporter gene constitutively via binding to its cis-acting element, called the GAL4 binding site. But there was a major obstacle. I was of course aware that if candidate A was not the real UPR-specific transcription factor, its partner protein candidate B could not be the real UPR-specific transcription factor either, and this would result in the loss of several precious months. For a while I struggled with whether I should conduct this two-hybrid screening or try something else.
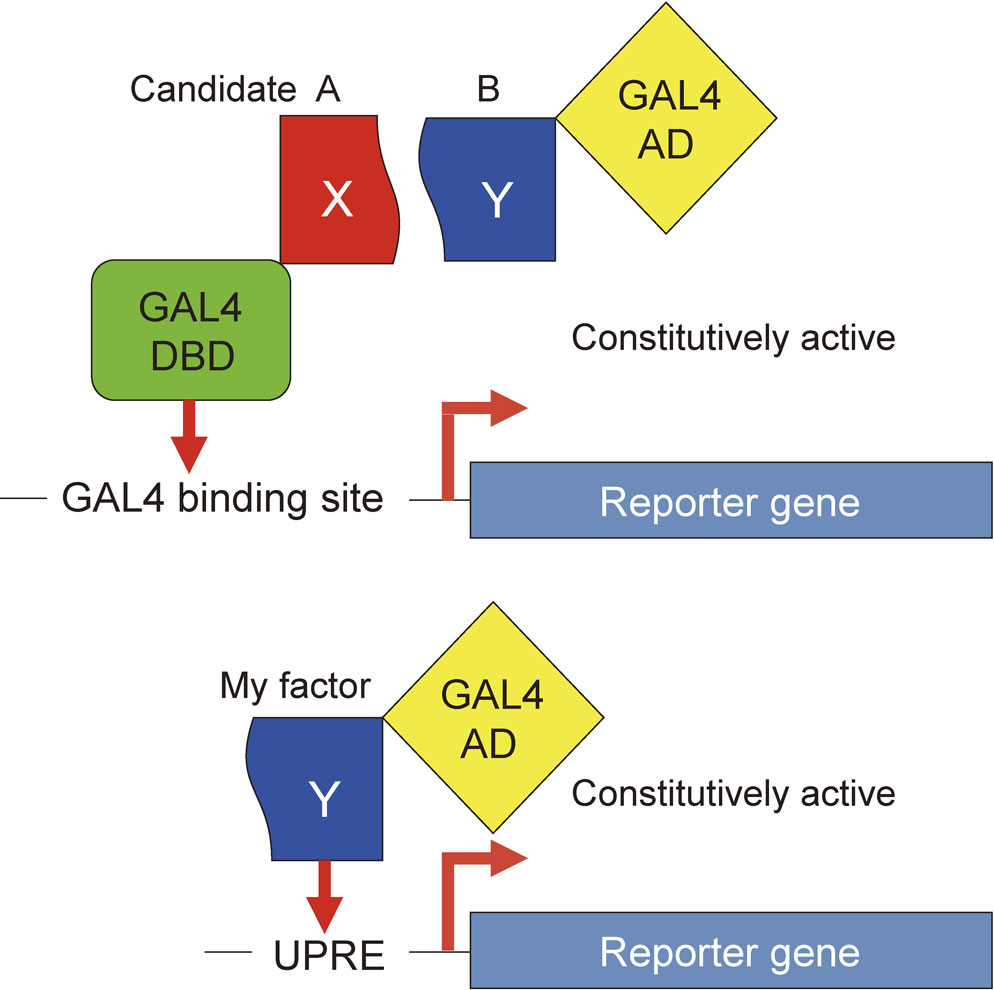
The impasse was solved one happy day in January, 1995 when a bright idea suddenly jumped into my mind. The yeast UPR-specific transcription factor I wanted to identify is somehow activated, by a mechanism unknown, only when the cell is burdened with ER stress. However, if the gene encoding my transcription factor, which should exist in the cDNA library, is fused to the gene encoding the GAL4 activation domain, which is made to be constitutively active, my factor should become constitutively active in a yeast strain regardless of the presence or absence of ER stress. Under this scenario, if the UPRE is placed upstream of the reporter gene instead of the GAL4-binding site used for two-hybrid screening, then my factor, fused to the GAL4 activation domain (one hybrid), would constitutively enhance transcription of the reporter gene (Fig. 3).
This one-hybrid screening, a modified version of two-hybrid screening, can only be used to identify transcription factors. Nevertheless, it worked out extremely very well for me. I was able to pull out the HAC1 gene, which encodes the basic leucine zipper-type transcription factor Hac1p. Disruption of the HAC1 gene abolished UPR signaling completely, as in cells lacking Ire1p. This confirmed it — yeast cells have only a single UPR-specific transcription factor! This also meant that if I had conducted two-hybrid screening, I would have failed to identify Hac1p because candidate B, the partner of candidate A, cannot be Hac1p. My success in identifying Hac1p can be ascribed without hesitation to this born-from-struggle, blockbuster of an idea for one-hybrid screening. This screening greatly helped me again later when I tried to identify mammalian UPR-specific transcription factors capable of binding to ER stress response element responsible for the transcriptional induction of mammalian ER chaperone genes.16) Had Takashi allowed me to employ multicopy suppressor screening, I might have been able to identify Hac1p (as Peter did) but Hiderou Yoshida and I would never have identified ATF6 and XBP1, two critical mammalian UPR-specific transcription factors, because multicopy suppressor screening works only for yeast genes. The idea of one hybrid screening pushed my research dramatically forward. Now as then, my appreciation to Takashi for his critical suggestion has never flagged.
Mechanism to connect Ire1p with Hac1p
I submitted my paper describing the identification and characterization of Hac1p to ‘Cell’ but it was rejected. The reviewers requested that we also reveal how Ire1p activation is connected to Hac1p production. At that time I had already made the very interesting observation that HAC1 mRNA is spliced in response to ER stress in an Ire1p-dependent manner. So, I published my paper of Hac1p with characterization of the UPRE in 1996,21) and then Tetsushi Kawahara and I worked hard to obtain the data required for submission of the splicing paper to ‘Cell’. One day in October, 1996, somebody called to say that Peter Walter talked in a meeting about his identification of Hac1p as a yeast UPR-specific transcription factor as well as his discovery of Ire1p-dependent HAC1 mRNA splicing. Peter obtained the HAC1 gene by multicopy suppressor screening, which explains why he was faster than me. This phone call pushed me over the edge into a bottomless pit. Strangely, however, the caller continued to tell me Peter’s interpretation of how HAC1 mRNA splicing results in the production of Hac1p, which seemed completely at odds with my interpretation at the time.
Peter’s paper was published in the November 1 1996 issue of ‘Cell’.23) It turned out that both Peter and I had made three identical observations (Fig. 4). First, Hac1p is not detected in lysates of unstressed cells but is clearly detected in lysates of ER-stressed cells. Second, HAC1 mRNA is constitutively expressed as a 1.4 kb band, which is converted to a shorter 1.2 kb band in ER-stressed cells due to removal of an intron consisting of 252 nucleotides. These changes in Hac1p level and HAC1 mRNA size do not occur in cells deficient in Ire1p. Third, because the 5′ end of the intron is located within the HAC1 open reading frame, the Hac1p C-terminus is replaced from 10aa to 18aa without affecting the Hac1p N-terminus of 220aa, as a result of mRNA splicing. Thus, Hac1p of 230aa (220aa + 10aa) is translated from HAC1 precursor mRNA (1.4 kb) but Hac1p of 238aa (220aa + 18aa) is translated from HAC1 mature mRNA (1.2 kb).
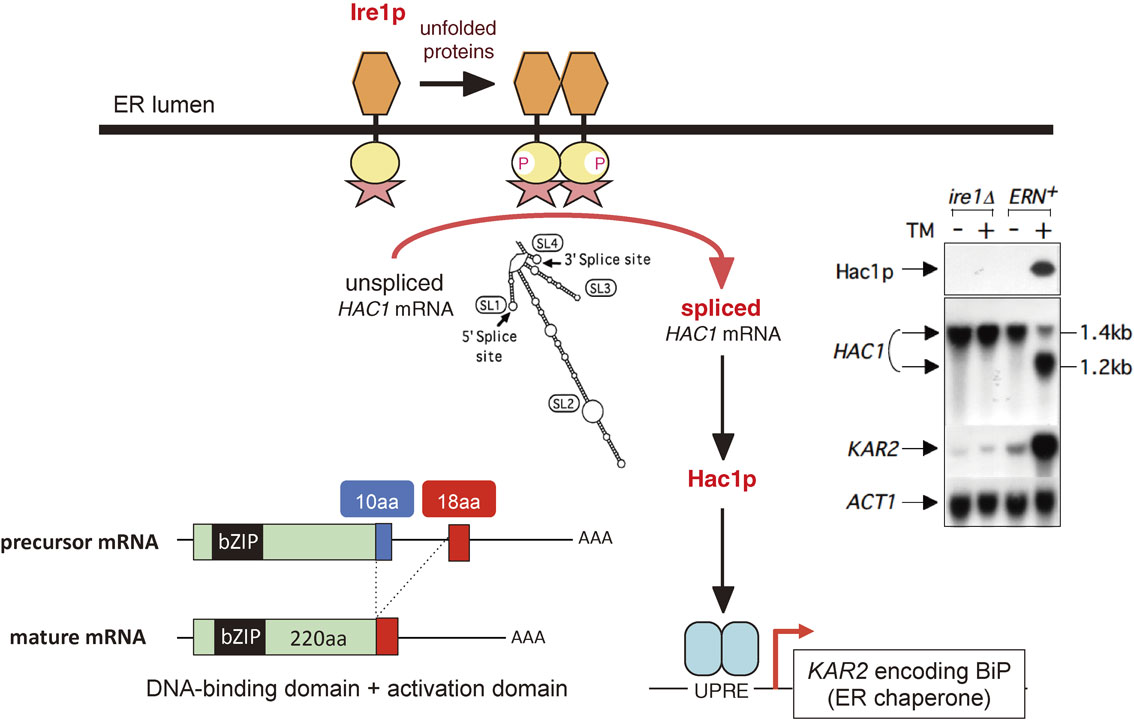
Obviously, there are two interpretations for these observations, depending on whether HAC1 precursor mRNA is translated or not. Peter considered that HAC1 precursor mRNA is constitutively translated but that its product, Hac1p of 230aa, is rapidly degraded due to the presence of C-terminal 10aa, whereas the translational product of HAC1 mature mRNA, Hac1p of 238aa, is stable because it lacks the 10aa, a destabilizer of Hac1p, resulting in the detection of Hac1p only in ER-stressed cells. In marked contrast, I considered that HAC1 precursor mRNA is not translated due to the presence of the intron, which has the ability to block translation, but that HAC1 mature mRNA lacking the intron is efficiently translated, resulting in the detection of Hac1p only in ER-stressed cells. Tetsushi and I conducted additional experiments and submitted a paper to ‘Molecular Biology of the Cell (MBC)’. It was not accepted but Associate Editor Randy Schekman wrote in his decision letter on January 16 in 1997 that “Some of the work in your manuscript is clearly at variance with one of the conclusions of the Walter group. If there were good reasons to favor your view, this manuscript would be quite appropriate for MBC”. This comment really encouraged me. Tetsushi and I continued working harder than ever and were eventually able to demonstrate that both the Hac1p of 230aa and Hac1p of 238aa were highly unstable, with half-lives of 2 min, and that the intron of 252 nucleotides indeed blocked translation of HAC1 precursor mRNA, probably due to its bulky stem-loop structures (SL1–SL4) (Fig. 4). My paper was published in the October 1997 issue of ‘MBC’.24)
In December, 1996, Peter and Mary-Jane organized a symposium at the American Society of Cell Biology meeting held at San Francisco. I replaced Mary-Jane, who had suddenly become sick, and gave my interpretation.25) Although Peter was quite confident of his interpretation at that time, he revised it in a paper published in the November 1 1997 issue of Current Biology.26) Furthermore, Peter showed that base-pairing interaction between the intron (SL2) and the 5′ untranslated region is required and sufficient to block mRNA translation.27) Now Peter and I agree that ER stress-induced splicing of HAC1 precursor mRNA permits synthesis of the yeast UPR-specific transcription factor Hac1p.
What is the biological significance of mRNA splicing-mediated replacement of the Hac1p C-terminus from 10aa to 18aa, given that it does not change the stability of Hac1p? By constructing and analyzing fusions with the GAL4 DNA-binding domain, I demonstrated that the 18aa functioned as a transcriptional activation domain.28) Thus, removal of the intron allows not only the synthesis of Hac1p but also the production of a highly active transcription factor, because the DNA-binding domain included in the 220aa region is fused to the activation domain. In other words, this regulation provides a “fail-safe” mechanism, as follows. HAC1 intron-mediated translational block might be released under certain circumstances in the absence of Ire1p activation, resulting in the production of Hac1p of 230aa. Its low transcriptional activity is rational in this case because the ER is not stressed, as evidenced by the lack of Ire1p activation, and the cell does not need to activate the transcription of ER chaperone genes. In this connection I showed that enhanced expression of ER chaperones in the absence of ER stress (which can be achieved, for example, by constitutive expression of Hac1p of 238aa) is toxic to the cell.28) Hac1p of 230aa may have its own function.
Mechanism of HAC1 mRNA splicing
mRNA splicing is usually carried out by the spliceosome in the nucleus; however, the sequences around the 5′ and 3′ splice sites in HAC1 precursor mRNA do not match the consensus sequence (GT-AG or AT-AC). Indeed, Peter showed that the splicing of HAC1 precursor mRNA is not affected by conditional mutation of two components of the spliceosome (prp2ts and prp8ts), and that Rlg1, a tRNA ligase, is directly involved in the final step of splicing, namely joining the two exons after ER stress-induced cleavage of HAC1 precursor mRNA29) (Fig. 5).
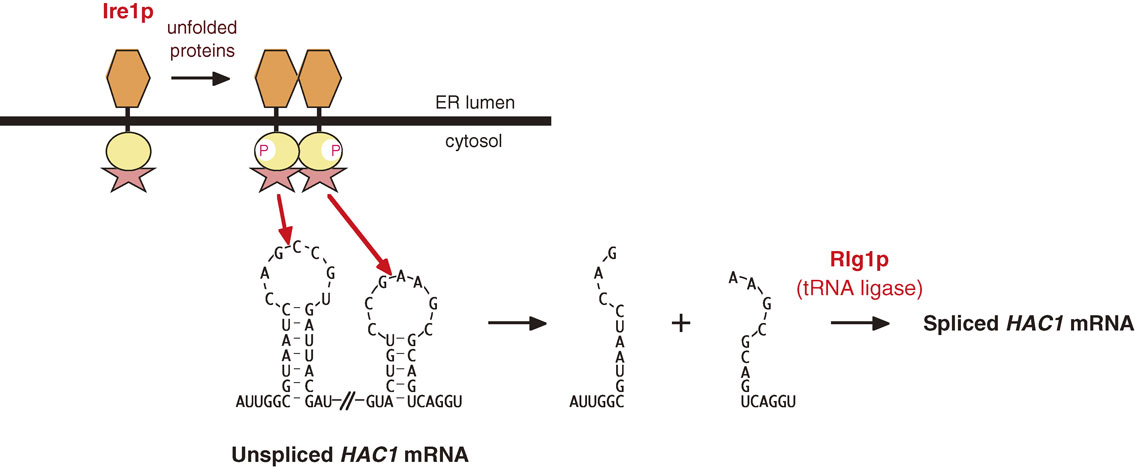
Both the 5′ and 3′ splice sites in HAC1 precursor mRNA form stem-loop structures (SL1 and SL4, respectively; Fig. 4). I demonstrated by mutational analysis in vivo that the cleavage occurs between the third and fourth nucleotides of this loop (between G and C at the 5′ site and between G and A at the 3′ site)24) (Fig. 5). I also showed that at least the first, third, and sixth nucleotides of the loop are strictly recognized for cleavage reaction, and that the cleavage of the 5′ and 3′ splice sites occurs with no obligate order, unlike spliceosome-mediated splicing, in which the 5′ and 3′ splice sites are cleaved sequentially.30)
The above results predicted that ER stress-induced cleavage of HAC1 precursor mRNA involves a novel type of endonuclease. Surprisingly, Peter unraveled that the endonuclease is Ire1p itself. He noticed that a region of Ire1p immediately C-terminal to the protein kinase domain (tail domain) is similar in sequence to mammalian RNase L, and showed that a purified cytoplasmic region containing the protein kinase and tail domains directly cleaves HAC1 precursor mRNA at the characteristic stem-loop structures in vitro31) (Fig. 5). Thus, the yeast UPR is both simple and unique: it is executed by only three gene products, namely Ire1p, HAC1 mRNA, Rlg1p, and Hac1p, and employs an extremely unconventional method of signal transduction, namely spliceosome-independent splicing of HAC1 mRNA, which is initiated by cleavage by Ire1p and terminated by ligation by Rlg1p.
To mammals and beyond
Establishment of the yeast UPR as the Ire1p-Hac1p pathway in the mid 1990’s opened up a greatly rewarding path to the mechanism of the mammalian UPR, which had remained obscure since 1977. Randy Kaufman and David Ron identified IRE1α (ubiquitously expressed in mice) and IRE1β (mainly expressed in the gut in mice), respectively, as mammalian homologues of yeast Ire1p in 1998.32),33) I and David Ron identified XBP1 as a functional mammalian homologue of yeast Hac1p.34),35) ER stress-induced and IRE1-mediated splicing of XBP1 mRNA causes replacement of the XBP1 C-terminus, as in yeast, resulting in production of the highly active transcription factor pXBP1(S). Because the XBP1 intron removed by this splicing is only 26 nucleotide’ long, it cannot block translation, and therefore XBP1 precursor mRNA is constitutively translated to produce pXBP1(U), which functions as a negative regulator of pXBP1(S).36) In addition to IRE1, David Ron and I identified PERK and ATF6α/β, respectively, as ER stress sensors (all three ubiquitously expressed in mice) in mammals in 1999.37)–39) Thus, the mammalian UPR has evolved into a highly sophisticated system which copes with ER stress much more efficiently and effectively. Please refer to my review for information on development from yeast to mammals40) and my recent paper for how ATF6α/β deal with ER stress occurring physiologically during early embryonic development in medaka fish.41) Obviously, the ER chaperone BiP is essential in yeast, mice and medaka fish.12),13),41),42) My studies on the UPR clearly showed that ER chaperone levels must be adjusted according to need in the ER, which is as critical as an ER chaperon function.41) The importance of the UPR is now established in many diverse fields. Retrospectively, this is most impressive to me, in the sense that analysis of the UPR started with yeast in 1989, and that the Lasker Basic Medical Research Award, the most prestigious medical award in U.S.A., was given to this field 25 years later. The UPR has grown up pretty much as Peter and I had hoped — indeed, it might have exceeded our expectations.
Profile

Kazutoshi Mori was born in Okayama Prefecture in 1958. He graduated from the Faculty of Pharmaceutical Sciences, Kyoto University in 1981. He was a master course student from 1981 to 1983 and a doctoral course student from 1983 to 1985 at the Graduate School of Pharmaceutical Sciences, Kyoto University, and received his Ph.D. degree from Kyoto University in 1987. He worked as an Instructor at Gifu Pharmaceutical University from 1985 to 1989, and then as a postdoctoral fellow at the University of Texas Southwestern Medical Center in Dallas from 1989 to 1993 under the supervision of Drs. Mary-Jane Gething and Joe Sambrook. He was Deputy Research Manager from 1993 to 1996 and Research Manager from 1996 to 1999 at the HSP Research Institute in Kyoto (Director: Dr. Takashi Yura). He was Associate Professor at the Graduate School of Biostudies, Kyoto University from 1999 to 2003, and became Professor at the Graduate School of Science, Kyoto University in 2003. He first encountered the Unfolded Protein Response (UPR) in Texas in 1989, and has been working on it ever since. He unraveled the basic mechanisms of the yeast and mammalian UPR. He continues working on the biology, physiology and evolution of the UPR. For his accomplishments, he was awarded the Wiley Prize (U.S.A.) in 2005, the Investigator Award of the Japanese Biochemical Society in 2006, the Osaka Science Award in 2008, the Canada Gairdner International Award in 2009, the Medal of Honor with Purple Ribbon from the Emperor in 2010, the 2011 Uehara Prize, the 2013 Asahi Prize, and the Shaw Prize (Hong Kong) and Lasker Basic Medical Research Award (U.S.A.) in 2014.
References
- 1) Gething, M.J. and Sambrook, J. (1992) Protein folding in the cell. Nature 355, 33–45.
- 2) Helenius, A., Marquardt, T. and Braakman, I. (1992) The endoplasmic reticulum as a protein folding compartment. Trends Cell Biol. 2, 227–231.
- 3) Brodsky, J.L. and McCracken, A.A. (1997) ER-associated and proteasome-mediated protein degradation: how two topologically restricted events came together. Trends Cell Biol. 7, 151–156.
- 4) Mori, K. (2000) Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 101, 451–454.
- 5) Stone, K.R., Smith, R.E. and Joklik, W.K. (1974) Changes in membrane polypeptides that occur when chick embryo fibroblasts and NRK cells are transformed with avian sarcoma viruses. Virology 58, 86–100.
- 6) Isaka, T., Yoshida, M., Owada, M. and Toyoshima, K. (1975) Alterations in membrane polypeptides of chick embryo fibroblasts induced by transformation with avian sarcoma viruses. Virology 65, 226–237.
- 7) Shiu, R.P., Pouyssegur, J. and Pastan, I. (1977) Glucose depletion accounts for the induction of two transformation-sensitive membrane proteins in Rous sarcoma virus-transformed chick embryo fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 74, 3840–3844.
- 8) Hunter, T. and Sefton, B.M. (1980) Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc. Natl. Acad. Sci. U.S.A. 77, 1311–1315.
- 9) Haas, I.G. and Wabl, M. (1983) Immunoglobulin heavy chain binding protein. Nature 306, 387–389.
- 10) Munro, S. and Pelham, H.R. (1986) An Hsp70-like protein in the ER: identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell 46, 291–300.
- 11) Kozutsumi, Y., Segal, M., Normington, K., Gething, M.J. and Sambrook, J. (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332, 462–464.
- 12) Rose, M.D., Misra, L.M. and Vogel, J.P. (1989) KAR2, a karyogamy gene, is the yeast homolog of the mammalian BiP/GRP78 gene. Cell 57, 1211–1221.
- 13) Normington, K., Kohno, K., Kozutsumi, Y., Gething, M.J. and Sambrook, J. (1989) S. cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell 57, 1223–1236.
- 14) Mori, K., Sant, A., Kohno, K., Normington, K., Gething, M.J. and Sambrook, J.F. (1992) A 22 bp cis-acting element is necessary and sufficient for the induction of the yeast KAR2 (BiP) gene by unfolded proteins. EMBO J. 11, 2583–2593.
- 15) Kohno, K., Normington, K., Sambrook, J., Gething, M.J. and Mori, K. (1993) The promoter region of the yeast KAR2 (BiP) gene contains a regulatory domain that responds to the presence of unfolded proteins in the endoplasmic reticulum. Mol. Cell. Biol. 13, 877–890.
- 16) Yoshida, H., Haze, K., Yanagi, H., Yura, T. and Mori, K. (1998) Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins; involvement of basic-leucine zipper transcription factors. J. Biol. Chem. 273, 33741–33749.
- 17) Mori, K., Ma, W., Gething, M.J. and Sambrook, J. (1993) A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 74, 743–756.
- 18) Nikawa, J. and Yamashita, S. (1992) IRE1 encodes a putative protein kinase containing a membrane-spanning domain and is required for inositol phototrophy in Saccharomyces cerevisiae. Mol. Microbiol. 6, 1441–1446.
- 19) Cox, J.S., Shamu, C.E. and Walter, P. (1993) Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206.
- 20) Mori, K. (1993) Mechanism of induction of yeast KAR2 (BiP) by stress. J. UOEH 15, 42–49.
- 21) Mori, K., Kawahara, T., Yoshida, H., Yanagi, H. and Yura, T. (1996) Signalling from endoplasmic reticulum to nucleus: transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells 1, 803–817.
- 22) Mori, K., Ogawa, N., Kawahara, T., Yanagi, H. and Yura, T. (1998) Palindrome with spacer of one nucleotide is characteristic of the cis-acting unfolded protein-response element in Saccharomyces cerevisiae. J. Biol. Chem. 273, 9912–9920.
- 23) Cox, J.S. and Walter, P. (1996) A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87, 391–404.
- 24) Kawahara, T., Yanagi, H., Yura, T. and Mori, K. (1997) Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol. Biol. Cell 8, 1845–1862.
- 25) Pahl, H.L. and Baeuerle, P.A. (1997) Endoplasmic reticulum-induced signal transduction and gene expression. Trends Cell Biol. 7, 50–55.
- 26) Chapman, R.E. and Walter, P. (1997) Translational attenuation mediated by an mRNA intron. Curr. Biol. 7, 850–859.
- 27) Ruegsegger, U., Leber, J.H. and Walter, P. (2001) Block of HAC1 mRNA translation by long-range base pairing is released by cytoplasmic splicing upon induction of the unfolded protein response. Cell 107, 103–114.
- 28) Mori, K., Ogawa, N., Kawahara, T., Yanagi, H. and Yura, T. (2000) mRNA splicing-mediated C-terminal replacement of transcription factor Hac1p is required for efficient activation of the unfolded protein response. Proc. Natl. Acad. Sci. U.S.A. 97, 4660–4665.
- 29) Sidrauski, C., Cox, J.S. and Walter, P. (1996) tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 87, 405–413.
- 30) Kawahara, T., Yanagi, H., Yura, T. and Mori, K. (1998) Unconventional splicing of HAC1/ERN4 mRNA required for the unfolded protein response: sequence-specific and non-sequential cleavage of the splice sites. J. Biol. Chem. 273, 1802–1807.
- 31) Sidrauski, C. and Walter, P. (1997) The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039.
- 32) Tirasophon, W., Welihinda, A.A. and Kaufman, R.J. (1998) A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824.
- 33) Wang, X.Z., Harding, H.P., Zhang, Y., Jolicoeur, E.M., Kuroda, M. and Ron, D. (1998) Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 17, 5708–5717.
- 34) Yoshida, H., Matsui, T., Yamamoto, A., Okada, T. and Mori, K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891.
- 35) Calfon, M., Zeng, H., Urano, F., Till, J.H., Hubbard, S.R., Harding, H.P., Clark, S.G. and Ron, D. (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96.
- 36) Yoshida, H., Oku, M., Suzuki, M. and Mori, K. (2006) pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J. Cell Biol. 172, 565–575.
- 37) Harding, H.P., Zhang, Y. and Ron, D. (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274.
- 38) Haze, K., Yoshida, H., Yanagi, H., Yura, T. and Mori, K. (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799.
- 39) Haze, K., Okada, T., Yoshida, H., Yanagi, H., Yura, T., Negishi, M. and Mori, K. (2001) Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem. J. 355, 19–28.
- 40) Mori, K. (2009) Signalling pathways in the unfolded protein response: development from yeast to mammals. J. Biochem. 146, 743–750.
- 41) Ishikawa, T., Okada, T., Ishikawa-Fujiwara, T., Todo, T., Kamei, Y., Shigenobu, S., Tanaka, M., Saito, T.L., Yoshimura, J., Morishita, S., Toyoda, A., Sakaki, Y., Taniguchi, Y., Takeda, S. and Mori, K. (2013) ATF6alpha/beta-mediated adjustment of ER chaperone levels is essential for development of the notochord in medaka fish. Mol. Biol. Cell 24, 1387–1395.
- 42) Luo, S., Mao, C., Lee, B. and Lee, A.S. (2006) GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell. Biol. 26, 5688–5697.
- 43) Shamu, C.E. and Walter, P. (1996) Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 15, 3028–3039.