Reviews
The past, present, and future of disease-modifying therapies for Alzheimer’s disease
2017 年 93 巻 10 号 p. 757-771
詳細
2017 年 93 巻 10 号 p. 757-771
The development of disease-modifying therapies for Alzheimer’s disease (AD) is an urgent issue. Progress in the understanding of AD pathophysiology based on the amyloid hypothesis has led to the development of numerous candidate disease-modifying therapies over the past 15 years. The therapeutic target, amyloid β (Aβ), starts to accumulate in AD brains long before the onset of cognitive decline. γ-secretase inhibitors, γ-secretase modulators, and β-secretase inhibitors aim to reduce the production of toxic Aβ species by modifying the processing of amyloid precursor protein. Another strategy is to eliminate accumulated Aβ by active or passive immunotherapeutic approaches. Therapeutic strategies targeting tau protein are also currently emerging. Despite these efforts, successful disease-modifying therapies for AD have not yet been developed. Recently, very early interventional trials targeting preclinical stages of AD have begun; the paradigm shift in AD therapies from cure to prevention could be key to the success of disease modification.
The number of people with dementia (referred to as “ninchi-sho” in Japanese) is steadily increasing worldwide in association with extended life expectancy. Today, 47 million people are living with dementia around the globe, and this number is estimated to increase to 131 million by 2050.1) The monetary cost of dementia is also huge, an estimated 818 billion US$ worldwide, and continues to grow.1) In Japan, an estimated 4.62 million people were living with dementia as of 2012, a number expected to reach 8.5–11.5 million in 2060 in association with the rapidly aging population.2)
Alzheimer’s disease (AD), characterized by memory impairment followed by progressive cognitive decline, is the most prevalent cause of dementia. As of August 2017, three types of acetylcholinesterase inhibitors and one N-methyl-D-aspartate receptor antagonist have been approved as therapeutic drugs, but these are symptomatic drugs that do not stop or delay the progression of the disease’s pathology. Because advanced patients incur greater costs, the development of potent disease-modifying therapies for AD is urgently needed to slow disease progression. In this article, we describe the history, current status, and future prospects of the development of disease-modifying therapies for AD.
Amyloid hypothesis — rationale for disease-modifying therapies.In 1907, Alois Alzheimer initially described a patient with the two main pathological features of AD: senile plaques, the extracellular plaque-like structures observed outside neuronal cells, and neurofibrillary tangles (NFT), the bundles of fibrous proteins deposited within the cytoplasm of neurons (Fig. 1). The principal pathological concept of AD has remained unchanged, but the molecular basis of senile plaque formation underwent dramatic revision in the 1980s; β-amyloid (Aβ) was identified as the primary component of senile plaque amyloid,3),4) and hyperphosphorylated tau was identified as the main component of NFT.5),6) Since the 1990s, amyloid precursor protein (APP)7) and presenilin-1 (PSEN1)8) have been identified as the genes responsible for familial AD, and mutations in these genes have been shown to cause increased production or deposition of aggregated Aβ.

Therapeutic strategies for Alzheimer’s disease based on the amyloid hypothesis. Aβ is produced by a two-step sequential cleavage of APP by β-secretase and γ-secretase. Toxic Aβ fragments aggregate and finally form plaques. Tau-medicated neurotoxicity and NFT formation are considered to be downstream events in the amyloid cascade. Most DMTs currently under development target steps in the amyloid cascade. (Images are provided by Dr. Ryoko Ihara, the University of Tokyo.)
Based on the accumulated evidence, the “amyloid hypothesis” was proposed as the pathogenetic mechanism of AD9) (Fig. 1). According to the amyloid hypothesis, increased production and/or decreased clearance resulting in the accumulation of Aβ, particularly the toxic Aβ42 species, is the primary cause of AD.10)–12) Aβ is produced by a two-step sequential cleavage of APP, which is a single-pass transmembrane protein of unknown biological function. First, the extracellular domain of APP is cleaved by β-secretase to produce soluble APPβ (sAPPβ) and a membrane-associated C-terminal fragment of APP termed C99. The activity of β-cleavage is considered to determine the total amount of Aβ production. Second, C99 is successively cleaved from the C-terminal side by γ-secretase to yield Aβ peptides ranging in length from 38 to 43 amino acids.13),14) The most abundant Aβ species is Aβ40, consisting of 40 amino acids, whereas Aβ42, only comprising approximately 10% of total produced Aβ, is the most aggregable and toxic isoform of Aβ.15) The efficiency of the successive γ-cleavage strongly affects the production ratio of toxic Aβ42 to total Aβ. Soluble Aβ monomers secreted into the extracellular space assemble into oligomers and fibrils, finally forming insoluble plaques. Oligomers are considered to be the most toxic species among the various forms of Aβ.
Although the link of amyloid accumulation to tau pathology remains unknown, tau pathology is generally believed to be downstream in the amyloid cascade. Tau is a microtubule-associated protein that binds to microtubules and contributes to their stabilization. Self-aggregation of tau, together with its hyperphosphorylation, may lead to microtubular destabilization and neuronal dysfunction, eventually resulting in the manifestation of clinical AD symptoms. Disease-modifying therapies have been developed targeting different steps of the amyloid cascade, including tau pathology. To date, more than 100 clinical trials have been conducted on disease-modifying therapies for AD, most of which have been based on the amyloid hypothesis.
New clinical staging of AD based on pathophysiology.Clinically, dementia is defined as the impairment of daily life due to the progress of decline in cognitive function. Prior to the onset of dementia, cognitive function is slightly reduced but does not impair daily living function; this state between dementia and normal cognition is termed mild cognitive impairment (MCI) (Fig. 2).16) While MCI encompasses the prodromal phase of various dementia-causing diseases, MCI with the main symptom of episodic memory disorder is termed amnestic MCI. The background pathology of amnestic MCI is often AD, and these cases are termed MCI due to AD or prodromal AD. However, the accumulated evidence from autopsies and recent dramatic advances in in vivo imaging techniques, e.g., amyloid positron emission tomography (PET), has shown that amyloid deposition is already present even prior to MCI, 10–20 years before the onset of dementia.17),18) Importantly, recent failures in AD clinical trials at the dementia stage have underscored the idea that the preclinical stages of AD should be defined as part of the disease process and targeted for treatment at a very early stage.
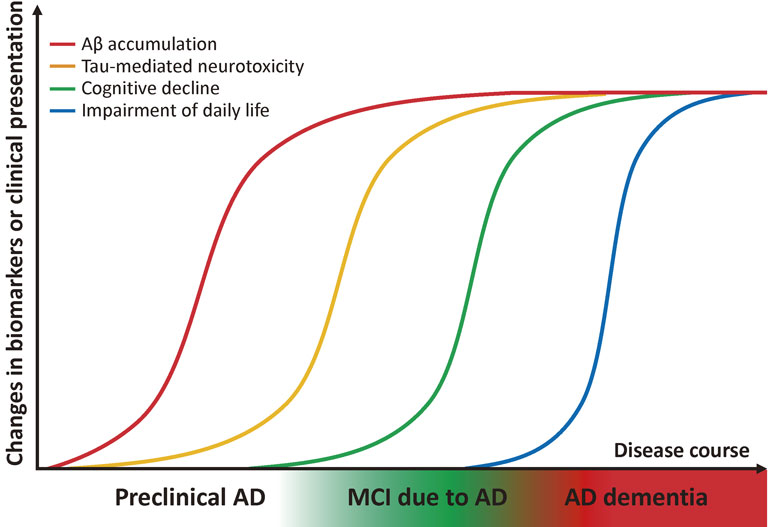
Hypothetical model of biomarker changes and proposed new clinical staging of AD. Accumulation of Aβ and subsequent manifestation of tau-mediated neurotoxicity begin roughly 10–20 years prior to the onset of dementia. MCI due to AD is a prodromal phase of AD, i.e., an intermediate phase between normal cognitive functioning and dementia. Preclinical AD is a stage earlier than MCI in which cognitive function is normal but the presence of amyloid pathology is suggested by biomarkers.
The novel AD diagnostic guideline proposed by the National Institute on Aging-Alzheimer’s Association in 2011 defines “preclinical AD” as a stage preceding MCI due to AD (Fig. 2). According to the guideline, preclinical AD is the state in which amyloid deposition is suggested based on various biomarkers but is clinically asymptomatic. Notably, preclinical AD is currently meant to be used for clinical research or trials, not in general clinical practice. Recent evidence supports the idea that amyloid deposition is a strong risk factor for the future development of AD dementia,19)–22) although it remains to be determined how many of individuals with preclinical AD progress to MCI or dementia within their lifetime, as well as whether amyloid deposition precedes neurodegeneration (including tau pathology) and dysfunction progress in various areas of cerebral cortices.
The candidate drugs currently under clinical development, together with their identification numbers in individual clinical trial registries (e.g., ClinicalTrials.gov, the ISRCTN registry, and The European Union Clinical Trials Register) are shown in Table 1, as well as in the text. During the process of drug development, drug safety and efficacy are verified through a series of clinical trials. Phase 1 studies are usually performed on healthy volunteers to verify a drug’s safety and find an appropriate dosage. Phase 2 studies are then conducted to test the efficacy and safety of a drug on small number of patients. Phase 3 trials, the final step before drug approval, are conducted for a large number of patient groups to demonstrate efficacy and safety.
| Mechanism-based classification | Name | Developer | Current status | |
|---|---|---|---|---|
| Anti-amyloid strategies |
Aβ vaccination | AN1792 | Elan | Discontinued |
| CAD106 | Novartis | Phase 2/3, being tested in API GENERATION | ||
| ACI-24 | AC immune | Phase 1/2 | ||
| ABvac40 | Araclon Biotech | Phase 2 | ||
| Affitope AD02 | Affilis AG | Phase 2 | ||
| ACC-001 | Janssen | Discontinued | ||
| Anti-Aβ antibodies | Bapineuzumab | Pfizer, Janssen | Discontinued | |
| Gantenerumab | Roche/Chugai | Phase 3, being tested in DIAN-TU | ||
| Crenezumab | Genentech/AC immune/Roche | Phase 3, being tested in API-ADAD | ||
| Solanezumab | Eli Lilly | Phase 3 | ||
| Aducanumab | Biogen | Phase 3 | ||
| γ-secretase inhibitor | Semagacestat | Eli Lilly | Discontinued | |
| Notch-sparing γ-secretase inhibitor |
Avagacestat | Bristol-Myers Squibb | Discontinued | |
| γ-secretase modulator | Tarenflurbil | Myriad Genetics & Laboratories | Discontinued | |
| CHF5074 | CereSpir™ Incorporated/ Chiesi Pharmaceuticals Inc. |
Phase 2 | ||
| PF-06648671 | Pfizer | Phase 1 | ||
| β-secretase inhibitor | LY2811376 | Eli Lilly | Discontinued | |
| LY2886721 | Eli Lilly | Discontinued | ||
| MK-8931 (Verubacestat) | Merck | Phase 3 | ||
| AZD3293/LY3314814 | AstraZeneka/Eli Lilly | Phase 2/3 | ||
| CNP520 | Novartis/Amgen | Phase 2/3, being tested in API GENERATION | ||
| E2609 (Elenbecestat) | Eisai/Biogen | Phase 3 | ||
| JNJ-54861911 | Janssen | Phase 3 (EARLY trial) | ||
| Anti-tau strategies |
Tau aggregation inhibitor | LMTM (TRx0237) | TauRx Therapeutics Ltd | Phase 3 |
| Tau kinase inhibotor | Lithium | University of Dundee | Phase 3 | |
| NP031112 (Tideglusib) | Noscira | Discontinued | ||
| Active vaccination for tau | ACI-35 | AC immune/Jansenn | Phase 1b | |
| AAD-vac1 | Axon Neuroscience SE | Phase 2 | ||
Immunotherapy for AD was initiated by the monumental report by Schenk and colleagues, which showed that active immunization with Aβ42 attenuated amyloid pathology in a transgenic mouse model of AD.23) The first clinical trial of active Aβ vaccination in humans used a synthetic full-length Aβ peptide, AN1792, as the immunogen. Disappointingly, the phase 2a trial of this treatment was terminated after 2 or 3 doses because of its high incidence of T-cell mediated meningoencephalitis.24) Long-term follow up analysis revealed no evidence of improvement in survival or time to severe dementia, although AN1792 significantly reduced the Aβ load.25) These studies provided both positive and negative lessons: immunization removed amyloid from the brain, but the removal of amyloid alone was insufficient to slow the progression of cognitive decline. Despite these failures, vaccine development is still ongoing using the Aβ N-terminal portion as the immunogen, based on findings showing that meningoencephalitis may have been caused by the Th1-activating peptide sequence of Aβ15-42.
CAD106, containing multiple copies of Aβ1-6 peptide derived from the N-terminal B cell epitope of Aβ, was the next active immunogen tested after AN1792. In 2008, a one-year phase 1 study in Sweden showed that this immunogen produced an acceptable antibody response and a favorable safety profile without meningoencephalitis.26) Continued safety and favorable tolerability are reported for long-term treatment in patients with mild AD.27)
ACI-24 is a liposomal vaccine consisting of multiple Aβ1-15 sequences attached with palmitoylated lysines on both ends, aiming to mimic the β-sheet conformation of aggregated Aβ. Preclinical studies using a mouse model showed the induction of high titers of anti-Aβ IgG2b antibodies, which involve a noninflammatory Th2 helper cell response.28) A phase 1/2a study in patients with mild to moderate AD was started in 2008 (eudract_number: 2008-006257-40). ACI-24 is also used in a phase 1b trial for the prevention of cognitive decline in patients with Down syndrome (NCT02738450). Several other vaccines, such as ABvac40 and Affitope AD02, are currently under clinical trials, but results have yet to be reported.
Passive immunization by humanized anti-Aβ antibodies.Following the failure of AN1792, the mainstream development of AD immunotherapy shifted to passive immunotherapy, based on observations that the passive transfer of anti-Aβ antibodies eliminated Aβ in the brains of APP-transgenic mice.29) The mechanism of how anti-amyloid antibodies eliminate Aβ deposits in AD brains remains unclear. The “peripheral sink hypothesis” postulates that the sequestration of Aβ by anti-Aβ antibodies in the peripheral blood alters the equilibrium of Aβ transportation and promotes the efflux of Aβ from brains.30),31) However, the “central nervous system clearance hypothesis,” which assumes the partial penetration of anti-Aβ antibodies across the blood brain-barrier to act in the brain parenchyme by inhibiting the toxic oligomerization of Aβ or promoting the microglial phagocytic clearance of amyloid plaques, is currently becoming widely supported.29),32),33)
The first passive immunotherapy tested in a late-phase clinical trial was bapineuzumab, a humanized monoclonal antibody against amyloid-β that recognizes the N terminus of Aβ and binds to its aggregated form.34) Although prespecified efficacy endpoints were not achieved in the phase 2 study, different therapeutic effects were observed when participants were stratified according to gene carrier status for the apolipoprotein E (APOE) ε4 allele, a major genetic risk factor of AD. Possible beneficial effects were observed in ε4 noncarriers, while ε4 carriers more frequently developed amyloid-related imaging abnormalities (ARIA) than did noncarriers. Amyloid PET scans in 28 patients showed reduced brain amyloid deposition in the treatment group compared with both baseline and placebo measurements.35) A phase 3 study began in 2007 for patients with mild to moderate AD, with different doses applied depending on their APOE status (NCT00575055 and NCT00574132). However, clinical development of bapineuzumab was terminated in Aug. 2012 because the first two phase 3 clinical trials did not show clinical benefits.36)
The clinical trial of bapineuzumab brought increased awareness of ARIA. ARIA was first reported in the phase 1 study of bapineuzumab and was subsequently reported in phase 2 and 3 studies. Similar findings were reported with other anti-Aβ antibodies, underscoring ARIA as a common adverse event related to amyloid-modifying therapy. ARIA includes two types of signal abnormalities in MRI imaging. One is ARIA-E, which appears as a hyperintensity in fluid-attenuated inversion recovery imaging sequences, possibly representing vasogenic edema and sulcal fluid effusions. The other is ARIA-H, which appears as a focal low intensity spot in T2*-weighted gradient-recalled echo (GRE) sequences, supposedly attributed to microhemorrhages and hemosiderosis. Increased clearance of amyloid from the parenchymal plaque to perivascular space is implicated in ARIA-E. Infiltration of perivascular amyloid into blood vessels and the direct removal of vascular amyloid may affect the vascular integrity and result in leakage of blood components. ARIA can present as a wide variety of clinical symptoms depending on its location and the extent of the lesion. Most ARIA events are asymptomatic, but they may sometimes cause headache, confusion, nausea, vomiting, and consciousness disturbance. The Alzheimer’s Association Research Roundtable Workgroup published a recommendation on the management of ARIA in 2011, suggesting that the emergence of asymptomatic ARIA does not necessarily require the discontinuation of a study and may be considered an indication of effective amyloid removal.37)
Gantenerumab is a humanized IgG1 antibody with a similar profile to bapineuzumab that recognizes the N-terminal and central portions of Aβ and preferentially binds to aggregated Aβ. A phase 1 study showed favorable safety and tolerability profiles, but the incidence of ARIA was reported.38) A phase 2/3 study in patients with prodromal AD was launched in 2010 (NCT01224106) but halted in Dec. 2014 following the recommendation of preplanned interim futility analysis. Although no significant differences in primary endpoints between treatment arms were observed in this study, gantenerumab showed possible clinical benefits in patients who progressed faster, together with dose-dependent reductions in brain Aβ standard uptake value ratio (SUVR) and both phosphorylated tau and total tau in cerebrospinal fluids.39) Gantenerumab was also adopted in the Dominantly Inherited Alzheimer Network-Trials Unit (DIAN-TU) study (NCT01760005) began in 2012, which aims to verify the preventive effect of gantenerumab against AD in asymptomatic individuals who have a parent with autosomal-dominantly inherited AD.
Crenezumab is a humanized IgG4 antibody that binds to a variety of Aβ species, including fibrillary, oligomeric, and monomeric forms of Aβ. Although two phase 2 studies in patients with mild to moderate AD failed to meet their primary endpoints, a phase 3 study with increased dose began in 2016 in individuals with MCI or prodromal AD (NCT02670083). Crenezumab is also being tested in a primary prevention trial for autosomal-dominant AD (ADAD) that began in 2013, known as the API-ADAD trial (NCT01998841), in a large pedigree of autosomal-dominantly inherited familial AD with the PSEN1 E280A mutation.
Solanezumab is a humanized monoclonal IgG1 antibody that recognizes the middle region of Aβ and binds preferentially to soluble monomeric Aβ over aggregated Aβ. Phase 1 and 2 studies confirmed the safety and tolerability of solanezumab, including its low incidence of ARIA.40) Two phase 3 studies (EXPEDITION and EXPEDITION 2) were conducted from 2009 to 2012, each of which followed patients with mild to moderate AD for 1.5 years, but the cognitive and functional primary outcomes were not achieved.41) However, post hoc analysis suggested possible beneficial effects on cognition and function in a mild AD population. Based on these results, an additional phase 3 trial (EXPEDITION 3) began in 2013, but the cognitive primary endpoint (Alzheimer’s Disease Assessment Scale-cognitive subscale 14 (ADAS-Cog 14) did not meet statistical significance at 18 months (p = 0.095). However, several secondary endpoints evaluating severity of dementia (e.g., Clinical Dementia Rating sum-of-box), cognitive function (e.g., Mini Mental State Examination), and activity of daily living (e.g., Alzheimer’s Disease Cooperative Study (ADCS)-instrumental activities of daily living) showed statistically significant differences, although the effect sizes remained too low (within the 10%–15% range) to support clinical significance. Solanezumab continues to be tested in prevention trials for individuals at risk of AD, i.e., A4 (NCT02008357) and DIAN-TU (NCT01760005).
Aducanumab is a fully human IgG1 monoclonal antibody that selectively binds to aggregated forms of Aβ, especially insoluble fibrils. This antibody was generated by cloning B-cell lines that secrete naturally occurring anti-Aβ antibodies, screened from blood of cognitively normal aged people or those with unusually slow cognitive decline. The rationale of this strategy was that the immune systems of those people who successfully resisted AD, and thus the transplantation of their antibodies, may exert anti-Aβ effects. In March 2015, the results of a 1-year analysis of a phase 1b study were released, which showed that aducanumab successfully reduced amyloid deposition in a dose-dependent manner and slowed decline in Mini Mental State Examination and Clinical Dementia Rating sum-of-box scores. However, ARIA frequently occurred at higher doses and in APOE ε4 carriers. Approximately one-third of these ARIA cases were symptomatic, presenting with mild to moderate headache, visual disturbances, or confusion.42) In August 2015, two identical large-scale phase 3 studies began in individuals with MCI due to AD or mild AD as ascertained by a positive amyloid PET scan (NCT02477800 and NCT02484547).
γ-secretase inhibitors and modulators.Inhibitors of APP-cleaving enzymes have been anticipated as potential drugs for AD. Inhibition of γ-secretase aims to decrease production of Aβ, including the toxic Aβ42. Semagacestat was the first γ-secretase inhibitor (GSI) tested in a global clinical trial, but its phase 3 study was prematurely terminated in 2010 because it had no significant effect on cognitive function and high incidence of skin cancer.43) The adverse effect of this GSI was supposedly caused by the concomitant inhibition of the Notch signaling pathway, which is an essential cell signaling system in cell differentiation. The phase 2 study of avagacestat, a relatively Notch-sparing GSI, was discontinued in 2011 because of a high frequency of adverse effects, including skin cancer, and a lack of effect on cognitive function.44) γ-secretase modulators (GSM), which allosterically activate γ-secretase to reduce Aβ42 production, remain under development. GSMs are thought to promote the sequential cleavage of APP by γ-secretase and increase the ratio of nontoxic shorter species of Aβ (e.g., Aβ38). The development of the first-generation GSM tarenflurbil was discontinued due to its ineffectiveness on cognitive decline and the loss of activities of daily living shown in a phase 3 study in patients with mild AD,45) partly because of the poor brain penetrance due to its hydrophilic structure as a nonsteroidal anti-inflammatory drug. Novel GSMs, such as CHF5074 and PF-06648671, are currently under clinical trials.
β-secretase inhibitors.Inhibition of β-secretase is expected to reduce the total production of Aβ. β-site APP-cleaving enzyme 1 (BACE1), which accounts for the majority of β-secretase activity in the central nervous system, was simultaneously cloned by five independent groups in 1999.46)–50) BACE1 predominantly cleaves APP between amino acid residues Met671 and Asp672. Intriguingly, APP mutations close to the β-cleavage site strongly influence the risk of AD by affecting the efficiency of β-cleavage: pathogenic genetic mutations of familial AD, i.e., LysMet670/671AspLeu and Ala673Val, increase the efficiency of β-cleavage and result in overproduction of Aβ.51),52) In contrast, whole-genome sequence analysis of 1,795 Icelanders revealed that Ala673Thr mutation in APP lowers the risk of AD53) and decreases production of Aβ. These lines of genetic evidence provide strong support for the potent therapeutic significance of BACE1 inhibitors.
The development of BACE1 inhibitors took much longer than did that of γ-secretase inhibitors. The BACE1 inhibitors developed initially were mainly peptidomimetic inhibitors mimicking the sequences of BACE1 substrates, which suffered from problems in pharmacological properties, such as low bioavailability and low penetrance across the blood-brain barrier. Although the design and screening of nonpeptidic, small-molecule BACE1 inhibitors were delayed because of the relatively wide and shallow structure of the catalytic pocket of BACE1, several BACE1 inhibitors are currently under clinical development.
At the early stage of clinical development, BACE1 inhibitors were assumed to be safer than other treatments because BACE1 knockout mice were viable and fertile without any obvious abnormalities.54) However, BACE1 has gradually been shown to have a number of potential substrates, of which the inhibition of BACE1 cleavage exhibit abnormal phenotypes.55)
MK-8931 (verubacestat) is a small-molecule BACE1 inhibitor that showed optimal tolerability and safety profiles in preclinical studies using rodents and monkeys. In the following phase 1/2 studies in humans, verubacestat exhibited a dose-dependent reduction in Aβ42 and Aβ40 in cerebrospinal fluids without serious adverse effects.56) According to a press release from the developer (Merck) on February 14, 2017, a phase 2/3 trial in mild to moderate AD initiated in 2012 was terminated by the decision of an external data analysis committee, which identified virtually no chance of finding a positive effect according to the results of an interim analysis. Another phase 3 clinical trial in prodromal AD is ongoing, and the results are expected to be disclosed in 2019 (NCT01953601).
AZD3293/LY3314814 is a BACE1 inhibitor that showed uniquely slow off-rate kinetics from BACE1 in preclinical studies.57) A phase 1 clinical trial showed a favorable tolerance over the dose range and prolonged Aβ suppression in plasma and cerebrospinal fluid with once-weekly dosing.58) Phase 3 studies targeting MCI (NCT02245737) and mild AD (NCT02245737, NCT02783573) are currently underway.
Several other BACE1 inhibitors are being developed in prodromal AD or cognitively normal people at high risk for AD. CNP520, a highly selective BACE1 inhibitor, is being tested in a prevention trial called the Alzheimer’s Prevention Initiative (API) Generation Study, (NCT02565511 and NCT03131453), which evaluates efficacy on cognitively normal people with genetic risk for developing AD.
In Dec. 2016, two phase 3 studies using E2609 (elenbecestat) began in patients with MCI due to AD and mild AD with biomarker-confirmed amyloid pathology, respectively (NCT02956486, NCT03036280). The phase 2/3 EARLY trial (NCT02569398) tests the efficacy of JNJ-54861911 on individuals who are asymptomatic and at risk for developing AD, as confirmed by biomarkers.
Anti-tau strategies.Tau is the main protein component of NFT, the other important pathological hallmark of AD, but it has received less focus as a therapeutic target. Several possible reasons exist for this lack of attention. First, NFT has long been regarded as the end product of the disease process, rather than the cause of AD, because NFT forms in cerebral cortices after amyloid deposition. Second, tau deposition is not specific to AD, and a number of other neurological disorders, such as frontotemporal dementia, corticobasal degeneration, and progressive supranuclear palsy, also present with tau deposition, collectively referred to as tauopathy. Third, no mutation in the tau gene (MAPT: microtubule associated protein tau) has been identified in familial AD. However, the identification of missense mutations in frontotemporal dementia with parkinsonism-17 strongly suggested that tau deposition is a cause of neuronal death, which may also be the case in AD brains. Other findings also support anti-tau therapies: the degree of cognitive decline in AD correlates with the severity of tau deposition, rather than that of amyloid. Additionally, tau deposition may start earlier than amyloid deposition in degeneration-prone areas, including the hippocampus.59),60) Recent significant progress of in vivo imaging of tau deposition (tau PET) has also encouraged the development of tau-targeted therapeutics.
Methylthioninium chloride (methylene blue) is a deep blue dye that has been clinically used for methemoglobinemia. The speculated mechanism of this drug is the inhibition of tau aggregation by blocking the tau-tau binding interaction through the repeat domain.61),62) A phase 2 trial of methylthioninium chloride (Rember®) in patients with mild or moderate AD reported significant beneficial effects in cognitive scales.63) The clinical development of methylthioninium chloride was followed by that of LMTM (TRx0237), the stabilized and reduced form of methylthioninium chloride, which has improved bioavailability and tolerability. Multiple phase 3 studies were conducted in patients with AD or frontotemporal dementia (NCT01689233, NCT01689246, NCT01626378), although some questions have been raised about their conduct, including a lack of study blindness due to urinary coloring. Recently, the results of NCT01689246 were reported and failed to demonstrate any clinical benefits of LMTM as an add-on treatment for patients with mild to moderate AD.64)
The function of normal tau is to stabilize microtubules by interacting with tubulin. The abnormal tau proteins accumulated in AD brains are excessively phosphorylated, supposedly causing the destabilization of microtubules and formation of NFTs. Based on this hypothesis, the inhibition of abnormal phosphorylation by kinase inhibitors has been proposed. Among a number of kinases that can phosphorylate tau in vitro, glycogen synthase kinase 3 (GSK3) has been most intensively studied as a therapeutic target. NP031112 (tideglusib), a GSK3b inhibitor, has been developed as a therapeutic drug for AD and progressive supranuclear palsy. A phase 2 study conducted in patients with mild to moderate AD showed an acceptable safety profile but no clinical benefit.65) Lithium, a widely used drug for bipolar disorder, has also been shown to act as a GSK3b inhibitor. A small-scale phase 2 trial of this drug was conducted in patients with mild AD but did not meet the primary and secondary outcome measures.66) A phase 3 study targeting individuals with MCI has been registered but has not yet begun (NCT02601859).
Vaccination against tau is another strategy for removing tau from the brain. AADvac1 consists of a synthetic peptide derived from amino acids 294 to 305 of human tau, which is the structural determinant of the tau protein essential for pathological tau-tau interaction in NFTs. In a preclinical study, AADvac1 improved sensorimotor function and reduced NFT pathology in transgenic rats expressing human truncated tau and displayed favorable safety and tolerability profiles in rodents and larger animals.67) A phase 1 study has been completed in patients with mild to moderate AD, showing that AADvac1 has a favorable safety profile and excellent immunogenicity.68) No meningoencephalitis or vasogenic edema were observed. The enrollment for a phase 2 study in mild to moderate AD began in March 2016 (NCT02579252).
ACI-35 is a liposome-based anti-phosphorylated-tau vaccine that contains a synthetic tau peptide phosphorylated at Ser396 and Ser404. In a preclinical study using wild-type and Pro301Leu mutant tau transgenic mice, ACI-35 elicited a robust antibody response specifically against phosphorylated tau and not against nonphosphorylated tau. ACI-35 was also reported to improve motor phenotype and extend the lifespan of mutant mice, and no signs of an inflammatory response were observed.69) A phase 1b study in patients with mild to moderate AD began in 2013 (ISRCTN13033912).
Numerous clinical trials of disease-modifying therapies for AD have been conducted over the past 15 years, most of which have failed, leading to a difficult situation in the clinical development of AD therapies. Besides the difficulty in engaging the drug targets of AD, several problems specific to AD clinical trials hamper their successful outcomes, including a lack of highly reliable biomarkers and the difficulty of objectively and quantitatively evaluating cognitive decline. A crucial problem discussed in recent years is the timing of treatment. As previously mentioned, the deposition of Aβ has recently been shown to start long before the onset of cognitive decline. This observation has led to the idea that starting anti-Aβ treatment after the manifestation of clinical symptoms may be too late. Thus, clinical trials targeting the preclinical AD stage have been highlighted, shifting the concept of AD therapy from “treatment of symptoms” to “prevention of pathophysiology.” The following early intervention studies on cognitively normal elderly populations with a high risk of AD have begun worldwide.
Dominantly Inherited Alzheimer’s Network-trials unit (DIAN-TU) (NCT01760005).This interventional study is being conducted in conjunction with the DIAN observational study (NCT00869817) on cognitively normal or mildly impaired individuals who have a parent with a mutated gene known to cause dominantly inherited AD, i.e., mutations in PSEN1, PSEN2, and APP. DIAN-TU has enrolled over 200 mutation carriers worldwide, and treatment intervention is being conducted using solanezumab or gantenerumab.
Anti-Amyloid Treatment in Asymptomatic Alzheimer’s study (A4) (NCT02008357).Normal elderly individuals are screened using amyloid PET, and those with positive amyloid accumulation are treated with solanezumab. The study plans to enroll 1150 people worldwide. Primary outcome measures include changes in the ADCS-Preclinical Alzheimer Cognitive Composite (ADCS-PACC), a composite of existing cognitive tests designed to capture subtle cognitive decline in preclinical AD participants.70),71) Notably, the University of Tokyo Hospital joined A4 as the only site in non-English speaking nations in Sep. 2016; 149 individuals have been screened and 10 positive individuals have been randomized as of Aug. 2017.
Alzheimer’s Prevention Initiative (API).This prevention study includes two projects. The API-ADAD targets asymptomatic individuals belonging to the PSEN1 Glu280Ala mutation family in Columbia. Participants are treated using crenezumab (NCT01998841). The API-Generation Study targets cognitively normal individuals with genetic risk factors. Generation S1 (NCT02565511) is testing the efficacy of an Aβ vaccine (CAD 106) or BACE1 inhibitor (CNP 520) on carriers of the homozygous APOE ε4 allele. Another phase 2/3 trial of CNP520 targeting a broader population will also be launched, recruiting not only APOE ε4 homozygotes but also heterozygotes with evidence of elevated brain amyloid proved by biomarkers (Generation S2, NCT03131453).
EARLY trial (NCT02569398).This phase 2/3 trial of the BACE1 inhibitor JNJ-54861911 conducted by Janssen is targeting amyloid-positive participants who are asymptomatic but at risk for developing AD dementia. As with the A4 study, the primary outcome includes changes from baseline in the ADCS-PACC to 54 months.
Although these studies differ in their target groups and interventional drugs, their underlying idea is common: to explore whether early intervention targeting Aβ in groups at high risk for AD can be established as a preventive therapy. Targeting subjects who have not yet developed obvious cognitive impairment is a challenging endeavor for many reasons, including ethical aspects. Because changes in cognitive function and the progression of biomarkers in preclinical AD are estimated to be slight, higher sensitivity and accuracy are needed in the cognitive tests, clinical evaluation scales, and imaging and fluid biomarkers. To establish biomarkers and evaluations methods in preclinical AD, a number of longitudinal observational studies are being conducted worldwide, and the results are being shared in open databases, e.g., the AD Neuroimaging Initiative (ADNI),72) Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL),73) and Japanese-ADNI (J-ADNI).74) Collaborative efforts are also underway to construct a platform for global sharing of data from large cohort studies.75)
Finally, patient registries are also being established to further the efficient recruitment of participants at risk of developing AD for clinical trials. These registries include the Brain Health Registry, Global Alzheimer’s Platform (GAP),76) and European Prevention of Alzheimer’s Dementia consortium (EPAD);77) in Japan, the Orange Registry78) and Integrated Registry Of Orange Plan (IROOP)79) have been developed. The main objective of these registries is to establish an efficient large-scale system for screening candidates from the elderly population and form a “trial-ready cohort” relevant to different types of clinical trials.
AD is a slowly progressive disease with a very long time course, and the onset of dementia may occur at its final stage of pathophysiology. Although it is important to manage various risk factors from the earliest stages to prevent AD, especially those related to lifestyle, the paradigm shift of AD therapy from cure to prevention may be key to the successful development of disease-modifying therapies. By taking advantage of recent progress in the elucidation of AD pathophysiology that has accelerated the discovery of novel therapeutic targets, current innovative AD clinical trials will surely lead to the successful development of disease-modifying therapies against AD in the very near future.

Kazushi Suzuki was born in 1974 in Saitama prefecture and graduated from Gunma University School of Medicine in 1999. After two years of residency training in internal medicine, he joined Department of Neurology in the University of Tokyo in 2001 and has worked both as clinical neurologist and as neuroscience researcher thereafter. In 2008, He received Ph.D. degree from Graduate School of Medicine, the University of Tokyo for his work on transcriptional dysregulation of genes as a pathophysiology of polyglutamine diseases. After obtaining Ph.D. degree, he worked as distinguished assistant professor of Center for Epidemiology and Preventive Medicine in the University of Tokyo Hospital. In 2012, he studied abroad as a postdoctoral researcher at Oregon Health & Science University in the US and engaged in basic research on differentiation of motor neurons. After returning to Japan in 2015, he is appointed as project assistant professor both in Department of Neurology and in Unit for Early and Exploratory Clinical Development in the University of Tokyo Hospital. He is currently being engaged in many clinical trials of neurological disorders, especially Alzheimer’s disease. He is also appointed as site principal investigator of the University of Tokyo in several multicenter clinical studies of Alzheimer’s disease such as global Dominantly Inherited Alzheimer Network. Throughout his research career, his main goal is to realize a cure for neurodegenerative diseases which have no treatment. He currently focuses on preclinical stage of Alzheimer’s disease as promising therapeutic window and actively working on clinical research of therapeutic drugs and biomarkers.