2. Molecules regulating lymphocyte migration across blood vessels
A) HEVs in peripheral lymph nodes.
In the early 1980s, Yamaguchi and Schoefl documented that circulating lymphocytes are able to selectively recognize and adhere to the lumen of HEVs and that this mechanism is influenced by the circulating lymphocyte level.5) They found through meticulous electron microscopic analyses that, whereas about 40% of the lymphocytes interacting with the HEV ECs are in the process of transmigrating in normal mice, about 90% of the interacting cells are in the process of transmigrating in lymphopenic mice in which circulating lymphocytes were depleted by thoracic duct cannulation,5) indicating that lymphocyte transmigration is upregulated when the circulating lymphocyte level is low. They also found in the lymphocyte-depleted mice that intravenously injected lymphocytes swiftly adhere to HEVs and penetrate the HEV wall (Fig. 1) and that the speed and intensity of lymphocyte binding/transmigration are both greatly enhanced compared with normal mice.5) These observations indicated that lymphocyte trafficking across HEVs is homeostatically regulated by the number of lymphocytes in the blood. It could be that a humoral factor(s) produced under lymphopenic conditions acts on lymphocytes and/or HEV ECs to enhance lymphocyte transmigration.

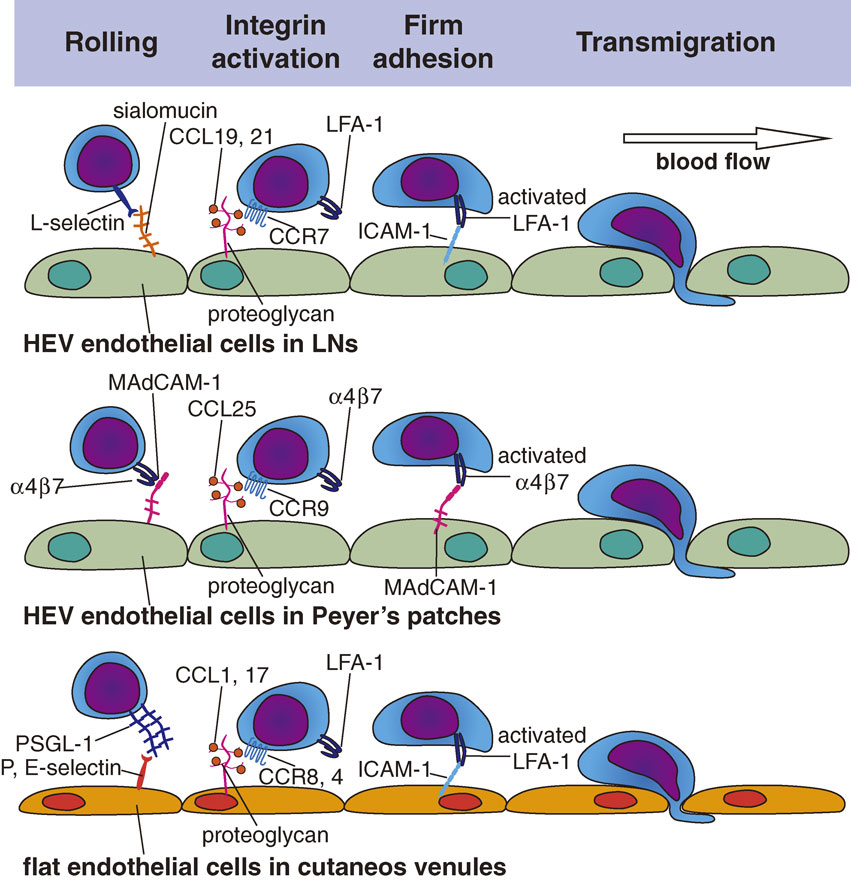
It was subsequently found that the lymphocytes’ interaction with HEV ECs is directed by a site-specific adhesion cascade involving several specific molecules and chemokines that act in sequence (Fig. 2).1),2),6) This adhesion cascade is initiated by leukocyte tethering/rolling, followed by the firm arrest of rolling lymphocytes, and finally by the transvenular migration (extravasation) of lymphocytes.2),3),7) This entire process is very similar to that observed in the lumen of inflamed blood vessels,8) although some distinct and specific molecules are used in HEVs, as detailed below.
i) Tethering/Rolling.
As naïve lymphocytes flow into HEVs, they decelerate rapidly and exhibit weak, transient, on-and-off adhesive interactions (tethering) with the HEV ECs and roll along the inner surface of the HEV wall (rolling). This process is mediated by CD62L (L-selectin) on lymphocytes and by sialomucins expressed on HEV ECs (Fig. 2). L-selectin is a lectin-type cell adhesion molecule that recognizes sugars and is expressed on all leukocytes. It binds to specific O-glycans expressed on HEV sialomucins, a group of heavily glycosylated proteins (mucins) whose carbohydrate moieties contain sialic acid. The critical recognition determinant on the O-glycans is 6-sulfo sialyl Lewis X (sLex), which serves as a capping structure on core-2 and extended core-1 branches, and is specifically recognized by the MECA-79 monoclonal antibody. HEVs in the peripheral lymph nodes express at least five different sialomucins, including GlyCAM-1,9) CD34,10) podocalyxin,11) endomucin,12) and nepmucin/CD300g.13) To synthesize the L-selectin-binding MEC-79-reactive sLex structures, HEV ECs express a set of glycosyltransferases, including α1,3-fucosyltransferases IV and VII, Core1-β3GlcNAcT (also known as β3GlcNAcT-3 or Core1-GlcNAcT), Core2-β1,6-GlcNAcT (Core2-GlcNAcT), GlcNAc6ST-1, and GlcNAc6ST-2.14)
The HEV sialomucins are also called peripheral node addressins (PNAds), because they function as “address code” molecules for peripheral node HEVs. These sialomucins share a common sugar epitope, i.e., the 6-sulfo sLex structure mentioned above. Binding of the MECA-79 antibody to this epitope abrogates L-selectin-PNAd interactions. In mesenteric lymph node HEVs, MAdCAM-1, a sialomucin bearing two immunoglobulin-like domains,15) serves as a vascular addressin, and α4β7 integrin serves as the cognate lymphocyte receptor that mediates rolling and adhesion,16) as described below. Within the HEV lumen, all L-selectin-expressing leukocytes undergo rolling/tethering, but as described below, only lymphocytes show firm adhesion to HEV ECs2) (Fig. 2).
ii) Firm arrest/adhesion.
After tethering/rolling, lymphocytes further decelerate and undergo a shear-resistant firm arrest/adhesion to the HEV wall, which is primarily mediated by an interaction between the β2 integrin LFA-1 (CD11a/CD18) on lymphocytes and ICAM-1/ICAM-2 on HEV ECs (Fig. 2). While L-selectin is constitutively active, integrins generally need to be activated to mediate adhesion. This integrin activation is induced by G-protein coupled receptor (GPCR) signaling in the lymphocytes at HEVs, which is initiated by the lymphocytes’ interaction with chemokines displayed on ECs via specific receptors. Chemokines are generally positively charged, and hence bind to negatively charged molecules such as certain glycosaminoglycan chains that are abundantly expressed on HEV ECs; this binding prevents the chemokines from getting washed off by the blood flow.2),17) In the peripheral lymph nodes, LFA-1 on T cells is activated when HEV-associated chemokines including CCL21/CCL19 and CXCL12 interact with their specific receptors on T cells, CCR7 and CXCR4, respectively, whereas LFA-1 on B cells is activated by interactions with CXCL13 via CXCR5. Because these chemokine receptors are expressed preferentially on lymphocytes, only lymphocytes exhibit firm adhesion via activated LFA-1, which then binds to the immunoglobulin-like-domain-containing adhesion molecules, ICAM-1 and ICAM-2 on the surface of HEV ECs.
iii) Intraluminal crawling and transmigration.
Upon undergoing firm adhesion, naïve lymphocytes start to crawl along the luminal surface of HEVs (intraluminal crawling) and slowly migrate to distant emigration sites, where they then transmigrate at certain hot spots (“exit ramps”)4) along the HEV wall (Fig. 2). The HEV basal lamina has numerous pores, through which lymphocytes pass to reach the abluminal side of HEVs, without disrupting the basal lamina. The basal lamina consists of type IV collagen, fibronectin, and laminin, which allow chemokine immobilization mainly via electrostatic interactions. Hence, the HEV basal lamina binds locally produced lymphoid chemokines, including CCL21, CCL19, CXCL12, and CXCL13, creating a chemokine-rich environment. The HEV basal lamina also functions as a guidance structure for the directional trafficking of lymphocytes from HEVs into the lymphoid tissue parenchyma.2)
Although lymphocytes were reported to cross the endothelial barrier using both paracellular (between adjacent ECs) and transcellular (through the cytoplasm of ECs) migration routes, Schoefl clearly showed, using electron microscopic and mathematical analyses, that lymphocytes predominantly use the paracellular route to exit HEVs.18)
In addition to the mechanisms described above, a variety of other adhesion molecules expressed on HEV ECs have been implicated in lymphocyte transmigration, although their modes of action remain ill defined. These molecules include CD31, VCAM-1, JAM-A, JAM-B, JAM-C, ESAM, VE-cadherin, and nepmucin (CD300g).19)
A specific lysophospholipid, lysophosphatidic acid (LPA) plays a particularly important role in the lymphocyte transmigration across HEVs. LPA is a naturally occurring bioactive lysophospholipid, consisting of a phosphate, a glycerol, and a fatty acid (Fig. 3a). LPA is mainly derived from its precursor, lysophosphatidylcholine, which is abundant in plasma, by the enzymatic action of the lysophospholipase D, autotaxin (ATX). When LPA binds to one of its specific receptors, LPA1–LPA6, which are all GPCRs, multiple signaling pathways are activated with various downstream physiological and pathological effects. Approximately ten years ago, we20) and Steve Rosen’s group21) found that the primary LPA-producing enzyme ATX is transcribed abundantly in HEV ECs. Subsequent analyses showed that the ATX expressed in HEVs regulates lymphocyte trafficking by locally generating LPA. Local inhibition of the ATX/LPA axis substantially blocks the lymphocyte transmigration across HEVs, while a local administration of LPA abrogates this effect.22) At the HEV lumen, LPA acts directly on HEV ECs via its receptors LPA4 and LPA6, although its actions through these receptors are different, given that LPA4 deficiency causes delayed lymphocyte transmigration across the HEV wall, whereas LPA6 deficiency also compromises lymphocyte transmigration but to a much lesser extent.23) In an in vitro transmigration assay, ATX inhibition impairs the release of lymphocytes that migrate underneath HEV ECs, and this defect is abrogated by adding LPA; LPA appears to contribute to lymphocyte de-adhesion (or release) from ECs by regulating the myosin II activity in HEV ECs (Fig. 3b).22)

Under physiological conditions, not only naïve lymphocytes but also DC precursors, plasmacytoid DCs (pDCs), and central memory T cells extravasate from HEVs. While the DC precursor migration into lymph nodes is thought to follow basically the same steps as that of naïve lymphocytes,24) its precise mechanism remains to be fully explored. The pDCs show robust transmigration underneath HEV ECs but not non-HEV ECs, using adhesion molecules very similar to those used by naïve lymphocytes.25) The pDCs also require CCR7 to enter the lymph nodes via HEVs.26),27) This is also the case for central memory T cells, which readily proliferate and differentiate into effector cells in response to their antigenic stimulation in lymph nodes. These cells characteristically express high levels of L-selectin and CCR7, which they use to interact with HEV ECs, just like naïve T cells do. However, to what extent these cells require the HEV-associated lysophospholipid LPA for their transmigration remains to be explored.
When sterile inflammation occurs locally, blood-borne neutrophils rapidly and abundantly migrate into the draining lymph nodes via HEVs. Under these conditions, IL-17-producing lymphocytes first migrate into the draining lymph nodes, where they produce IL-17, which induces the production of CXCL2, a chemokine ligand for CXCR2, in HEVs, leading to the HEV-mediated migration of CXCR2-expressing neutrophils from the blood into the draining lymph nodes. The effect of IL-17 on CXCL2 depends on IL-1β, which is also enhanced by IL-17.28) Thus, although neutrophils are prevented from entering lymph nodes via HEVs under physiological conditions, they can migrate into lymph nodes abundantly when HEVs undergo an inflammation-induced molecular switch, which initiates the neutrophils’ CXCR2 engagement by CXCL2 displayed on ECs.
B) HEVs in intestinal lymphoid tissues.
The lymphocyte trafficking to the small intestine is governed by two types of adhesion pathways. One is mediated by the interaction between lymphocyte L-selectin and HEV-expressed sialomucins/PNAds, which is mainly used by naïve lymphocytes, and the other is mediated by the interaction between lymphocyte integrin α4β7 and the vascular cell adhesion molecule MAdCAM-1, which is mainly used by lymphocytes that have been exposed to antigen-experienced DCs in the small intestine. When naïve lymphocytes that have migrated into the small intestine encounter DCs, they are exposed to high concentrations of DC-derived retinoic acid and start to upregulate their expressions of the integrin α4β7 and the chemokine receptor CCR9.29) The α4β7 specifically binds MAdCAM-1, and CCR9 is the receptor for the chemokine CCL25 secreted by small intestinal venules. Thus, these lymphocytes use α4β7 and CCR9 to recognize tissue-specific cues expressed on small intestinal ECs, i.e., MAdCAM-1 and CCL25. Recently, the orphan chemokine receptor GPR15 has been shown to control the localization of T effector cells in the colon.30)
C) Flat ECs in peripheral tissues.
Flat ECs found in non-specialized regular postcapillary venules in the skin also mediate constitutive immune cell trafficking under steady state conditions, albeit to a much lesser extent than do HEV ECs. These ECs support leukocyte rolling under non-inflamed conditions,31) due to a constitutive, low-level expression of E- and P-selectins. The rolling frequency is largely determined by P-selectin, with E-selectin playing a smaller role, and L-selectin is not involved.32) Most skin T cells express an E- and P-selectin–binding molecule, the cutaneous lymphocyte antigen (CLA), which is derived from the glycosylation of a lymphocyte sialomucin PSGL-1, and a chemokine receptor CCR8, whose expression is induced by keratinocyte-derived factors.33) Upon binding to EC-displayed selectins, the CLA-expressing T cells extravasate from dermal venules. The engagement of lymphocyte CCR8 with its ligand CCL1 constitutively expressed in the dermal venules is thought to promote this extravasation by activating lymphocyte integrins. Skin T cells also express another chemokine receptor CCR4, whose engagement with a dermal venule-expressed chemokine, CCL17, promotes T cell migration into the skin. In inflamed skin, activated T cells express high levels of CCR10, whose engagement with CCL27, which is produced by keratinocytes and is highly displayed on inflamed venules in the skin, is critical for T cell recruitment to the skin.34) Interestingly, skin DCs are able to produce the active vitamin D3 metabolite 1,25(OH)2D3 from sunlight-induced vitamin D3, and 1,25(OH)2D3 upregulates the CCR10 expression in T cells.35) However, CCR10 is largely absent from the T cells in uninflamed skin;33),36) hence, the CCL17/CCR10 axis appears to be important for T cell migration into inflamed skin but not for the constitutive T cell migration into normal skin.
Recent studies indicate that T cells are abundant in the skin and have unique immunological abilities.37) A careful study in humans indicated that 2 × 1010 T cells exist in the skin, which is almost twice the number of T cells in the entire circulation.38) Most of the skin T cells have the phenotype of effector memory T cells, and some are central memory T cells. The prevailing hypothesis is that effector memory T cells that arise upon the antigenic stimulation of naïve as well as central memory T cells in lymph nodes migrate into peripheral tissues via venules bearing flat ECs and return to the lymph nodes via lymphatics, whereas the central memory T cells that also arise in lymph nodes recirculate between the blood vascular and lymphatic vascular systems using HEVs, just like naïve T cells.39),40) However, the presence of CLA-expressing central memory (L-selectin+CCR7+) T cells in the skin (∼20% of normal skin T cells) in humans41) indicates that not all of the central memory T cells recirculate via the conventional route; some of them migrate into the periphery and then move to the lymph nodes via lymphatics. On the other hand, a substantial proportion of memory T cells in the skin appear to be sessile and do not leave the tissue; hence, they are now called resident memory T cells (TRMs).42) These cells provide effective protection against local antigen re-challenge. The failure of these cells to exit the tissue is currently thought to be due to their low expression of the transcription factor KLF2 and of S1PR1 and to their high expression of the C-type lectin CD69; as a result of these conditions, the cells fail to respond to an “exit cue” provided by S1P (sphingosine-1-phosphate), which is released mainly from lymphatic ECs. S1P’s involvement in lymphocyte egress will be discussed below.
Regulatory T cells (Tregs) are also found in the skin, where they comprise 10∼20% of the normal skin T cells in man38) and mouse.43) They constitutively migrate to the draining lymph nodes via lymphatics in the steady state.44) These cells show increased migration during cutaneous immune responses and return to the skin upon re-exposure to antigen. The migrating Tregs have a stronger immunosuppressive effect than lymph node-residing Tregs and appear to contribute to the downregulation of cutaneous immune responses.44) These Tregs express CD103, CCR4, and CCR5, but the molecular mechanism underlying their migration from the skin to lymph nodes remains unclear.
A recent study using transgenic mice expressing a photoconvertible fluorescent protein, Kaede, confirmed that DCs also continuously migrate from the skin to draining lymph nodes under steady-state conditions.44) DCs do not apparently require β2 integrins for their migration, since CD18−/− mice deficient in the β2 integrin subunit show uncompromised DC migration from the blood to normal or inflamed skin, or from the skin to draining lymph nodes.45) This finding was later verified by multiphoton microscopy, which showed that DCs crawl into lymphatics independent of integrins46) and that they are guided into lymphatics by a tissue-immobilized gradient of CCL21 in an integrin-independent but CCR7-dependent manner.47)
Innate lymphoid cells (ILCs) are a group of non-T, non-B lymphocytes that are important in innate immune responses and in the regulation of inflammation.48) In the skin, group 2 ILCs are relatively abundant and continuously patrol the tissue at speeds comparable to those described for dermal DCs, frequently interacting with perivascular mast cells.49) However, how these cells are recruited to the skin and whether they migrate from the skin into the draining lymph nodes remain unknown.
3. Molecules regulating DC migration into lymphatics
a) Chemokines and their receptors.
The trafficking of lymphocytes and DCs into the lymphatic vessels is an active process.50) Chemokines presented on lymphatic ECs are important for attracting and guiding DCs into lymphatics, and DCs interacting with the lymphatic ECs respond to these chemokines. For instance, a CCR7-ligand chemokine, CCL21, is abundantly expressed on lymphatic capillaries and induces the chemotaxis of CCR7-expressing DCs, allowing them to migrate toward and to enter into lymphatics. CCL21 binds a lymphatic EC marker podoplanin with high affinity, is expressed on the basal lamina of lymphatics, and is shed into the perivascular stroma,51) which may contribute to the formation of a peri-lymphatic CCL21 concentration gradient. In addition, a chemokine-scavenging molecule, CCRL1, is expressed by lymphatic ECs that line the ceiling but not the floor of the subcapsular sinus.52) CCRL1 sequesters and induces CCL21 degradation, which is thought to contribute to the formation of a CCL21 concentration gradient from the sinus toward the lymph node parenchyma, which helps direct DCs to migrate toward the inner areas of lymph nodes. Indeed, DCs have been shown to require the lymphatic EC-displayed CCL21 to enter the lumen of lymphatics, in a CCR7-dependent manner.53),54)
In addition, CCR7’s function can be modulated locally; CCR7 is upregulated by locally generated molecules including prostaglandin E255) and extracellular NAD+,56) both of which are released from damaged or inflamed cells. Notably, however, while DC migration across the subcapsular sinus lymphatic EC layer is CCR7-dependent, T cell migration across the sinus lymphatic ECs is completely independent of CCR7 signaling.57) Thus, the CCL21-CCR7 axis does not appear to be the only regulator of immune cell trafficking across the lymph node subcapsular sinus.
Under inflamed conditions, the chemokine CX3CL1 (fractalkine) appears on lymphatic ECs and is actively secreted, and the soluble rather than membrane-anchored chemokine promotes DC migration toward lymphatics and EC transmigration.58) CXCL12 is also induced on the surface of lymphatic ECs upon an inflammatory stimulus, and DCs migrate across these cells in a CXCL12/CXCR4-dependent manner.59) In the case of lymphocytes, CXCL12 acts in synergy with CCR7 ligands to promote cell migration by sensitizing the cells through CXCR4, thus enabling them to respond to lower concentrations of CCR7 ligands.60) Given that mature DCs also express both CCR7 and CXCR4 at levels comparable to those on lymphocytes, chemokine-induced synergy may also enhance DC recruitment into lymphatics under certain conditions.
Interestingly, CCL1 is expressed by the subcapsular sinus lymphatic ECs of skin-draining lymph nodes but not by capillary lymphatic vessels in the skin, and inhibiting the CCL1/CCR8 interaction leads to impaired DC migration into the lymph node parenchyma, indicating that the CCL1/CCR8 axis functions downstream of the DC entry into lymphatics by regulating entry into the subcapsular sinus of the lymph nodes.61) Notably, malignant melanoma cells often express high levels of CCR8 and use CCL1-CCR8 to enter into the lymph node parenchyma, forming lymph node metastases.62) Thus, certain types of tumor cells can coopt the normal mechanism of CCL1-CCR8-dependent immune cell trafficking to metastasize to the lymph node.
b) Adhesion molecules and their receptors.
As mentioned above, integrins are not required for the DC migration into afferent lymphatics in the steady state.46) Podoplanin expressed on lymphatic ECs can capture CCL21 (see above) and also binds a lectin-type molecule CLEC-2. Podoplanin’s binding to CLEC-2 promotes the formation of actin-rich protrusions on DCs, which allow the DCs to spread along stromal scaffolds and support DC motility.63) While integrin-binding molecules including ICAM-1 and VCAM-1 are expressed at only low levels in lymphatic ECs, their expression is strongly upregulated during inflammation, and they contribute to DC migration by interacting with β2 integrins (LFA-1 and Mac-1)64) and β1 integrin VLA-4,65) respectively.
Lymphatic ECs produce an immunomodulatory molecule, semaphorin 3A. This molecule promotes actomyosin contraction in the trailing edge of DCs by binding plexin A1 on DCs and may induce the disassembly of adhesive components at the trailing edge as well, thus promoting DC transmigration.66)
5. Lymphocyte egress from lymphoid tissues
Lymphocyte egress from lymphoid tissues is currently thought to be regulated primarily by sphingosine-1-phosphate (S1P), which is structurally similar to LPA. S1P acts on a family of five G-protein-coupled receptors (S1P1–S1P5) and is rapidly degraded into a biologically inactive form by S1P phosphatases and S1P lyase in vivo.
S1P is released from erythrocytes and vascular ECs, causing a high S1P concentration in the peripheral blood. In contrast, S1P is relatively scarce in lymphoid tissues, due to an abundance of S1P lyase. This differential concentration of S1P between the blood and lymphoid tissue is currently thought to drive lymphocyte emigration from lymphoid tissues. Lymphocytes within the lymphoid tissues express high levels of the S1P receptor S1P1 and thus undergo chemotaxis in response to the S1P gradient. In contrast, peripheral blood lymphocytes express low levels of S1P1, due to its downregulation by internalization in response to the high S1P concentration in the blood. When blood-borne lymphocytes enter lymphoid tissues, S1P1 is upregulated due to the paucity of S1P in the tissue. Within the lymph nodes, the lymphocytes are then transported to the cortical sinuses, the medullary sinus, and finally to the efferent lymphatics, by sensing the S1P concentration gradient in an S1P1-dependent manner. This cyclical change in lymphocyte S1P1 expression, which has been proposed to direct lymphocyte egress from the lymph nodes,69) may similarly regulate lymphocyte egress from the thymus.
However, this widely accepted hypothesis cannot be readily reconciled with the following findings. First, S1P1 transcripts are also abundant in the ECs and vascular smooth muscle cells surrounding blood vessels, and strong S1P1 activation is detected in both the lymphatic and vascular ECs in lymphoid tissues, where most lymphocytes show no evidence of S1P1 activation under homeostatic conditions.70) Second, S1P1 is also expressed at high levels on macrophages, DCs, and natural killer cells, but only lymphocytes exit the lymph nodes in response to physiological concentrations of S1P. These findings indicate that S1P’s role in regulating lymphocyte egress is more complex than described so far.
S1P also appears to regulate the barrier function of the HEVs in antigen-stimulated lymph nodes. Herzog et al.71) reported that platelets migrate across HEVs together with lymphocytes in mesenteric lymph nodes and are activated by specific interactions between the platelet cell surface lectin CLEC-2 and podoplanin, expressed on the surrounding FRCs. The activated platelets then secrete S1P, which stimulates the HEVs to maintain their vascular integrity. However, the effect of the podoplanin-CLEC-2 interaction on HEV integrity can only be detected in mesenteric lymph nodes, where exogenous antigens are abundant, and not in peripheral lymph nodes unless the mice are immunized, implying that this mechanism is important under inflammatory conditions.