1. Introduction
Peroxisomes are single-membrane-bounded ubiquitous organelles containing a hundred different enzymes that catalyze various metabolic pathways, including β-oxidation of very long-chain fatty acids, the synthesis of ether lipids such as plasmalogens, and bile-acid metabolism1) (Table 1). They were discovered in 1954, named peroxisomes in 1965, and defined that peroxisomes contain one or more enzymes that use molecular oxygen to remove hydrogen atoms and form hydrogen peroxide from organic substrates.2) Catalase, a typical marker enzyme of peroxisomal matrix, degrades hydrogen peroxides.
Table 1. Functions of peroxisome
a
| 1. |
Respiration: H2O2-producing oxidases, catalase |
| 2. |
Fatty acid β-oxidation: acyl-CoA oxidase, bifunctional protein (hydratase-dehydrogenase), thiolase |
| 3. |
Ether glycerolipid (plasmalogen) biosynthesis: DHAP-ATPase, alkyl-DHAP synthase |
| 4. |
Transamination and oxidation (gluconeogenesis): serine-pyruvate aminotransferase (alanine-glyoxylate aminotransferase) |
| 5. |
Purine catabolism |
| 6. |
Polyamine catabolism |
| 7. |
Bile acid biosynthesis |
| 8. |
Pipecolic acid catabolism |
| 9. |
Phytanic acid catabolism |
aRepresentative functions in mammalian peroxisomes are listed, where peroxisomal enzymes involved in the functions (1 to 4) are also described.
Peroxisomes are thought to form by the division of pre-exiting peroxisomes after the import of newly synthesized proteins.3) Molecular mechanisms of peroxisome biogenesis, including peroxisomal import of newly synthesized matrix and membrane proteins, have been extensively investigated basically by several eukaryotic systems. Studies at the molecular level on both peroxisome assembly and peroxisome biogenesis disorders (PBDs, Table 2) rapidly progressed in the last three to four decades. The identification and isolation of over 30 essential genes termed PEXs encoding peroxisome biogenesis factors named peroxins, have been performed by means of the genetic phenotype-complementation of peroxisome assembly-deficient cell mutants, named pex mutants impaired in PEX genes. Such mutants from Chinese hamster ovary (CHO) cells (Table 3; see below),4),5) several yeast species including Saccharomyces cerevisiae,6) Pichia pastoris,7),8) Hansenula polymorpha,9) and Yarrowia lipolytica10) (also see reviews11)–16)), and plant Arabidopsis thaliana17) have been successfully contributing to the investigations of peroxisome biogenesis and protein traffics in eukaryotes.18),19) I herein review and address peroxisome biogenesis and human deficiency disorders by making use of mammalian model cell systems.
Table 2. Peroxisomal disease
| Peroxisome biogenesis disorders (PBDs) |
| Zellweger spectrum disorders |
| Zellweger syndrome (ZS) |
| Neonatal adrenoleukodystrophy (NALD) |
| Infantile Refsum disease (IRD) |
| Rhizomelic chondrodysplasia punctata (RCDP) |
| |
| Single-enzyme deficiencies |
| Adrenoleukodystrophy (ALD) |
| Acyl-CoA oxidase deficiency |
| D-Bifunctional protein deficiency |
| 3-Ketoacyl-CoA thiolase deficiency |
| Refsum disease (phytanyl-CoA hydroxylase deficiency), α-Methylacyl-CoA racemase deficiency |
| Hyperoxaluria type I (alanine glyoxylate aminotransferase deficiency) |
| Mevalonate kinase deficiency |
| Glutaric aciduria 3 (glutaryl-CoA oxidase deficiency) |
| Acatalasemia |
Table 3. Complementation groups (CGs) and
PEX genes of peroxisome deficiencies
| Gene |
CG |
PBD |
CHO mutants |
Ps-memb.
biogenesisa |
Peroxin |
| US/EU |
Japan |
(kDa) |
Characteristics |
| PEX1 |
1 |
E |
ZS, NALD*, IRD* |
Z24, ZP107 |
+ |
143 |
AAA family |
| PEX2 |
10 |
F |
ZS, IRD* |
Z65 |
+ |
35 |
PMP, RING |
| PEX3 |
12 |
G |
ZS |
ZPG208 |
− |
42 |
PMP, PMP-DP |
| PEX5 |
2 |
|
ZS, NALD |
ZP105*, ZP139 |
+ |
68 |
PTS1 receptor, TPR family |
| PEX6 |
4(6) |
C |
ZS, NALD* |
ZP92 |
+ |
104 |
AAA family |
| PEX7 |
11 |
R |
RCDP |
ZPG207 |
+ |
36 |
PTS2 receptor, WD motif |
| PEX10 |
7(5) |
B |
ZS, NALD |
|
+ |
37 |
PMP, RING |
| PEX11β |
16 |
|
ZS |
|
+ |
28 |
PMP |
| PEX12 |
3 |
|
ZS, NALD, IRD |
ZP109 |
+ |
40 |
PMP, RING |
| PEX13 |
13 |
H |
ZS, NALD* |
ZP128 |
+ |
44 |
PMP, PTS1-DP, SH3 |
| PEX14 |
15 |
K |
ZS |
ZP110 |
+ |
41 |
PMP, PTS1-DP, PTS2-DP |
| PEX16 |
9 |
D |
ZS |
|
− |
39 |
PMP, PMP-DP |
| PEX19 |
14 |
J |
ZS |
ZP119 |
− |
33 |
CAAX motif, PMP receptor |
| PEX26 |
8 |
A |
ZS, NALD*, IRD* |
ZP124, ZP167 |
+ |
34 |
PMP, Pex1p-Pex6p recruiter |
| |
|
|
|
ZP114 |
+ |
|
|
*, temperature-sensitive phenotype. aPeroxisomal membrane assembly is normal (+) or impaired (−).
PBD, peroxisomal biogenesis disorders; ZS, Zellweger syndrome; IRD, infantile Refsum disease; NALD, neonatal adrenoleukodystrophy; RCDP, rhizomelic chondrodysplasia punctata; DP, docking protein; PMP, peroxisome membrane protein; TPR, tetratricopeptide repeat.
2. Genetic approaches to studying mammalian peroxisome biogenesis
Two mutually complementary genetic approaches used for isolation of PEX genes encoding peroxins were genetic phenotype-complementation of peroxisome assembly-defective mutants of CHO cells and a combination of the human orthologue isolation by homology search on the human expressed sequence tag database using yeast PEX genes and cells from patients with PBDs of more than a dozen different genotypes, i.e., complementation groups (CGs) (Table 3; see below).4),5),20)–22)
2.1. Mammalian cell mutants deficient of peroxisome.
2.1.1. Cell lines from patients with PBDs.
The PBDs include Zellweger syndrome (ZS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), which are called Zellweger syndrome spectrum, and rhizomelic chondrodysplasia punctata (RCDP)23),24) (Table 2). Patients with ZS show severe neurological abnormalities, characteristic dysmorphism and hepatomegaly, and rarely survive with an average life of only 6 months. NALD patients have the symptoms similar to ZS patients, but they survive a little longer, early childhood. By contrast, patients with IRD do not manifest significant abnormalities in the central nervous system, and survive with the longest average life, 3–11 years.23) RCDP patients show distinct phenotypic characteristics such as severe growth failure and rhizomelia. Genetic heterogeneity consisting of 14 CGs has been identified in PBDs by cell-fusion CG analysis using fibroblast cell lines derived from PBD patients5),20),25)–27) (Table 3), where the primary cause for PBDs was revealed to be the impaired biogenesis of peroxisomes.5),20)
2.1.2. Isolation of CHO cell lines.
Two methods were developed for the isolation of mammalian somatic cell mutants defective in peroxisome biogenesis: (i) colony autoradiographic screening with a phenotypic marker, dihydroxyacetonephosphate acyltransferase (DHAP-ATase) deficiency;28),29) and (ii) the photo-sensitized selection method using 9-(1′-pyrene)nonanol (P9OH) and an exposure to long wave-length ultraviolet (UV) light which kills wild-type cells incorporating P9OH as a fatty alcohol into plasmalogens and survive cell mutants deficient in such activity.30),31) We so far isolated 12 CGs of peroxisome-deficient CHO cell mutants by these methods (Table 3). All of CHO cell mutants showed a typical phenotype of deficiency in peroxisome biogenesis, such as the impaired protein import, no catalase latency, severely affected DHAP-ATase activity, as noted in fibroblasts from PBD patients.5),29)
A complete set of CG analyses by cell-fusion between 12 CGs of CHO cell mutants and 13 CGs of fibroblasts from patients with PBDs revealed that 11 CGs of CHO mutants represent the human PBD CGs5),29) (Table 3). A PBD patient of the 14th CG, CG16, was recently identified.32) Together, genetic heterogeneities comprising 15 CGs are currently reported in mammals including humans and CHO cells.
2.2. Peroxisome biogenesis genes.
2.2.1. Genetic phenotype-complementation screening.
PEXs were isolated by genetic phenotype-complementation of peroxisome biogenesis-deficient mutants of mammalian somatic cells such as CHO cells (Fig. 1A) and of several species of yeast including S. cerevisiae, P. pastoris, Y. lipolytica, and H. polymorpha.12),24),33),34) Forward genetics method using animal somatic cell mutants such as CHO cell mutants was shown to be a highly effective approach for isolating essential genes including the peroxin genes, PEXs.
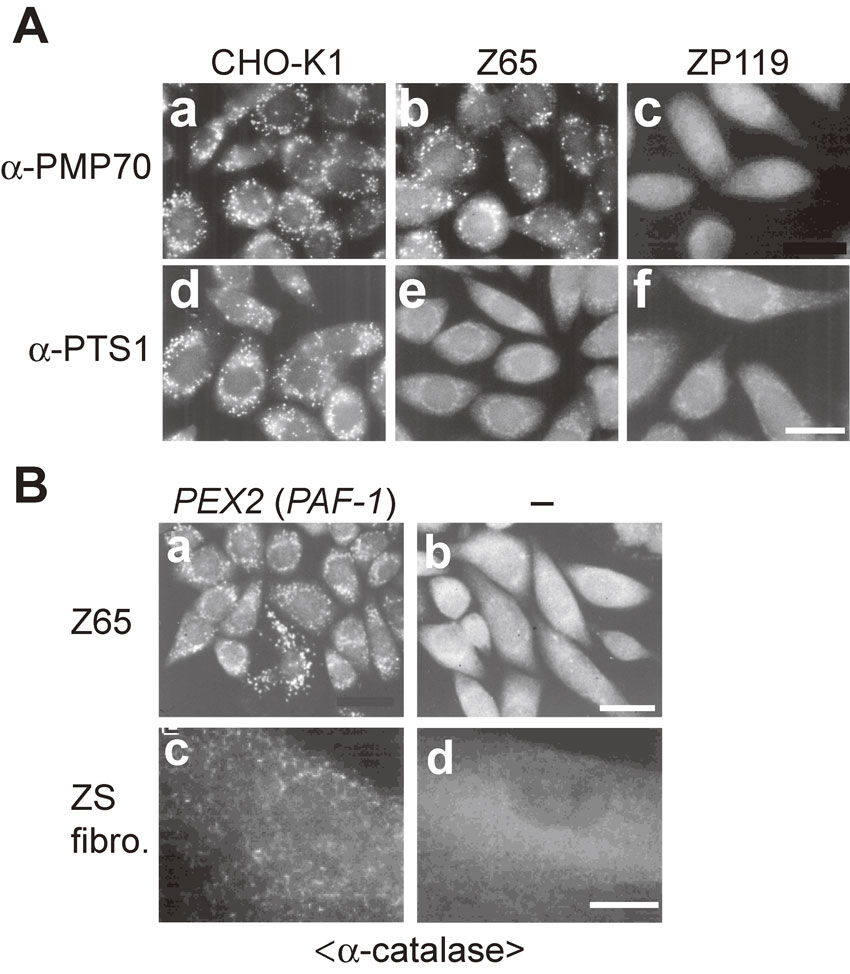
We searched for the gene encoding a factor complementing the impaired peroxisome biogenesis of one, Z65, of the CHO cell mutants by transfecting a rat liver cDNA library.35) Transfectants were selected by the 12-(1′-pyrene) dodecanoic acid (P12)/UV method,36) showing peroxisomes as verified by staining with anti-catalase antibody (Fig. 1B). An open reading frame encoded a novel 35-kDa peroxisomal integral membrane protein with two membrane-spanning segments and a RING finger motif, C3HC4,37) termed peroxisome assembly factor-1 (PAF-1)35) (Table 3; Fig. 1B). PAF-1 was unified as PEX2 in 1996.33) Expression of PEX2 (called Zellweger gene) in fibroblasts from a ZS patient of CG10 (F) complemented the impaired peroxisome biogenesis38) (Fig. 1B). Dysfunction of PEX2 caused by a homozygous nonsense point mutation at R119ter was shown for the first time to be responsible for ZS, a prototype of the PBDs.38)
A more practical approach, i.e., a transient expression assay skipping the revertant selection by P12/UV,39) was also developed for further isolation of PEX cDNAs including nine others, PEX1, PEX3, PEX5, PEX6, PEX12, PEX13, PEX14, PEX19, and PEX2621),34),40)–48) (Table 3; Fig. 2). Human PEX5,49),50) PEX14,51) and PEX19 (PXF)52) were earlier identified. These PEXs were shown to be the pathogenic genes involved in PBDs of nine CGs22),24),34),53),54) (Table 3).
2.2.2. Expressed sequence tag homology search.
An alternative strategy, i.e., the homology search by screening the expressed sequence tag database using yeast PEX genes, successfully made it feasible to isolate human orthologue genes responsible for PBDs:22),24),34) PEX1,55),56) PEX3,57) PEX5,58) PEX6,59) PEX7,60)–62) PEX10,63),64) PEX12,65) PEX13,66) PEX14,67) and PEX16.68),69)
A PBD patient of the 14th CG, CG16, was recently identified with pathogenic gene PEX11β.32) Therefore, 14 PEXs are now shown to be responsible for PBDs of 14 distinct CGs5),22),24),27),53),70) (Table 3).
2.2.3. Genotypes of RCDP.
Several recent findings classified RCDP into five genotypes, of which responsible pathogenic genes are delineated. RCDP type 1 is caused by mutation of PEX7 encoding the peroxisome-targeting signal 2 (PTS2) receptor;60)–62) types 2, 3, and 4 are impaired in three genes, DHAPAT, ADAPS, and FAR1, encoding peroxisomal enzymes, DHAP-ATase, alkyl-DHAP synthase, and fatty acyl-CoA reductase, respectively, involved in the synthesis of plasmalogens;71)–73) type 5 manifesting a mutation of Pex5pL74) that transports PTS1 proteins and Pex7p-PTS2 protein complex to peroxisomes.75)
2.2.4. Genotype–Phenotype Relationships.
Patients with milder form of PBDs, NALD and IRD, tend to manifest less severe biochemical abnormalities, whose specimen including skin fibroblasts likely contain residual peroxisomes, occasionally termed mosaicism. However, clinical severity or prognosis of patients with PBDs cannot be readily predicted only on the basis of biochemical analyses. Various types of mutations such as nonsense point mutations, missense mutations, insertion and deletion of nucleotides mostly with concomitant frameshifts, splicing defects, etc. in both homozygotic and heterozygotic alleles have been identified in PBD patients. Patients with severe ZS tend to carry severe mutation such as nonsense mutations, frameshifts, and deletions, while many patients with NALD or IRD patients frequently harbor missense mutations.76),77) There is also a relationship between severe phenotype and absence of peroxisomal ghosts. Defects of PEX3, PEX16 and PEX19 encoding membrane-assembly peroxins lead to absence of ghosts and cause ZS phenotypes.22),24),34) Many cell lines from milder PBD patients, those with NALD and IRD with missense PEX mutations, showed a temperature-sensitive (ts) phenotype, restoration of peroxisome biogenesis at 30 °C78)–81) (Table 3).
Search for pathogenic genes responsible for all PBD CGs is accomplished.82) Prenatal DNA diagnosis using PEX genes is now possible for PBDs of all 14 CGs.
3. Biogenesis of peroxisomes
3.1. Membrane biogenesis.
3.1.1. Peroxins essential for membrane assembly of peroxisomes.
Three mammalian peroxins, Pex3p, Pex16p, and Pex19p, exclusively required for peroxisomal membrane assembly were isolated by the functional phenotype-complementation assay on pex3 and pex19 CHO cell mutants41),47) and the EST database search using yeast PEX genes.52),57),68),69) Malfunctions of Pex3p, Pex16p, and Pex19p, causes the most severe PBD, ZS, of three CGs, CG12 (G), CG9 (D), and CG14 (J), respectively22),24),34),83) (Table 3).
Pex3p, Pex16p, and Pex19p were identified as essential factors for assembly of peroxisomal integral membrane proteins (PMPs) in several species including humans25),47),68),69),84)–89) (Fig. 2). They function as essential factors in the transport process of membrane proteins and membrane vesicle assembly in a concerted manner. Pex19p is 33-kDa farnesylated protein harboring farnesylation CAAX box motif and localized mostly in the cytosol and only partly anchored to peroxisomal membranes.47) Pex19p has a chaperone-like role in the cytosol or at the peroxisome membrane and/or functions as a cycling import receptor for newly synthesized PMPs.90),91) Pex19p forms stable Pex19p-PMP complexes except for Pex3p in the cytosol with a broad PMP-binding specificity.91)–93) Pex3p, 42-kDa integral membrane protein of peroxisomes, serves as the membrane-anchoring site for Pex19p-PMP complexes, termed Class I pathway.91),94) Very recently, we demonstrated that translocation of PMPs including topologically distinct PMPs such as multi-membrane spanning PMPs and an N-terminally signal-anchored protein via the class I pathway is a common event in mammalian cells.91)
Pex16p, a protein absent in most yeasts,69),95) functions as the receptor for Pex19p complexes with newly synthesized Pex3p,94) named Class II pathway (Fig. 2). The function of Pex16p is not conserved between different species. It is noteworthy that C-tailed anchor-type peroxin Pex26p, the recruiter of Pex1p-Pex6p complex, is transported in a class I pathway,96) which is distinct from the GET3-dependent topogenesis of yeast Pex15p, a functional orthologue of Pex26p.97)
At the step of docking of a cytosolic Pex19p-PMP complex onto Pex3p, Pex19p unloads the cargo PMP and shuttles back to the cytosol for a next round of PMP transport, while the released PMP integrates into the membrane. The membrane insertion of PMPs proceeds in the absence of ATP.96),98)–100) Pex19p and Pex3p apparently facilitate the insertion of transmembrane domains in a concerted manner.101),102) Investigation of molecular mechanisms underlying the membrane integration of the cargo PMPs is under way.
3.1.2. ER is involved in peroxisome biogenesis?
In regard to peroxisomal membrane assembly, the concepts of the Pex19p- and Pex16p-dependent direct import as well as the ER-dependent indirect import have recently emerged.94),103) ER was postulated to provide the initial ‘seed’ for recruiting Pex3p and Pex16p required for peroxisome assembly.104)–106) Several groups suggested a different view of peroxisomal membrane biogenesis that peroxisomes are formed from ER107),108) upon induction of Pex3p;104),109),110) another study111) proposed that all peroxisomal membrane proteins are transported via ER. Several peroxisomal membrane proteins might be transported to peroxisomes via ER,112)–114) implying a semi-autonomous property of peroxisomes. A recent proximity-specific ribosome profiling suggested that many PMPs are translated at the ER in both mammalian and yeast cells, implying that they are plausible substrates for the indirect route.115) Interestingly, several PMPs seem to target to peroxisomes both directly from the cytosol91),94),96),116) and indirectly via the ER.103),117),118)
However, the significance of such observations remains under debate. A study119) suggested that peroxisomes are generally formed by growth and division under normal conditions and that only under a condition where no peroxisome is present in a cell, they can be formed from the ER after the expression of the complementing PEX gene. Meanwhile, we demonstrated that Pex3p, the membrane receptor for Pex19p-complexes with PMPs including Pex16p, is directly targeted to peroxisomes in a Pex19p-Pex16p dependent class II pathway in mammalian cells.94) Moreover, we very recently provided several lines of evidence that most, if not all, mammalian PMPs are indeed authentic substrates for the Pex19p- and Pex3p-mediated class I direct pathway.91) At any event, future investigations on whether the two distinct routes exist simultaneously in cells and when cells use these routes are required for comprehensive understanding of PMP biogenesis.24),83),105),106)
3.2. Matrix protein import.
Ten peroxins including Pex1p, Pex2p, Pex5p, Pex6p, Pex7p, Pex10p, Pex12p, Pex13p, Pex14p, and Pex26p are involved in protein import into peroxisomal matrix24),34),90) (Fig. 2).
3.2.1. PTS import receptors.
PTS1 and PTS2 proteins are recognized by Pex5p and Pex7p, respectively, in the cytoplasm. Two isoforms of Pex5p, Pex5pS and Pex5pL with an internal 37-amino-acid insertion, are identified in mammals. PTS1 proteins are transported by homo- and hetero-oligomers of Pex5pS and Pex5pL to peroxisomes, where Pex14p of an 800-kDa complex functions as the initial Pex5p-docking site (Fig. 2). Pex5pL translocates the Pex7p-PTS2 protein complex to Pex14p.120),121) After releasing the cargoes, Pex5p and Pex7p translocate to a 500-kDa ‘translocation complex’ consisting of the RING peroxins, Pex2p, Pex10p, and Pex12p.121) Both Pex5p and Pex7p finally translocate back to the cytosol.121)–126) At the terminal step of the protein import reaction, AAA peroxins, Pex1p and Pex6p, recruited to Pex26p (Pex15p in yeast) on peroxisomes catalyze the ATP-dependent export of Pex5p.121),124),127)
3.2.2. Peroxisome-cytoplasmic shuttling of import receptors.
Mono-ubiquitination via the thioester bond of the conserved cysteine residue at position 11 in the N-terminal region of Pex5p (Ub-Pex5p) is a prerequisite for the Pex5p recycling, i.e., in the export step from peroxisomes to the cytosol,128)–131) as in yeast132),133) (Fig. 2). Moreover, a cytosolic factor, AWP1/ZFAND6 involved in the export of Ub-Pex5p is identified in mammals;131) USP9X and Ubp15 are suggested as a potential deubiquitinase in mammals134) and yeast,135) respectively. A distinct redox state may affect the recycling of Pex5p requiring Cys-ubiquitination, thereby being as a possible cause to the phenotype of deficiency in matrix protein import in PEX-defective cells.136)

