Abstract
Favipiravir (T-705; 6-fluoro-3-hydroxy-2-pyrazinecarboxamide) is an anti-viral agent that selectively and potently inhibits the RNA-dependent RNA polymerase (RdRp) of RNA viruses. Favipiravir was discovered through screening chemical library for anti-viral activity against the influenza virus by Toyama Chemical Co., Ltd. Favipiravir undergoes an intracellular phosphoribosylation to be an active form, favipiravir-RTP (favipiravir ribofuranosyl-5′-triphosphate), which is recognized as a substrate by RdRp, and inhibits the RNA polymerase activity. Since the catalytic domain of RdRp is conserved among various types of RNA viruses, this mechanism of action underpins a broader spectrum of anti-viral activities of favipiravir. Favipiravir is effective against a wide range of types and subtypes of influenza viruses, including strains resistant to existing anti-influenza drugs. Of note is that favipiravir shows anti-viral activities against other RNA viruses such as arenaviruses, bunyaviruses and filoviruses, all of which are known to cause fatal hemorrhagic fever. These unique anti-viral profiles will make favipiravir a potentially promising drug for specifically untreatable RNA viral infections.
1. Introduction
Significant progress has been made in anti-infection therapies since the middle of the 20th century with the advent of antibiotics, vaccines and other anti-microbial agents. Mainstay of anti-viral therapy has been preventive vaccines and therapeutic anti-viral agents, which are, however, efficacious for a certain type of viruses. There are still emerging or re-emerging viral infections, to which effective drug or vaccine therapies are not available. Resistance to existing anti-viral drugs has become a serious problem.
Influenza is one of the most prevalent infections caused by influenza virus, and epidemic every year in the world. Highly pathogenic avian influenza virus A(H5N1) in humans was identified in Hong Kong in 1997 and continuously causes outbreaks.1) Avian influenza A(H7N9) in 2013 in China,2) and pandemic influenza A(H1N1) in 2009 which caused 17,700 deaths in 2009, are of public health concern.3) Epidemic of A(H1N1) influenza virus resistant to oseltamivir, a neuraminidase (NA) inhibitor, occurred in the 2008 to 2009 season4) and amantadine, an M2 protein inhibitor, showed a high rate of resistance after administration. Due to limited diversity of the mechanism of action of currently available anti-influenza agents, there is a strong need to discover an anti-influenza drug with a novel mechanism of action.
Recently, viruses mediated by arthropod or wild animals are spreading in the world. Outbreaks of Ebola and Lassa viruses in Western Africa in 2014 raised a social concern over prevention and therapy if they occur.5),6) These viruses are highly pathogenic and cause fatal infections.5),7)
Favipiravir (Fig. 1) was discovered by chemical modification of a pyrazine analog initially screened by in vitro anti-influenza virus activity in cells. Favipiravir is a selective and potent inhibitor of influenza viral RNA polymerase,8) and effective against all subtypes and strains of influenza viruses including ones sensitive or resistant to marketed neuraminidase and M2 inhibitors. Favipiravir demonstrated anti-viral activities against other RNA viruses.9) These data clearly suggest that favipiravir is a promising drug for the treatment of infections by not only influenza virus but also a wide range of RNA viruses. On the other hand, favipiravir has a risk for teratogenicity and embryotoxicity. Therefore, the Ministry of Health, Labor and Welfare granted conditional marketing approval with strict regulations for its production and clinical use.10)

In this review, we will describe the mechanisms of action of favipiravir, a broad spectrum of anti-viral activities in vitro, and therapeutic potential in animal infection models of a wide range of RNA viruses.
2. Mechanisms of action of favipiravir
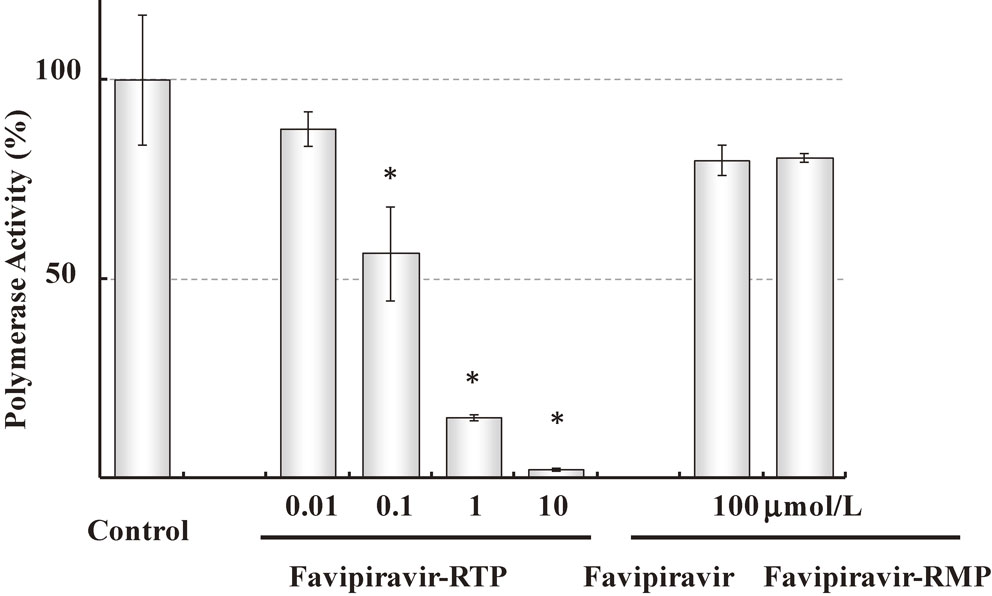
Favipiravir inhibited the replication of viral genome, which was the most manifested in the middle of viral proliferation cycle in a time-of-drug-addition test.8) Anti-viral activity of favipiravir was attenuated in the presence of purine nucleosides or purine bases, indicating competition of favipiravir with purine nucleosides rather than pyrimidine nucleosides.8) Madin Darby Canine Kidney (MDCK) cells are well used as an in vitro assay of influenza virus. MDCK cells were treated with favipiravir, and cellular metabolites were analyzed by HPLC. Favipiravir ribofuranosyl-5′-triphosphate (favipiravir-RTP), favipiravir ribofuranose (favipiravir-R) and favipiravir ribofuranosyl-5′-monophosphate (favipiravir-RMP) were detected.8) These results suggest that the activation of favipiravir takes place once it is incorporated into cells. Favipiravir-RTP was chemically synthesized and was tested for the inhibition of RNA polymerase activity of influenza virus as assessed by incorporation of 32P-GTP. Favipiravir-RTP inhibited the viral RNA polymerase activity in concentrations ranging from nanomolar to micromolar.8) None of favipiravir and favipiravir-RMP affected influenza RNA-dependent RNA polymerase (RdRp) at 100 µmol/L (Fig. 2). These results indicate that favipiravir exerts its anti-viral activity as a pro-drug, since favipiravir is intra-cellularly phosphoribosylated to be an active form, favipiravir-RTP, which inhibits the viral replication by interacting with viral RNA polymerase.8)
The mechanism of the interaction of favipiravir-RTP with RdRp molecule has not been fully elucidated. It is hypothesized that favipiravir may be misincorporated in a nascent viral RNA, or it may act by binding to conserved polymerase domains, thus preventing incorporation of nucleotides for viral RNA replication and transcription. It was recently demonstrated that favipiravir induced lethal mutagenesis during influenza virus infection, and reduced viral titer either at a low (0.0001 PFU/cell) or high (10 PFU/cell) multiplicity of infection in vitro. Sequence analysis of various nucleoprotein (NP) clones revealed an increase in the number of detectable G→A and C→T or C→U transition mutations, along with an increased mutation frequency, and a concomitant shift in the nucleotide profiles of the NP gene analyzed from various clones.11),13) It is important to note that no viable favipiravir resistant mutants were isolated in these studies.9),11)–14) Previous studies suggest that favipiravir has virucidal effects, implicating the mutagenesis by favipiravir may be engaged in various types of RNA viruses.
We undertook a primer extension assay using influenza H1N1 extracts as sources for RdRp and template viral RNAs. Addition of 5′Cap1 RNA to the assay mixture made cap-snatching and transcription take place. In the presence of favipiravir-RTP, a single molecule of favipiravir-RTP was incorporated into the nascent RNA, and the strand extension was blocked (Fig. 3).15) Jin et al.16) used synthetic RNA templates designed to form the panhandle structure, and recombinant PA/PB1/PB2 complex as RdRp. They demonstrated that favipiravir-RTP was efficiently incorporated into a nascent RNA strand, the incorporation of single molecule of favipiravir-RTP partially prevented the extension of RNA strand, and double incorporation of favipiravir-RTP molecules completely blocked the further extension. These two studies indicate that favipiravir-RTP efficiently inhibits the extension of viral RNA strand, once incorporated into a viral RNA strand.

Ribavirin is a guanosine analog, and has anti-viral activity against a wide range of RNA viruses. Ribavirin undergoes phosphorylation in cells to generate ribavirin monophosphate (ribavirin-MP), and further its triphosphate form. Although multiple mechanisms of action of ribavirin are documented,17) the best known is inhibition of inosine monophosphate dehydrogenase (IMPDH) by ribavirin-MP, leading to reduced GTP levels.18) The inhibition of IMPDH was compared between favipiravir-RMP and ribavirin-MP, whose 50% inhibitory concentration (IC50) values were 601 and 3.9 µmol/L, respectively.8) The concentration of GTP in MDCK cells treated with ribavirin-MP was much lower than that with favipiravir-RMP.19) These data suggest unlikely that IMPDH is the target for favipiravir.
Nucleic acid synthesis is essential for life in viruses and humans. Unlike RNA virus, humans do not have RdRp, but have DNA-dependent RNA polymerase (DdRp) and DNA-dependent DNA polymerase. Favipiravir-RTP was tested for the inhibition of those polymerase activities.20) Favipiravir-RTP inhibited influenza RdRp with an IC50 of 0.341 µmol/L, but did not human DNA polymerases α, β, γ at up to 1000 µmol/L. Favipiravir slightly inhibited human RNA polymerase II, which belongs to DdRp, with an IC50 of 905 µmol/L.9) These results are consistent with evidence that favipiravir did not inhibit DNA and RNA synthesis at 637 µmol/L in MDCK cells.8)
Schematic representation of intracellular activation and mechanism of actions of favipiravir is depicted based on findings of favipiravir obtained to date (Fig. 4). Once incorporated into host cells, favipiravir undergoes phosphoribosylation and further phosphorylation to become favipiravir-RTP, which blocks viral RdRp. This mechanism of action of favipiravir is quite unique, since marketed influenza drugs inhibit either entry or release of the virus. Selective inhibition of viral RdRp by favipiravir implicates a wider anti-viral spectrum. Further investigation is needed to clarify the correlation between anti-viral activity of RNA viruses by favipiravir and inhibition of their RdRp activities.
3. Anti-viral activities of favipiravir
It was demonstrated that favipiravir inhibited proliferation of RNA viruses but not of DNA viruses.21) In vitro anti-viral activities of favipiravir cited from published studies are enlisted in Table 1.
Table 1.
In vitro anti-viral activities of favipiravir
| Group |
Family |
Virus |
EC50 (µg/mL) |
Reference |
RNA (−)
strand |
Orthomyxoviridae |
Influenza A (seasonal) |
0.01–0.94 |
Furuta, Y. et al. 200221)
Sleeman, K. et al. 201022) |
| Influenza A (H5N1) |
0.2–1.9 |
Sidwell, R.W. et al. 200725)
Sleeman, K. et al. 201022) |
| Influenza A (H1N1)pdm09 |
0.13–3.53 |
Sleeman, K. et al. 201022) |
| Influenza A (H7N9) |
1.4 |
Watanabe, T. et al. 201361) |
| Influenza B |
0.04–0.8 |
Furuta, Y. et al. 200221)
Sleeman, K. et al. 201022) |
| Influenza C |
0.03–0.06 |
Furuta, Y. et al. 200221) |
| Bunyaviridae |
La Crosse |
5 |
Gowen, B.B. et al. 200729) |
| Punta Toro |
8.6–30 |
Gowen, B.B. et al. 2007, 201029),34) |
| Rift Valley fever |
4.2–5.0 |
Gowen, B.B. et al. 2007, 201029),34) |
| Sandfly fever |
4.7–18 |
Gowen, B.B. et al. 2007, 201029),34) |
| Dobrava |
15 |
Buys, K.K. et al. 201135) |
| Maporal |
10 |
Buys, K.K. et al. 201135) |
| Crimean-Congo hemorrhagic fever |
1.1 |
Oestereich, L. et al. 201436) |
| Prospect Hill |
10 |
Buys, K.K. et al. 201135) |
| Severe fever thrombocytopenia syndrome |
0.71–1.3 |
Tani, H. et al. 201641) |
| Arenaviridae |
Junin |
0.8–3.0 |
Gowen, B.B. et al. 2007, 201029),34)
Mendenhall, M. et al. 201131) |
| Pichinde |
0.9–3.9 |
Gowen, B.B. et al. 2007, 201029),34) |
| Tacaribe |
0.9–4.1 |
Gowen, B.B. et al. 2007, 201029),34) |
| Guanarito |
2.4 |
Mendenhall, M. et al. 201131) |
| Machupo |
2.2 |
Mendenhall, M. et al. 201131) |
| Lassa |
1.7–11.1 (EC90) |
Safronetz, D. et al. 201532)
Oestereich, L. et al. 201633) |
| Filoviridae |
Ebola |
10.5 |
Oestereich, L. et al. 201453)
Smither, S.J. et al. 201454) |
| Rhabdoviridae |
Rabies |
5.1–7.0 |
Yamada, K. et al. 201658) |
| Paramyxoviridae |
Human metapneumovirus |
1.3–6.3 (EC90) |
Jochmans, D. et al. 201660) |
| Respiratory syncytial virus |
41 |
Furuta, Y. et al. 200221) |
RNA (+)
strand |
Flaviviridae |
West Nile |
53 |
Morrey, J.D. et al. 200844) |
| Yellow fever |
42 |
Julander, J.G. et al. 200943) |
| Zika virus |
3.5–3.8 |
Zmurko J. et al. 201645) |
| Togaviridae |
Western equine encephalitis |
1.2, 49 (EC90) |
Delang, L. et al. 201447)
Julander, J.G. et al. 200946) |
| Venezuelan equine encephalitis |
1.7 |
Delang, L. et al. 201447) |
| Eastern equine encephalitis |
2.8 |
Delang, L. et al. 201447) |
| Barmah forest |
2.8 |
Delang, L. et al. 201447) |
| Ross river |
0.5 |
Delang, L. et al. 201447) |
| Mayaro |
2.5 |
Delang, L. et.al. 201447) |
| Chikungunya |
0.3–9.4 |
Delang, L. et al. 201447) |
| Picornaviridae |
Polio |
4.8 |
Furuta, Y. et al. 200221) |
| Rhino |
23 |
Furuta, Y. et al. 200221) |
| Enterovirus 71 |
23 |
Wang, Y. et al. 201650) |
| Caliciviridae |
Noro |
19–39 |
Rocha-pereira, J. et al. 201251) |
Favipiravir demonstrated anti-viral activities against all subtypes of influenza virus strains, including type A, B and C in studies using laboratory strains of influenza virus with 50% effective concentration (EC50) values ranging from 0.014 to 0.55 µg/mL.21) Favipiravir was evaluated in vitro for the ability to block the proliferation of representative influenza viruses, including seasonable strains A(H1N1), A(H1N1)pdm09, A(H3N2), and B; highly pathogenic avian influenza virus A(H5N1) isolated from human. These strains contain ones resistant to oseltamivir or zanamivir, and several ones resistant to both NA inhibitors. It is noted that favipiravir demonstrated anti-viral activities against all strains tested (Fig. 5).22) Favipiravir was not cytotoxic for MDCK cells with 50% cytotoxic concentration (CC50) of >1000 µg/mL, demonstrating a high antiviral index.21)
3.2 Efficacy of favipiravir in vivo.
Favipiravir was assessed in murine models of influenza virus infection (Fig. 6). Mice were infected with a lethal dose of influenza virus either of H3N2 (A/Victoria/3/75), H3N2 (A/Osaka/5/70) or H5N1 (A/Duck/MN/1525/81). Favipiravir was orally administered 1 hr post-infection. Survival rate was significantly improved in mice dosed with favipiravir at more than 30 mg/kg/day, regardless of twice a day (b.i.d.) or four times a day, whereas none of mice survived in the control group.9)

The effect of favipiravir on the viral load was investigated in mice infected with influenza virus H1N1 (A/California/04/09). Oral treatment with favipiravir at 60 and 300 mg/kg/day decreased the viral load in the lung 3 and 6 days post-infection. Favipiravir demonstrated anti-viral efficacy equal to or more potent than oseltamivir and zanamivir in the murine infection model with influenza virus A(H1N1)pdm09.23) Furthermore, favipiravir was significantly more efficacious than oseltamivir in the murine infection models with the virus challenge of 100 times higher titer with influenza viruses A/PR/8 (H1N1), or of delayed dosing (up to 96 hrs post-infection) in A/Duck/MN/1525/81 (H5N1) infectious murine model.24),25)
Combination of anti-viral agents with a different mechanism of action is utilized to enhance therapeutic effects or to reduce the emergence of resistant virus clones. Synergistic effects were demonstrated by the combination of favipiravir with oseltamivir in a murine model of influenza virus.26) Combination of favipiravir and oseltamivir significantly improved the survival rate of mice infected with influenza virus A/Victoria/3/75 (H3N2), while either of single agent alone showed limited effects. Similar combination effects were demonstrated in mice infected with influenza viruses A/NWS/33 (H1N1) or A/Duck/MN/1525/81 (H5N1). These results provide us with wider therapeutic options for the treatment for epidemic influenza virus resistant to existing anti-influenza agents or for severe patients.
4. Efficacy of favipiarvir in other viruses
Favipiravir has the characteristic of acting specifically on RNA viruses.21) Among infections by RNA viruses, arena-, bunya-, flavi- and filoviruses cause hemorrhagic fever and/or encephalitis with high case fatality rate. Neither vaccines nor approved anti-viral agents are available for those viral infections, underscoring urgent needs for effective broad spectrum anti-viral agents. Ribavirin is the only drug effective for arenaviral hemorrhagic fever, and used off-label. Its therapeutic effects are based on the comparison with historical data.27)
The next section is the review on the efficacy of favipiravir for hemorrhagic fever viruses and related ones, including picorna-, noro- and rabies virus.
4.1 Arenaviridae.
Many of arenaviruses are known to cause severe diseases in human.28) No agents are approved for the treatment of arenavirus infections besides ribavirin with toxicity concerns. In vitro activity of favipiravir against pathogenic arenaviruses was compared with ribavirin. EC50 values of favipiravir in cytopathic effect (CPE) assay using Vero cells were 0.79–0.94 µg/mL for Junin virus, Pichinde virus (PICV) and Tacaribe virus. In the virus yield reduction assay with Vero cells on days 3 and 5 post-infection, the 90% effective concentration (EC90) values against Lassa virus (LASV) were 1.7 and 11.1 µg/mL, respectively.32) In addition, favipiravir showed higher selectivity for tested viruses than ribavirin (Table 2).
Table 2.
In vitro inhibitory effects of favipiravir and ribavirin against Arenaviruses (Based on Gowen
et al., 2007
29))
| Virusa |
strain |
Favipiravirb |
Ribavirinb |
| CC50 |
EC50 |
SI |
CC50 |
EC50 |
SI |
| JUNV |
Candid 1 |
188 |
0.79 |
239 |
51 |
2.7 |
19 |
| PICV |
An 4763 |
175 |
0.94 |
186 |
38 |
3.2 |
12 |
| TCRV |
TRVL 11573 |
214 |
0.94 |
227 |
68 |
2.4 |
28 |
aJUNV, Junin virus; PICV, Pichinde virus; TCRV, Tacaribe virus.
b50% cytotoxic concentration (CC50) and 50% effective concentration (EC50) values are in µg/mL for JUNV, PICV and TCRV determined by CPE reduction assays using Vero cells, and SI (selectivity index) is calculated as CC50/EC50.
In the PICV infected hamster model, oral treatment with favipiravir suppressed deaths, reduced viral loads in the blood and tissues, and blocked the damage in the liver at 60 mg/kg/day b.i.d. for 7 days beginning 4 hr post-infection.29) Administration of favipiravir 4–6 days post-infection significantly increased the survival rate (P < 0.001) at 100 mg/kg/day b.i.d. for 7 days in the PICV infected hamster model.30) In a guinea pig model of PICV infection, therapeutic effects of oral favipiravir were demonstrated, even after symptoms emerged.31)
It was recently reported that oral favipiravir was efficacious in the guinea pig and mouse infection models of LASV.32),33) Therapeutic effects were shown by favipiravir, when dosing commenced 2 days post-infection with LASV (Fig. 7A). Subcutaneous treatment of favipiravir at 300 mg/kg/day once a day decreased fever, blocked loss of body weight and increased the survival rate in infected guinea pigs. These effects were more manifested than those achieved by ribavirin at 50 mg/kg/day. Favipiravir improved the survival rate of guinea pigs infected with LASV, even when the treatment was initiated 5, 7 and 9 days post-infection (Fig. 7B).32)
4.2 Bunyaviridae.
La Crosse virus (LACV), Rift Valley fever virus (RVFV), Crimean-Congo hemorrhagic fever virus (CCHFV), Severe fever with thrombocytopenia syndrome virus (SFTSV) and hantavirus are in the family of Bunyaviridae, and cause severe diseases such as hemorrhagic fever, fever with thrombocytopenia, and fever with renal or pulmonary syndromes. In in vitro study, favipiravir demonstrated more potent and selective anti-viral activities against these viruses in Vero cells than ribavirin (Table 3).34),35),41) EC50 values were 0.9–30 µg/mL for LACV, Punta Toro virus (PTV), RVFV, Sandfly fever virus, SFTSV and Dobrava, Maporal and Prospect Hill Hantaviruses in the assays of CPE or focus forming unit (FFU) reduction.
Table 3.
In vitro inhibitory effects of favipiravir and ribavirin against Bunyaviruses (Based on Gowen
et al., 2007,
29) Buys
et al., 2011
35) and Tani
et al., 2016
41))
| Virusa |
strain |
Favipiravirb |
Ribavirinb |
| CC50 |
EC50 |
SI |
CC50 |
EC50 |
SI |
| LACV |
— |
>1000 |
5.0 |
>199 |
877 |
17 |
51 |
| PTV |
Adames |
>1000 |
30 |
>33 |
898 |
42 |
21 |
| RVFV |
MP-12 |
>980 |
5.0 |
>196 |
>906 |
13 |
>70 |
| SFNV |
Naples |
>1000 |
18 |
>55 |
>729 |
22 |
>33 |
| DOBV |
Sotkamo |
757 |
15 |
52 |
296 |
18 |
17 |
| MPRLV |
HV9021050 |
753 |
10 |
74 |
256 |
11 |
22 |
| PHV |
MP40 |
600 |
10 |
58 |
248 |
5.6 |
44 |
| SFTSV |
SPL010 |
>157 |
0.9 |
>167 |
>157 |
7.7 |
>20 |
aLACV; La Crosse virus, PTV; Punta Toro virus, RVFV; The Rift Valley fever virus, SFNV; Sandfly fever virus, DOBV; Dobrava virus, MPRLV; Maporal virus, PHV; Prospect Hill virus UNV, SFTSV; Severe Fever with Thrombocytopenia Syndrome virus.
b50% cytotoxic concentration (CC50) and 50% effective concentration (EC50) values are in µg/ml as determined by CPE reduction assays using Vero cells, and SI (selectivity index) is calculated as CC50/EC50.
Oral treatment with favipiravir b.i.d. blocked the lethality, reduced viral loads in the blood and the tissues, and suppressed hepatic lesions in the infection models of PTV in mice and hamsters.29),34) Oral treatment with favipiravir b.i.d. improved the survival rates in mice infected with CCHFV, when the dosing commenced 2 days post-infection.36) In hamsters infected with RVFV, oral favipiravir b.i.d. blocked deaths, and decreased viral titer in the serum and in the tissues.37)
Severe fever with thrombocytopenia syndrome (SFTS) is an emerging viral disease caused by SFTSV, which has recently been identified in China, Korea and Japan.38)–40) Seasonality of SFTSV is throughout the year, but more common between spring to autumn. Favipiravir inhibited the replication of SFTSV with EC50 values of 0.71–1.3 µg/mL in FFU reduction assays using Vero cells.41) Therapeutic effects of favipiravir was demonstrated in SFTSV infection model in mice lacking interferon alpha receptors (IFNAR−/−). Oral treatment with favipiravir survived all mice (P < 0.001) at 300 mg/kg once a day for 5 days initiating 3 days post-infection, whereas initiating 4 and 5 days post-infection significantly increased survival rates (P < 0.01 and P < 0.05, respectively) over the placebo group (Table 4). Upon these pre-clinical study results, clinical research on SFTS has commenced in Japan.42)
Table 4. Therapeutic effects of favipiravir in the SFTSV infected IFNAR
−/− mice model (Based on Tani
et al., 2016
41))
| Group |
Treatment |
Treatment period |
Survival rate
survivor/total |
Maximum body weight change (%)
mean ± SD |
| Control |
vehicle |
day 0–4 |
1/10 |
77.3 ± 8.9 |
| Favipiravir |
300 mg/kg/day |
day 1–5 |
6/6*** |
97.9 ± 4.7 |
| |
|
day 2–6 |
6/6*** |
88.3 ± 5.1 |
| |
|
day 3–7 |
6/6*** |
83.3 ± 2.2 |
| |
|
day 4–8 |
5/6** |
80.2 ± 4.6 |
| |
|
day 5–9 |
3/6* |
75.5 ± 4.9 |
Six or 10 IFNAR−/− male mice in each group were inoculated subcutaneously with 1.0 × 106 TCID50 of SFTSV (SPL010 strain). Mice were treated with favipiravir at a dose of 300 mg/kg/day. Treatment was commenced 1, 2, 3, 4, or 5 days post-infection. Favipiravir was administered once daily until death or for 5 days. Survival was determined using Kaplan-Meier analysis and GraphPad Prism6. Significance was determined relative to results for the placebo group: ***, P < 0.001; **, P < 0.01; *, P < 0.05. Relative weight is shown as means with standard deviations (Tani et al., 201641)).
Favipiravir inhibited the replication of several pathogenic flaviviruses, including yellow fever virus (YFV) and West Nile virus (WNV).43),44) Higher concentrations of favipiravir were needed to achieve the same efficacy for flavivirus as for influenza virus. The EC90 of favipiravir for YFV was 51.8 µg/mL in a yield-reduction assay using Vero cells.43) In YFV-infected hamsters, oral treatment with favipiravir at 200 or 400 mg/kg/day for 8 days significantly decreased fatality rate when the treatment was initiated 4 hr before infection.43) Complete protection was achieved when oral treatment with favipiravir at 400 mg/kg/day was initiated within 2 days post-infection.
In vitro and in vivo anti-viral efficacy of favipiravir was demonstrated for WNV.44) Favipiravir inhibited the proliferation of WNV with an EC50 of 53 µg/mL in Vero cells. In WNV infected mice, oral administration of favipiravir at 400 mg/kg/day b.i.d. initiating 4 hr subcutaneous infection protected nine out of ten mice from mortality (p < 0.01), and reduced the expression of viral proteins and RNA in brain tissues on 6 days post-infection. Similar efficacy was demonstrated in the second species. Hamsters were orally dosed at 400 mg/kg/day b.i.d. commencing 4 hr after subcutaneous infection. Like mice, favipiravir significantly improved the survival rate (p < 0.01). It is of note that envelope protein of WNV was not detected in the brains of hamsters dosed with favipiravir when examined on day 7 post-infection.
Zika virus (ZIKV) is an emerging arbovirus of Flaviviridae, which is mainly transmitted by mosquito bites. Evidence to date suggests the association between fetal infection and microcephaly. Favipiravir inhibited the replication of ZIKV in Vero cells with an EC50 of 3.5–3.8 µg/mL, suggesting a therapeutic potential for the ZIKV infection.45)
4.4 Togaviridae.
Favipiravir showed anti-viral activity against Western equine encephalitis virus (WEEV) cultured in Vero cells with an EC90 of 49 µg/mL.46) In WEEV infected mice, oral treatment with favipiravir significantly improved the survival rate (p < 0.01) and prolonged time to death at 400 mg/kg/day b.i.d. for 7 days initiating 4 hr before infection. Viral titer in the brain on day 4 post-infection decreased to one tenth although with no statistical significance. Signs of disease were relatively modest, which was not eliminated by favipiravir.
Favipiravir demonstrated anti-viral activity against Chikungunya virus (CHIKV) in Vero cells with an EC50 of 0.3–9.4 µg/mL. In CHIKV infected mice, oral administration of favipiravir improved the survival rate at 300 mg/kg/day b.i.d. initiating 24 hr before or 4 hr post-infection.47)
4.5 Picornaviridae.
Replication of foot-and-mouth disease virus was inhibited by favipiravir in vitro with an EC50 of 14 µg/mL.48),49) Favipiravir blocked the replication of poliovirus in Vero cells and rhinovirus in HeLa cells with EC50s of 4.8 and 23 µg/mL, and with selectivity index of 29 and >43, respectively.21) Replication of Enterovirus was inhibited by favipiravir with an EC50 of 23 µg/mL.50)
4.6 Caliciviridae.
Favipiravir was active against murine norovirus with an EC50 of 39 µg/mL in CPE assay using murine leukaemia macrophage RAW 264.7 cells. Real-time PCR revealed that favipiravir inhibited viral RNA synthesis with an EC50 of 19 µg/mL.51) In persistent norovirus infected murine model, oral treatment with favipiravir significantly decreased viral titer in feces and the ratio of norovirus antigen positive mice at 600 mg/kg/day b.i.d. for 8 weeks beginning 4 weeks post-infection.14) It was demonstrated that favipiravir-RTP inhibited RNA polymerase activity of human Norovirus.52)
4.7 Filoviridae.
Favipiravir demonstrated anti-viral activity against Zaire Ebola virus (Mayinga 1976 strain) in Vero E6 cells with an EC50 of 10.5 µg/mL. In IFNAR−/− C57BL/6 mice infected with Mayinga 1976 strain, oral treatment with favipiravir protected all mice from deaths and decreased viral titer in the blood at 300 mg/kg/day b.i.d. for 8 days beginning 6 days post-infection, while all mice died in the placebo group.53) Similarly, in IFNAR−/− A129 mice infected with E718 strain, oral treatment with favipiravir completely protected infected mice from deaths at 300 mg/kg/day b.i.d. for 14 days beginning 1 hr post-infection.54)
Outbreak of Ebola virus disease (EVD) occurred in western Africa in 2014. French Institute of Health and Medical Research (INSERM) and Guinea Government carried out clinical study with favipiravir (JIKI study).55) Favipiravir was well tolerated, and demonstrated a trend to decrease the mortality of patients with the low viral load (cycle threshold < 20).56) Chinese group recently reported that favipiravir increased the survival rate with statistical significance (p < 0.05), and decreased the viral load in the patients with EVD in Sierra Leone.57)
4.8 Rhabdoviridae.
It has recently been reported that favipiravir was active against rabies virus (RABV) in murine neuroblastoma Neuro-2a cells with EC50s of 5.1–7.0 µg/mL.58) Favipiravir significantly improved the morbidity and mortality of mice infected with RABV, when oral administration at 300 mg/kg/day b.i.d. for 7 days was initiated 1 hr post-infection. Favipiravir showed no effects when administered after symptoms appeared.
These results suggest that favipiravir has a wide range of anti-viral spectrum among RNA viruses, and matches the medical needs for those specifically untreatable viral infections. Interferon α and ribavirin are possible drugs to use with wide anti-viral spectrum; however, debilitating side effects limit their use. Unlike these agents, favipiravir was well tolerated in clinical studies.
5. Conclusion
Favipiravir was discovered by phenotypic screening against influenza virus at Research Laboratories of Toyama Chemical Co., Ltd. We undertook further studies to investigate the mechanism of actions, and anti-viral effects against various types of viruses. Favipiravir is phosphoribosylated in the cells to be an active form, favipiravir-RTP, which is recognized as a purine nucleotide by RdRp, and inhibits the RdRp enzyme activity. Favipiravir-RTP exhibits no effects on DNA-dependent RNA or DNA polymerases. These characters explain that favipiravir favors RNA virus over DNA virus and mammalian cells. Favipiravir is efficacious in multiple types of Influenza viruses, regardless of sensitive or resistant to existing anti-influenza drugs. Of specific note is that favipiravir is active against a wide range of other RNA viruses in vitro and in vivo. In vitro studies indicate no emergence of resistance to favipiravir.
Favipiravir is currently approved for influenza infection in Japan, and at an advanced clinical development in US. Evidence to date from clinical studies shows that favipiravir is well tolerated in human. Wider anti-viral spectrum of favipiravir drove us to pursue clinical research for devastating virus infections such as Ebola and SFTS. We strongly believe that favipiavir with these unique profiles will be a promising therapeutic agent for unremedied infections by RNA viruses in the near future.
Profile

Yousuke Furuta was born in Gifu Prefecture in 1956. After receiving BS in Pharmacology from Kitasato University in Japan, he found a job as a researcher at Zenyaku Kogyo Company, Limited in Tokyo, Japan. He switched to Toyama Chemical Co., Ltd. in Toyama, Japan. In the following years, he developed his experience in pharmacology of cancer and virus through temporary transfer to Cancer Institute (HTLV, HIV) and Kitasato University (antiviral screening methods, infectious immunology). Based on the experience, he led a Toyama’s antiviral research team and invented Favipiravir (T-705), viral RNA polymerase inhibitor to influenza virus and T-1105, anti-FMDV agent. For these achievements, he obtained Ph.D. in Bioscience in Kitasato University.
Yousuke Furuta is a Senior Associate of Business Development Department at Toyama Chemical in Japan.
Profile

Takashi Komeno was born (1981) and grew up in Takaoka-city of Toyama Prefecture, Japan. He entered the Tokyo University of Science and received a MS degree from the Tokyo University of Science in 2005, he joined Toyama Chemical Co., Ltd. in Toyama, Japan as a researcher in pharmacology of bacteria and virus. He has engaged in development research of Favipiravir (T-705) since 2006. He is being transferred to Fujifilm since 2016.
Takashi Komeno is a researcher of Pharmaceutical and Healthcare research Laboratories at Fujifilm in Japan.
Profile

Takaaki Nakamura was born (1957) and grew up in Aichi Prefecture, Japan. After obtaining a BS degree from the University of Tokyo in 1981, he joined Fujisawa Pharmaceutical Company (currently Astellas) as a scientist in drug metabolism and cancer pharmacology in 1981. He moved to the research site at Pfizer in Japan in 1989, then to Fujifilm in 2008. In both companies his research centered on pharmacology in immunology, gastroenterology and cancer. He was transferred to Toyama Chemical Co., Ltd. in 2011. While working as a pharmacologist, he received a Ph.D. degree in Medical Sciences from the University of Osaka in 1988.
Takaaki Nakamura is currently Head of Research Laboratories and Leader of Inflammatory Field Project at Toyama Chemical in Japan.
Acknowledgement
This work was supported by Contracts NO1-AI-15435, NO1-AI-30048, NO1-AI-30063, NO1-AI-065357, HHSN272201000039I, HHSN272201100019I and U54 AI-065357 from NIAID, NIH.
We thank Drs. Heather Greenstone, Amy Krafft, Chris Tseng, Catherine Laughlin, Brian B. Gowen, Justin G. Julander, E. Bart Tarbet, Donald F. Smee, Dale L. Barnard, John D. Morrey, David Safronetz, Heinz Feldmann, Katrina Sleeman, Larisa V. Gubareva, Kimiyasu Shiraki, Emi Takashita, Takato Odagiri, Hideki Tani, Shigeru Morikawa, Masayuki Shimojima, Masayuki Saijo, Kenichi Sakamoto, Kentaro Yamada, Akira Nishizono. Maki Kiso, Yasushi Itoh, Tokiko Watanabe and Yoshihiro Kawaoka for their extensive collaboration.
YF and TN are employees of the Toyama Chemical Co., Ltd., the manufacturer of favipiravir. TK is an employee of the FUJIFILM Corporation. All authors declare no competing interests.
References
- 1) Poovorawan, Y., Pyungporn, S., Prachayangprecha, S. and Makkoch, J. (2013) Global alert to avian influenza virus infection: From H5N1 to H7N9. Pathog. Glob. Health 106, 217–223.
- 2) Li, Q., Zhou, L., Zhou, M., Chen, Z., Li, F., Wu, H., Xiang, N., Chen, E., Tang, F., Wang, D., Meng, L., Hong, Z., Tu, W., Cao, Y., Li, L., Ding, F., Liu, B., Wang, M., Xie, R., Gao, R., Li, X., Bai, T., Zou, S., He, J., Hu, J., Xu, Y., Chai, C., Wang, S., Gao, Y., Jin, L., Zhang, Y., Luo, H., Yu, H., He, J., Li, Q., Wang, X., Gao, L., Pang, X., Liu, G., Yan, Y., Yuan, H., Shu, Y., Yang, W., Wang, Y., Wu, F., Uyeki, T.M. and Feng, Z. (2014) Preliminary report: epidemiology of the avian influenza A(H7N9) outbreak in China. N. Engl. J. Med. 370, 520–532.
- 3) Writing Committee of the WHO Consultation on Clinical Aspects of Pandemic (H1N1) 2009 Influenza (2010) Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. N. Engl. J. Med. 362, 1708–1719.
- 4) CDC Health Advisory: CDC issues interim recommendations for the use of influenza antivirals in the setting of oseltamivir Resistance among circulating influenza A (H1N1) viruses, 2008–09 Season.
- 5) WHO Ebola Response Team (2014) Ebola virus disease in West Africa—the first 9 months of the epidemic and forward projections. N. Engl. J. Med. 371, 1481–1495.
- 6) Mylne, A.Q., Pigott, D.M., Longbottom, J., Shearer, F., Duda, K.A., Messina, J.P., Weiss, D.J., Moyes, C.L., Golding, N. and Hay, S.I. (2015) Mapping the zoonotic niche of Lassa fever in Africa. Trans. R. Soc. Trop. Med. Hyg. 109, 483–492.
- 7) Macher, A.M. and Wolfe, M.S. (2006) Historical Lassa fever reports and 30-year clinical update. Emerg. Infect. Dis. 12, 835–837.
- 8) Furuta, Y., Takahashi, K., Kuno-Maekawa, M., Sangawa, H., Uehara, S., Kozaki, K., Nomura, N., Egawa, H. and Shiraki, K. (2005) Mechanism of action of T-705 against influenza virus. Antimicrob. Agents Chemother. 49, 981–986.
- 9) Furuta, Y., Gowen, B.B., Takahashi, K., Shiraki, K., Smee, D.F. and Barnard, D.L. (2013) Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Res. 100, 446–454.
- 10) Nagata, T., Lefor, A.K., Hasegawa, M. and Ishii, M. (2015) Favipiravir: a new medication for the Ebola virus disease pandemic. Disaster Med. Public Health Prep. 9, 79–81.
- 11) Baranovich, T., Wong, S.S., Armstrong, J., Marjuki, H., Webby, R.J., Webster, R.G. and Govorkova, E.A. (2013) T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J. Virol. 87, 3741–3751.
- 12) Vanderlinden, E., Vrancken, B., Van Houdt, J., Rajwanshi, V.K., Gillemot, S., Andrei, G., Lemey, P. and Naesens, L. (2016) Distinct effects of T-705 (favipiravir) and ribavirin on influenza virus replication and viral RNA synthesis. Antimicrob. Agents Chemother. 60, 6679–6691.
- 13) de Ávila, A.I., Gallego, I., Soria, M.E., Gregori, J., Quer, J., Esteban, J.I., Rice, C.M., Domingo, E. and Perales, C. (2016) Lethal mutagenesis of Hepatitis C virus induced by Favipiravir. PLoS One 11, e0164691.
- 14) Arias, A., Thorne, L. and Goodfellow, I. (2014) Favipiravir elicits antiviral mutagenesis during virus replication in vivo. eLife 3, e03679.
- 15) Sangawa, H., Komeno, T., Nishikawa, H., Yoshida, A., Takahashi, K., Nomura, N. and Furuta, Y. (2013) Mechanism of action of T-705 ribosyl triphosphate against influenza virus RNA polymerase. Antimicrob. Agents Chemother. 57, 5202–5208.
- 16) Jin, Z., Smith, L.K., Rajwanshi, V.K., Kim, B. and Deval, J. (2013) The ambiguous base-pairing and high substrate efficiency of T-705 (Favipiravir) ribofuranosyl 5′-triphosphate towards influenza A virus polymerase. PLoS One 8, e68347.
- 17) Crotty, S., Cameron, C. and Andino, R. (2002) Ribavirin’s antiviral mechanism of action: lethal mutagenesis? J. Mol. Med. (Berl.) 80, 86–95.
- 18) Streeter, D.G., Witkowski, J.T., Khare, G.P., Sidwell, R.W., Bauer, R.J., Robins, R.K. and Simon, L.N. (1973) Mechanism of action of 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide (Virazole), a new broad-spectrum antiviral agent. Proc. Natl. Acad. Sci. U.S.A. 70, 1174–1178.
- 19) Smee, D.F., Hurst, B.L., Egawa, H., Takahashi, K., Kadota, T. and Furuta, Y. (2009) Intracellular metabolism of favipiravir (T-705) in uninfected and influenza A (H5N1) virus-infected cells. J. Antimicrob. Chemother. 64, 741–746.
- 20) Kiso, M., Takahashi, K., Sakai-Tagawa, Y., Shinya, K., Sakabe, S., Le, Q.M., Ozawa, M., Furuta, Y. and Kawaoka, Y. (2010) T-705 (favipiravir) activity against lethal H5N1 influenza A viruses. Proc. Natl. Acad. Sci. U.S.A. 107, 882–887.
- 21) Furuta, Y., Takahashi, K., Fukuda, Y., Kuno, M., Kamiyama, T., Kozaki, K., Nomura, N., Egawa, H., Minami, S., Watanabe, Y., Narita, H. and Shiraki, K. (2002) In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob. Agents Chemother. 46, 977–981.
- 22) Sleeman, K., Mishin, V.P., Deyde, V.M., Furuta, Y., Klimov, A.I. and Gubareva, L.V. (2010) In vitro antiviral activity of Favipiravir (T-705) against drug-resistant influenza and 2009 A(H1N1) viruses. Antimicrob. Agents Chemother. 54, 2517–2524.
- 23) Itoh, Y., Shinya, K., Kiso, M., Watanabe, T., Sakoda, Y., Hatta, M., Muramoto, Y., Tamura, D., Sakai-Tagawa, Y., Noda, T., Sakabe, S., Imai, M., Hatta, Y., Watanabe, S., Li, C., Yamada, S., Fujii, K., Murakami, S., Imai, H., Kakugawa, S., Ito, M., Takano, R., Iwatsuki-Horimoto, K., Shimojima, M., Horimoto, T., Goto, H., Takahashi, K., Makino, A., Ishigaki, H., Nakayama, M., Okamatsu, M., Takahashi, K., Warshauer, D., Shult, P.A., Saito, R., Suzuki, H., Furuta, Y., Yamashita, M., Mitamura, K., Nakano, K., Nakamura, M., Brockman-Schneider, R., Mitamura, H., Yamazaki, M., Sugaya, N., Suresh, M., Ozawa, M., Neumann, G., Gern, J., Kida, H., Ogasawara, K. and Kawaoka, Y. (2009) In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 460, 1021–1025.
- 24) Takahashi, K., Furuta, Y., Fukuda, Y., Kuno, M., Kamiyama, T., Kozaki, K., Nomura, N., Egawa, H., Minami, S. and Shiraki, K. (2003) In vitro and in vivo activities of T-705 and oseltamivir against influenza virus. Antivir. Chem. Chemother. 14, 235–241.
- 25) Sidwell, R.W., Barnard, D.L., Day, C.W., Smee, D.F., Bailey, K.W., Wong, M.H., Morrey, J.D. and Furuta, Y. (2007) Efficacy of orally administered T-705 on lethal avian influenza A (H5N1) virus infections in mice. Antimicrob. Agents Chemother. 51, 845–851.
- 26) Smee, D.F., Hurst, B.L., Wong, M.H., Bailey, K.W., Tarbet, E.B., Morrey, J.D. and Furuta, Y. (2010) Effects of the combination of favipiravir (T-705) and oseltamivir on influenza A virus infections in mice. Antimicrob. Agents Chemother. 54, 126–133.
- 27) McCormick, J.B., King, I.J., Webb, P.A., Scribner, C.L., Craven, R.B., Johnson, K.M., Elliott, L.H. and Belmont-Williams, R. (1986) Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med. 314, 20–26.
- 28) Moraz, M.L. and Kunz, S. (2011) Pathogenesis of arenavirus hemorrhagic fevers. Expert Rev. Anti Infect. Ther. 9, 49–59.
- 29) Gowen, B.B., Wong, M.H., Jung, K.H., Sanders, A.B., Mendenhall, M., Bailey, K.W., Furuta, Y. and Sidwell, R.W. (2007) In vitro and in vivo activities of T-705 against arenavirus and bunyavirus infections. Antimicrob. Agents Chemother. 51, 3168–3176.
- 30) Gowen, B.B., Smee, D.F., Wong, M.H., Hall, J.O., Jung, K.H., Bailey, K.W., Stevens, J.R., Furuta, Y. and Morrey, J.D. (2008) Treatment of late stage disease in a model of arenaviral hemorrhagic fever: T-705 efficacy and reduced toxicity suggests an alternative to ribavirin. PLoS One 3, e3725.
- 31) Mendenhall, M., Russell, A., Smee, D.F., Hall, J.O., Skirpstunas, R., Furuta, Y. and Gowen, B.B. (2011) Effective oral favipiravir (T-705) therapy initiated after the onset of clinical disease in a model of arenavirus hemorrhagic fever. PLoS Negl. Trop. Dis. 5, e1342.
- 32) Safronetz, D., Rosenke, K., Westover, J.B., Martellaro, C., Okumura, A., Furuta, Y., Geisbert, J., Saturday, G., Komeno, T., Geisbert, T.W., Feldmann, H. and Gowen, B.B. (2015) The broad-spectrum antiviral favipiravir protects guinea pigs from lethal Lassa virus infection post-disease onset. Sci. Rep. 5, 14775.
- 33) Oestereich, L., Rieger, T., Lüdtke, A., Ruibal, P., Wurr, S., Pallasch, E., Bockholt, S., Krasemann, S., Muñoz-Fontela, C. and Günther, S. (2016) Efficacy of Favipiravir alone and in combination with Ribavirin in a lethal, immunocompetent mouse model of Lassa fever. J. Infect. Dis. 213, 934–938.
- 34) Gowen, B.B., Wong, M.H., Jung, K.H., Smee, D.F., Morrey, J.D. and Furuta, Y. (2010) Efficacy of favipiravir (T-705) and T-1106 pyrazine derivatives in phlebovirus disease models. Antiviral Res. 86, 121–127.
- 35) Buys, K.K., Jung, K.H., Smee, D.F., Furuta, Y. and Gowen, B.B. (2011) Maporal virus as a surrogate for pathogenic New World hantaviruses and its inhibition by favipiravir. Antivir. Chem. Chemother. 21, 193–200.
- 36) Oestereich, L., Rieger, T., Neumann, M., Bernreuther, C., Lehmann, M., Krasemann, S., Wurr, S., Emmerich, P., de Lamballerie, X., Ölschläger, S. and Günther, S. (2014) Evaluation of antiviral efficacy of ribavirin, arbidol, and T-705 (favipiravir) in a mouse model for Crimean-Congo hemorrhagic fever. PLoS Negl. Trop. Dis. 8, e2804.
- 37) Scharton, D., Bailey, K.W., Vest, Z., Westover, J.B., Kumaki, Y., Van Wettere, A., Furuta, Y. and Gowen, B.B. (2014) Favipiravir (T-705) protects against peracute Rift Valley fever virus infection and reduces delayed-onset neurologic disease observed with ribavirin treatment. Antiviral Res. 104, 84–92.
- 38) Yu, X., Liang, M., Zhang, S., Liu, Y., Li, J., Sun, Y., Zhang, L., Zhang, Q., Popov, V.L., Li, C., Qu, J., Li, Q., Zhang, Y., Hai, R., Wu, W., Wang, Q., Zhan, F., Wang, X., Kan, B., Wang, S., Wan, K.L., Jing, H.Q., Lu, J.X., Yin, W.W., Zhou, H., Guan, X.H., Liu, J.F., Bi, Z.Q., Liu, G.H., Ren, J., Wang, H., Zhao, Z., Song, J.D., He, J.R., Wan, T., Zhang, J.S., Fu, X.P., Sun, L.N., Dong, X.P., Feng, Z.J., Yang, W.Z., Hong, T., Zhang, Y., Walker, D.H., Wang, Y. and Li, D.X. (2011) Fever with thrombocytopenia associated with a novel bunyavirus in China. N. Engl. J. Med. 364, 1523–1532.
- 39) Kim, K., Yi, J., Kim, G., Choi, S.J., Jun, K.I., Kim, N., Choe, P.G., Kim, N., Lee, J. and Oh, M. (2013) Severe fever with thrombocytopenia syndrome, South Korea, 2012. Emerg. Infect. Dis. 19, 1892–1894.
- 40) Takahashi, T., Maeda, K., Suzuki, T., Ishido, A., Shigeoka, T., Tominaga, T., Kamei, T., Honda, M., Ninomiya, D., Sakai, T., Senba, T., Kaneyuki, S., Sakaguchi, S., Satoh, A., Hosokawa, T., Kawabe, Y., Kurihara, S., Izumikawa, K., Kohno, S., Azuma, T., Suemori, K., Yasukawa, M., Mizutani, T., Omatsu, T., Katayama, Y., Miyahara, M., Ijuin, M., Doi, K., Okuda, M., Umeki, K., Saito, T., Fukushima, K., Nakajima, K., Yoshikawa, T., Tani, H., Fukushi, S., Fukuma, A., Ogata, M., Shimojima, M., Nakajima, N., Nagata, N., Katano, H., Fukumoto, H., Sato, Y., Hasegawa, H., Yamagishi, T., Oishi, K., Kurane, I., Morikawa, S. and Saijo, M. (2014) The first identification and retrospective study of severe fever with thrombocytopenia syndrome in Japan. J. Infect. Dis. 209, 816–827.
- 41) Tani, H., Fukuma, A., Fukushi, S., Taniguchi, S., Yoshikawa, T., Iwata-Yoshikawa, N., Sato, Y., Suzuki, T., Nagata, N., Hasegawa, H., Kawai, Y., Uda, A., Morikawa, S., Shimojima, M., Watanabe, H. and Saijo, M. (2016) Efficacy of T-705 (Favipiravir) in the treatment of infections with lethal severe fever with thrombocytopenia syndrome virus. mSphere 1, e00061–15.
- 42) Clinical study of favipiravir for patients with severe fever with thrombocytopenia syndrome. UMIN-CTR Clinical Trial, https://upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000025710.
- 43) Julander, J.G., Shafer, K., Smee, D.F., Morrey, J.D. and Furuta, Y. (2009) Activity of T-705 in a hamster model of yellow fever virus infection in comparison with a chemically related compound T-1106. Antimicrob. Agents Chemother. 53, 202–209.
- 44) Morrey, J.D., Taro, B.S., Siddharthan, V., Wang, H., Smee, D.F., Christensen, A.J. and Furuta, Y. (2008) Efficacy of orally administered T-705 pyrazine analog on lethal West Nile virus infection in rodents. Antiviral Res. 80, 377–379.
- 45) Zmurko, J., Marques, R.E., Schols, D., Verbeken, E., Kaptein, S.J. and Neyts, J. (2016) The viral polymerase inhibitor 7-deaza-2′-C-methyladenosine is a potent inhibitor of in vitro Zika virus replication and delays disease progression in a robust mouse infection model. PLoS Negl. Trop. Dis. 10, e0004695.
- 46) Julander, J.G., Smee, D.F., Morrey, J.D. and Furuta, Y. (2009) Effect of T-705 treatment on western equine encephalitis in a mouse model. Antiviral Res. 82, 169–171.
- 47) Delang, L., Segura Guerrero, N., Tas, A., Quérat, G., Pastorino, B., Froeyen, M., Dallmeier, K., Jochmans, D., Herdewijn, P., Bello, F., Snijder, E.J., de Lamballerie, X., Martina, B., Neyts, J., van Hemert, M.J. and Leyssen, P. (2014) Mutations in the chikungunya virus non-structural proteins cause resistance to favipiravir (T-705), a broad-spectrum antiviral. J. Antimicrob. Chemother. 69, 2770–2784.
- 48) Sakamoto, K., Ohashi, S., Yamazoe, R., Takahashi, K. and Furuta, Y. (2006) The inhibition of FMD virus excretion from the infected pigs by an antiviral agent, T-1105. FAO report of the research group of the standing technical committee of european commission for the control of Foot-and-Mouth Disease, Paphos, Cyprus. FAO Appendix 64, 418–424.
- 49) Furuta, Y., Takahashi, K., Shiraki, K., Sakamoto, K., Smee, D.F., Barnard, D.L., Gowen, B.B., Julander, J.G. and Morrey, J.D. (2009) T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. Antiviral Res. 82, 95–102.
- 50) Wang, Y., Li, G., Yuan, S., Gao, Q., Lan, K., Altmeyer, R. and Zou, G. (2016) In vitro assessment of combinations of enterovirus inhibitors against enterovirus 71. Antimicrob. Agents Chemother. 60, 5357–5367.
- 51) Rocha-Pereira, J., Jochmans, D., Dallmeier, K., Leyssen, P., Nascimento, M.S. and Neyts, J. (2012) Favipiravir (T-705) inhibits in vitro norovirus replication. Biochem. Biophys. Res. Commun. 10 (424), 777–780.
- 52) Jin, Z., Tucker, K., Lin, X., Kao, C.C., Shaw, K., Tan, H., Symons, J., Behera, I., Rajwanshi, V.K., Dyatkina, N., Wang, G., Beigelman, L. and Deval, J. (2015) Biochemical evaluation of the inhibition properties of Favipiravir and 2′-C-methyl-cytidine triphosphates against human and mouse norovirus RNA polymerases. Antimicrob. Agents Chemother. 59, 7504–7516.
- 53) Oestereich, L., Lüdtke, A., Wurr, S., Rieger, T., Muñoz-Fontela, C. and Günther, S. (2014) Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antiviral Res. 105, 17–21.
- 54) Smither, S.J., Eastaugh, L.S., Steward, J.A., Nelson, M., Lenk, R.P. and Lever, M.S. (2014) Post-exposure efficacy of oral T-705 (Favipiravir) against inhalational Ebola virus infection in a mouse model. Antiviral Res. 104, 153–155.
- 55) Mentré, F., Taburet, A.M., Guedj, J., Anglaret, X., Keïta, S., de Lamballerie, X. and Malvy, D. (2015) Dose regimen of favipiravir for Ebola virus disease. Lancet Infect. Dis. 15, 150–151.
- 56) Sissoko, D., Laouenan, C., Folkesson, E., M’Lebing, A.B., Beavogui, A.H., Baize, S., Camara, A.M., Maes, P., Shepherd, S., Danel, C., Carazo, S., Conde, M.N., Gala, J.L., Colin, G., Savini, H., Bore, J.A., Le Marcis, F., Koundouno, F.R., Petitjean, F., Lamah, M.C., Diederich, S., Tounkara, A., Poelart, G., Berbain, E., Dindart, J.M., Duraffour, S., Lefevre, A., Leno, T., Peyrouset, O., Irenge, L., Bangoura, N., Palich, R., Hinzmann, J., Kraus, A., Barry, T.S., Berette, S., Bongono, A., Camara, M.S., Munoz, V.C., Doumbouya, L., Harouna, S., Kighoma, P.M., Koundouno, F.R., Lolamou, R., Loua, C.M., Massala, V., Moumouni, K., Provost, C., Samake, N., Sekou, C., Soumah, A., Arnould, I., Komano, M.S., Gustin, L., Berutto, C., Camara, D., Camara, F.S., Colpaert, J., Delamou, L., Jansson, L., Kourouma, E., Loua, M., Malme, K., Manfrin, E., Maomou, A., Milinouno, A., Ombelet, S., Sidiboun, A.Y., Verreckt, I., Yombouno, P., Bocquin, A., Carbonnelle, C., Carmoi, T., Frange, P., Mely, S., Nguyen, V.K., Pannetier, D., Taburet, A.M., Treluyer, J.M., Kolie, J., Moh, R., Gonzalez, M.C., Kuisma, E., Liedigk, B., Ngabo, D., Rudolf, M., Thom, R., Kerber, R., Gabriel, M., Di Caro, A., Wölfel, R., Badir, J., Bentahir, M., Deccache, Y., Dumont, C., Durant, J.F., El Bakkouri, K., Uwamahoro, M.G., Smits, B., Toufik, N., Van Cauwenberghe, S., Ezzedine, K., Dortenzio, E., Pizarro, L., Etienne, A., Guedj, J., Fizet, A., de Sainte Fare, E.B., Murgue, B., Tran-Minh, T., Rapp, C., Piguet, P., Poncin, M., Draguez, B., Allaford Duverger, T., Barbe, S., Baret, G., Defourny, I., Carroll, M., Raoul, H., Augier, A., Eholie, S.P., Yazdanpanah, Y., Levy-Marchal, C., Antierrens, A., Van Herp, M., Günther, S., de, Lamballerie, X., Keïta, S., Mentre, F., Anglaret, X. and Malvy, D.; JIKI Study Group (2016) Experimental treatment with Favipiravir for Ebola virus disease (the JIKI Trial): A historically controlled, single-arm proof-of-concept trial in Guinea. PLoS Med. 13, e1001967.
- 57) Bai, C.Q., Mu, J.S., Kargbo, D., Song, Y.B., Niu, W.K., Nie, W.M., Kanu, A., Liu, W.W., Wang, Y.P., Dafae, F., Yan, T., Hu, Y., Deng, Y.Q., Lu, H.J., Yang, F., Zhang, X.G., Sun, Y., Cao, Y.X., Su, H.X., Sun, Y., Liu, W.S., Wang, C.Y., Qian, J., Liu, L., Wang, H., Tong, Y.G., Liu, Z.Y., Chen, Y.S., Wang, H.Q., Kargbo, B., Gao, G.F. and Jiang, J.F. (2016) Clinical and virological characteristics of Ebola virus disease patients treated with favipiravir (T-705)-Sierra Leone, 2014. Clin. Infect. Dis. 63, 1288–1294.
- 58) Yamada, K., Noguchi, K., Komeno, T., Furuta, Y. and Nishizono, A. (2016) Efficacy of Favipiravir (T-705) in rabies postexposure prophylaxis. J. Infect. Dis. 213, 1253–1261.
- 59) Sheu, T.G., Deyde, V.M., Okomo-Adhiambo, M., Garten, R.J., Xu, X., Bright, R.A., Butler, E.N., Wallis, T.R., Klimov, A.I. and Gubareva, L.V. (2008) Surveillance for neuraminidase inhibitor resistance among human influenza A and B viruses circulating worldwide from 2004 to 2008. Antimicrob. Agents Chemother. 52, 3284–3292.
- 60) Jochmans, D., van Nieuwkoop, S., Smits, S.L., Neyts, J., Fouchier, R.A. and van den Hoogen, B.G. (2016) Antiviral activity of Favipiravir (T-705) against a broad range of paramyxoviruses in vitro and against human metapneumovirus in hamsters. Antimicrob. Agents Chemother. 60, 4620–4629.
- 61) Watanabe, T., Kiso, M., Fukuyama, S., Nakajima, N., Imai, M., Yamada, S., Murakami, S., Yamayoshi, S., Iwatsuki-Horimoto, K., Sakoda, Y., Takashita, E., McBride, R., Noda, T., Hatta, M., Imai, H., Zhao, D., Kishida, N., Shirakura, M., de Vries, R.P., Shichinohe, S., Okamatsu, M., Tamura, T., Tomita, Y., Fujimoto, N., Goto, K., Katsura, H., Kawakami, E., Ishikawa, I., Watanabe, S., Ito, M., Sakai-Tagawa, Y., Sugita, Y., Uraki, R., Yamaji, R., Eisfeld, A.J., Zhong, G., Fan, S., Ping, J., Maher, E.A., Hanson, A., Uchida, Y., Saito, T., Ozawa, M., Neumann, G., Kida, H., Odagiri, T., Paulson, J.C., Hasegawa, H., Tashiro, M. and Kawaoka, Y. (2013) Characterization of H7N9 influenza A viruses isolated from humans. Nature 501, 551–555.


