Abstract
F-type ATPase is a ubiquitous molecular motor. Investigations on thermophilic F1-ATPase and its subunits, β and ε, by NMR were reviewed. Using specific isotope labeling, pKa of the putative catalytic carboxylate in β was estimated. Segmental isotope-labeling enabled us to monitor most residues of β, revealing that the conformational conversion from open to closed form of β on nucleotide binding found in ATPase was an intrinsic property of β and could work as a driving force of the rotational catalysis. A stepwise conformational change was driven by switching of the hydrogen bond networks involving Walker A and B motifs. Segmentally labeled ATPase provided a well resolved NMR spectra, revealing while the open form of β was identical for β monomer and ATPase, its closed form could be different. ATP-binding was also a critical factor in the conformational conversion of ε, an ATP hydrolysis inhibitor. Its structural elucidation was described.
1. Introduction
FoF1 ATP synthase is a ubiquitous molecular motor involved in H+-mediated energy conversion in organisms from bacteria to human and has been extensively investigated by a wide variety of methods.1)–9) A model image of a bacterial FoF1 ATP synthase is presented in Fig. 1. The H+-driven ATP synthase converts the energy of the transmembrane electrochemical potential generated by proton gradient to that of ATP. It consists of a water-soluble F1 sector and a membrane integrated Fo sector. The former has catalytic sites for ATP synthesis/hydrolysis, and the latter mediates H+ transport across membranes. The F1 and Fo sectors comprise multiple subunits. Basically, their compositions are α3β3γ1δ1ε1 and a1b2cn (n = 8–15), respectively. Mitochondrial ATP synthases have more subunits. F1 can be solubilized and the isolated F1 functions only as ATPase.

The first crystal structure of F1 from bovine heart mitochondria (MF1) provided atomic information of the α3β3γ complex (PDB ID: 1BMF).10) The catalytic site of F1-ATPase was found in the β subunit facing the α/β interface. The subassembly α3β3 formed a ring with long helices of the γ subunit in the center, consistent with Boyer’s rotary-catalysis model.1) The tertiary structure of the β subunit in the F1-ATPase differed according to the state of its active site, namely, an empty, MgAMPPNP (5′-adenylyl-imidodiphosphate, an ATP analog, with Mg2+) bound or MgADP (ADP with Mg2+) bound state (designated as βE, βTP, βDP, respectively). βTP and βDP took so-called closed forms because their hinged N- and C-terminal domains were closer to each other than those of βE, the active site of which adopted a relatively open conformation (open form). Actually, the rotation of the γ subunit in the fixed α3β3 complex on hydrolysis of ATP was directly demonstrated by epifluorescence microscopy for the α3β3γ complex from thermophilic Bacillus PS3 (TF1),11) which verified the Boyer’s model. This method (single molecule analysis) has been very powerful to understand the molecular mechanism of the rotational catalysis. The first observation revealed that the rotation of γ in F1 was carried out in a 120° step-wise manner. Then, the 120° rotation turned out to comprise 80° and 40° rotational substeps,12),13) and the former and latter were attributed to ATP-intake/ADP-release and hydrolysis/Pi-release, respectively.14),15) The pauses before the substep rotations were designated as ATP-binding dwell and catalytic dwell, respectively. In the case of FoF1-ATP synthase, the γεcn complex rotates in the α3β3δab2 stator coupled with H+ translocation across a membrane (Fig. 1).1)–9) Recently, single molecule analysis of human F1-ATPase revealed a three-substep rotation, namely, 65°, 25°, and 30°, suggesting that the mechanism underlying the rotational catalysis of mitochondrial F1-ATPase is different from the bacterial one.16)
In contrast to F1-ATPase, structure and functional mechanism of Fo is not yet well understood. It converts the electrochemical potential generated by the H+-gradient across membranes into rotation of the c-subunit ring and then into that of the γε-rotor in F1, or vice versa.17) The c-ring comprises 8–15 c-subunits, depending on the biological species.18)–23) The mechanism underlying the energy conversion at Fo is one of the major unresolved issues for understanding FoF1-ATP synthase.24)
Biochemical, genetic, crystallographic, and single molecular investigations have been successful in conceptual elucidation of structure and function of FoF1-ATP synthase. We have been working on this enzyme using nuclear magnetic resonance (NMR) to clarify the dynamic mechanism at atomic resolution on the basis of the aforementioned knowledge. NMR is a unique methodology that can provide information bridging atomic structure and function of proteins under physiological conditions. However, a serious drawback in NMR was the size limit. Since FoF1-ATP synthase is a huge protein complex, we started with the subunits and proceeded to F1. To overcome difficulty caused by the molecular size, we developed combined use of gene-engineering, isotope-labeling, and sophisticated solution NMR measurements. Although I do not touch on in this review, we also developed methodology in solid-state NMR to work on Foc-ring, a membrane protein complex. We have been working on TFoF1-ATP synthase in close collaboration with Prof. Masasuke Yoshida and his colleagues.
2. Targeting the heart of the β subunit; estimation of pKa of the putative catalytic carboxyl group
The isolated F1 hydrolyzes ATP to ADP and Pi. The smallest active complex of F1 is α3β3γ.2) The catalytic site is located in the β subunit, although residues from the α subunit are also required for ATP hydrolysis.25) Modification of Glu190 of the β subunit of TF1 (from thermophilic Bacillus PS3) with dicyclohexylcarbodiimide (DCCD)26) and substitution of this residue by glutamine27) resulted in complete loss of the activity. The crystal structure of mitochondrial F1-ATPase (MF1) actually revealed that the carboxylate of Glu188, corresponding to Glu190 of TF1β, was involved in a hydrogen bond with a bound water molecule in the vicinity of the γ-phosphate of a substrate analogue, AMPPNP.10) Therefore, it was suggested that this carboxyl group would activate the water for nucleophilic attack of the γ-phosphate. Therefore, pKa of Glu190 is an important parameter for understanding the mechanism underlying the catalytic reaction.
Since NMR has an atomic resolution even in a solution, it is one of the most powerful methods to determine pKa of a particular residue in a protein. However, it was not possible more than two decades ago because of high molecular masses of the β subunit (∼52 kDa) and F1-ATPase (∼370 kDa). To overcome molecular-size problem, we developed a specific isotope-labeling method using a combination of amino acid replacement and chemical modification. Since there was no Cys residue in TF1β, Glu190 was replaced with Cys, followed by carboxymethylation with iodo[1-13C]acetic acid.28) As can be seen in Fig. 2B, the labeled carboxylate signal could be clearly identified. The pH titration of this signal provided pKa = 5.7 ± 0.2 in the absence of nucleotide. Further correction on the difference in pKa between carboxymethylated Cys (CmCys) and glutamic acid gave the estimation of pKa of the carboxylate of Glu190 in TF1β as 6.8 ± 0.5. In the presence of ADP, pKa shifted to lower pH by less than one. A pKa value of regular Glu residue in proteins falls in the range 3–5.29) Therefore, the estimated pKa of Glu190 in the isolated TF1β is unusually high, even taking into account its large error range.

It was also possible to get a 13C-NMR spectrum of an active TF1 subcomplex, α(C193S/W463F)3β(E190[1-13C]CmCys)3γ, at p2H 8.1 in spite of its very high molecular mass (∼370 kDa) as can be seen in Fig. 2C. It suggested that at least one CmCys190 was under the chemical environment similar to that in the isolated β(CmCys190). Since ATPase activity of α3β(E190CmCys)3γ was 12% of that of α3β3γ,30) the role of the carboxylate at the 190 position in the F1-ATPase could be discussed. In the ground state structure of mitochondrial F1 at 1.9 Å resolution, the carboxyl group corresponding to this one formed a hydrogen bond with a water molecule in the AMPPNP bound βDP.31) The oxygen of the water molecule is located at 3.1 Å from the γ-phosphorus of AMPPNP in βDP. The authors indicated that the active site conformation of βDP was close to the transition state for γ-phosphate hydrolysis. The high pKa of Glu190 estimated in the TF1β monomer suggests that the carboxylate may be involved in the γ-phosphate hydrolysis of ATP. The lower activity of the α3β(E190CmCys)3γ complex might be due to the lower pKa of CmCys190 as well as the structural difference around the side chain. The exact mechanism underlying the ATP hydrolysis/synthesis is still under discussions.32),33)
3. Characterization of a nucleotide-induced conformational change of TF1β subunit
Crystallographic and single-molecule investigations have promoted general understanding of the rotational catalysis mechanism of FoF1 ATP synthase. However, dynamic mechanism at atomic resolution is not yet well established. Since the catalytic site is localized in the β subunit, intrinsic property of β monomer would be important to understand the rotational catalysis of F1. Although the molecular size of TF1β (52 kDa) was a serious problem, we started to work on TF1β monomer using NMR more than two decades ago.
The crystal structures of TF1α3β3 (PDB ID: 1SKY) complex and TF1α3β3γε (PDB ID: 4XD7) have been reported.34),35) The structure of the β subunit of the former took the open form and the latter included two open and one closed form structures. The open and closed forms were similar to those of MF1-ATPase. The β subunit is composed of roughly three domains, namely, the amino (N-) terminal, catalytic and carboxyl (C-) terminal domains (Fig. 5). Conformational difference between the open and the closed form of β in the crystal structures concentrates in the catalytic domain. At first, aromatic ring protons of Tyr and His residues (twelve each) were used for monitoring the conformational change of the TF1β monomer on nucleotide binding in solution, because of their relatively sharp NMR signals on deuteration of the other ring protons.36)–38) The residues only in the catalytic domain (Tyr-148, 199, 238, 307, and 341, and His-173, 179, 200, and 324) revealed chemical shift changes on AMPPNP/ATP binding. In the crystal structures, local conformations of a residue in the open and the closed form were different only in the catalytic domain. Therefore, the chemical shift perturbation might reflect a conformational change similar to that in MF1.
For elucidation of the origin of the chemical shift perturbations of aromatic proton signals, detailed information from all over the TF1β subunit was required. However, broadening and overlapping of the NMR signals due to its large molecular size (52 kDa, 473 amino acid residues) hampered identification of individual signals. To make observable signals less and sharper, we used segmental isotope-labeling and protein deuteration along with two-dimensional NMR at high magnetic field.39) Segmental labeling by either intein splicing reaction or chemical ligation40),41) was reported. However, it was a challenge, since no application to a really large molecule such as the β monomer was carried out.
We used an intein, PI-PfuI, from hyperthermophilic Pyrococcus furiosus for segmental isotope-labeling.39) The PI-PfuI and TF1β subunit genes were cut in the middle of the sequence, respectively. The N- and C-terminal fragments of the β subunit genes were ligated with those of the PI-PfuI genes in such a way that the β subunit fragments formed exteins by PCR (Fig. 3). The ligated genes of the intein-β subunit precursors (N and C) were cloned into different pET vectors and expressed in E. coli cell cultures separately. To label an N-terminal fragment of the β subunit, for example, the β-intein precursor (N) gene was expressed in a culture with 15NH4Cl and 13C-sucrose as the sole nitrogen and carbon sources, respectively. The N- and C-terminal precursors were mixed together in a buffer including denaturant. After removal of denaturant, the intein with a nick was renatured (Fig. 3). Then, peptide splicing was carried out at 70 °C by the intein. The ligated β subunit was once unfolded by urea, then refolded to the intact structure by removing it. The segmentally isotope-labeled β subunit included additional residues, Gly-Gly-Gly-Thr-Gly, at the ligation site.

We prepared four kinds of TF1β subunits labeled with 15N, 13C and 2H in the segments of residues 1–124, 1–271, 272–473, and 391–473, respectively. 1H-15N TROSY-HSQC (Transverse relaxation-optimized spectroscopy—Heteronuclear single quantum coherence) spectra of the four kinds of segmentally isotope-labeled TF1β monomers in the absence of nucleotide were presented in Fig. 4. TROSY-HSQC is a heteronuclear correlation spectroscopy specialized for a large molecule. The observed 1H/15N signals completely coincided with the corresponding ones in the spectrum of the uniformly 15N labeled authentic β subunit except for those of the inserted amino acid residues (GGGTG). Therefore, the ligated β subunits should take the intact structure. Sequential assignment was carried out using 3D TROSY-HNCO, 3D TROSY-HNCA, 3D TROSY-HN(CO)CA, 3D TROSY-HNCACB, 3D TROSY-HN(CO)CACB, and 3D 15N-edited NOESY (Nuclear Overhauser effect correlated spectroscopy) spectra. Here HNCO, for example, confers 1H, 15N, and 13C correlations of amide and carbonyl groups. NOESY provides the information on spatial proximity. To validate the sequential assignment, amino acid specific 15N labeling was performed for Tyr, Thr, Lys, Ser, and Gly. Thus, 89% of the NH (402/451), 89% of the Cα (417/473), 83% of the Cβ (357/431), and 90% of the CO (425/473) signals of TF1β were assigned.


The residues 156–163, 188–201, 301–310, and 316–322 (colored white in Fig. 5) could not be assigned. They are located in the active site region and are involved in the conformational change and/or ATP hydrolysis activity. The stretch 156–163 is a part of so-called P-loop, the binding site of nucleotide phosphate.10) That of 188–201 includes a putative catalytic site Glu190 and other important residues. The regions of 301–310 and 316–322 are involved in a significant conformational change on nucleotide binding, namely, formation of a new β sheet area (β3-β7, Fig. 5) and rearrangement of the surrounding area. Most unassigned signals (34 out of 49) were missing in the spectra. The major reason of loss of the signals should be the fast exchange of amide protons in the flexible regions. Those signals could not be recovered even on nucleotide binding. This fact strongly suggests that the four regions mentioned above are intrinsically flexible, which would be important for the global conformational change and the catalytic reaction.
A nucleotide-free TF1β solution was titrated with MgADP. The signals of the 136–139, 164–186, 209–212, 311–314, 328–352, and 410–430 residues shifted on the titration (highlighted in red in Fig. 5, all data in Fig. 7A). The residues of 328–352 and 410–430 formed the binding pocket for the adenine ring of ADP in the crystal structure, suggesting that the chemical shift perturbations could be ascribed to direct interaction with adenine.10) The chemical shift change of the other residues could be ascribed to conformational changes induced by nucleotide binding. Actually, all of these residues fell in the catalytic domain. This result was consistent with our idea on a nucleotide-induced conformational change obtained from 1H-NMR analysis of Tyr and His residues.37),38)
Then, the structures of nucleotide-free and nucleotide-bound TF1β monomers in solution should be characterized in the light of the crystal structures. The open and closed forms of β in the crystal structure of MF1 could be specified by the difference in relative orientation of the N- and C-terminal domains. NMR is one of excellent methods to confer orientation information of chemical groups in a protein in solution. For example, a residual dipolar coupling (RDC) of 15N and 1H reveals information on the orientation of 15N-1H bond relative to the static magnetic field, provided that the protein is partially aligned along the static magnetic field. If residues of interest are located in an α-helix, the orientation angle of the helix axis can be obtained. Judging from the secondary structures estimated from the assigned chemical shifts of 13Cα, 13Cβ, and carbonyl 13C (C′) of TF1β, the main folds of the N- and C-terminal domains are similar for the monomer and the crystal structure of TF1α3β3.34) Since there was no chemical shift change in the N- and C-terminal domains on nucleotide binding, their conformations would not be affected by it. Therefore, changes in the orientation of NH bonds should originate from a change in the domain orientation. Thus, TF1β subunits labeled in the N-terminal (1–124) and C-terminal (390–473) domains were used to determine their relative orientation. 1H-15N RDCs were measured in the absence and presence of MgADP in liquid-crystal, which could induce partial alignment of TF1β.39) In analysis of the RDC data of TF1β monomer, the β subunit structure in the TF1α3β3 crystal was used as the monomer structure model. To avoid flexibility problem, the residues located in either α-helices or β-sheets were used for the analysis. The number of RDCs used were 43 and 20 for the N- and C-terminal domains, respectively. The extent of alignment and the orientation angles of each domain relative to the reference frame were obtained on the fitting to the observed data.39) The results were similar for the N-terminal domains of the MgADP-bound and free β subunits. In contrast, it was not the case for their C-terminal domains. The relative orientations between the N- and C-terminal domains were determined by complete superposition of the N-terminal domains of interest. In the case of MgADP-free β, the orientation of helix 399–409 in the C-terminal domain (Fig. 6 left, red) was different by 5° for the obtained solution structure and TF1α3β3 crystal structure. In view of the error ranges, these structures were practically the same. Since the crystal structure of TF1β in the α3β3 complex is in good agreement with the open form of the β subunit in the MF1α3β3γ crystal structure (PDB ID: 1BMF), we can conclude that the β subunit monomer in solution also takes on the open form in the absence of a nucleotide.


For comparison of the conformations of the nucleotide-bound and nucleotide-free β monomers, the effect of MgADP on the relative orientations between the N- and C-terminal domains was examined. When an axis connecting the centers of mass of the N- and C-terminal domains (reference axis) was defined (vertical straight lines in Fig. 6), the angles between the reference and helix 399–409 axes were 110° and 144° for the MgADP-bound and free β subunits, respectively (Fig. 6 left). On the other hand, the corresponding angles in the open and closed forms in the MF1α3β3γ complex were 119° and 144°, respectively (Fig. 6 right). This result clearly revealed that the TF1β monomer took the open structure similar to that of MF1β in the MF1α3β3γ crystal structure,10) and the binding of MgADP induced a conformational change from the open to the closed form (a bending motion) in TF1β monomer in solution as expected from MF1α3β3γ and TF1α3β3γε35) crystal structures.
The conformational change of TF1β monomer was further examined with MgATP instead of MgADP.42) The angle between the reference and helix 399–409 axes was also 110° in good agreement with that with MgADP. On the other hand, the orientation of the helix axis was different for the MgATP- and MgADP-bound β monomers by 10° on the plane perpendicular to the reference axis. This reveals that there is a conformational variance even in the closed form depending on the bound nucleotide species. This was the first direct observation of the conformational change of the β subunit on MgATP binding because F1 could not be crystallized with MgATP. A similar conclusion was obtained from the chemical shift perturbation experiment with MgATP (Fig. 7B).42) Although small number of distinct differences were found, the general pattern of perturbed residues was similar to that with MgADP.
An important conclusion of this section is that the conformational change from the open to the closed form of a β subunit on nucleotide binding is its intrinsic property. Since the conformational change of β on nucleotide binding is one of the most important elements in the rotational catalysis, this intrinsic property can contribute to the driving force of the rotation of the γ subunit.
4. Stepwise mechanism driving the intrinsic bending motion of F1β subunit on nucleotide binding
Since the intrinsic conformational change is one of the essential driving forces of the rotational catalysis, elucidation of the atomic mechanism underlying the bending motion on nucleotide binding would be important for understanding F1-ATPase. This was carried out on TF1β monomer using combination of NMR, information of crystal structures and gene engineering.42) In the crystal structure of MF1, the phosphate groups of a nucleotide are tightly bound by the P-loop (GXXXGKT), which is the Walker motif A for the phosphate binding.43) In the presence of a bound nucleotide, the β sheet composed of β3 and β7 strands (Fig. 5) became longer than that in its absence through the formation of additional hydrogen bonds of Leu156/Ile306, Gly158/Ile306, and Gly158/Val308 in the P-loop region. With the conformational change induced by nucleotide binding, four new hydrogen bonds involving the P-loop residues and phosphate were generated besides those between the nucleotide and the P-loop backbone that would be too local to induce a global change. The first is the side chain of Lys164 (162 for MF1 numbering) forming hydrogen bonds with the backbone carbonyl group of Gly158 (156 for MF1) along with the phosphate group of the nucleotide. The second was that between the hydroxyl group of Thr165 (163 for MF1) and the side chain carboxyl group of Asp252 (256 for MF1) in the Walker motif B. The third was that between the carbonyl group of Ala160 (158 for MF1) and Nη-H of Arg333 (337 for MF1), and the latter further formed a hydrogen bond with Oδ of Asp311 (315 for MF1). The fourth was that between Arg191 and the γ-phosphate of AMPPNP. Are they involved in the mechanism driving the global change from the open to the closed forms?
Since the open and closed forms were similar for F1 and F1β monomer in terms of the relative orientation of the N- and C-terminal domains, we assumed that the hydrogen bonds essential for each form should be identical for the F1 and the β monomer. To identify the hydrogen bonds essential for the global conformational change of TF1β on nucleotide binding, we produced Ala-substituted proteins of β(K164A), β(T165A), β(R191A), β(D252A), β(D311A), and β(R333A).42) We used the aromatic proton signal of Tyr341 to monitor the nucleotide binding, because the aromatic ring of this residue stacked to the adenine ring of nucleotide in the crystal structure. For monitoring the conformational change of TF1β, those of Tyr148, Tyr199, and Tyr307 were used. The dissociation constant of AMPPNP (Kd) in the presence of Mg2+ was determined by the titration of Tyr341 signal, and was listed in Table 1 along with the ability of nucleotide-induced conformational change for the wild-type and mutant TF1β. While the binding affinity for β(D311A) and β(D252A) was similar to that of the wild-type β, it was reduced by two-, six- and 20-fold for β(T165A), β(R333A), and β(K164A), respectively. Therefore, the side chain of Lys164 should play a major role in the nucleotide binding as reported.44) The correlation between the nucleotide binding and the conformational change was examined by chemical shift changes of the conformation-relevant signals (Tyr148, Tyr199, and Tyr307) of the wild-type β as a function of the chemical shift change of Tyr341. In the case of Tyr148 and Tyr199, the chemical shift change was a linear function of the amount of the bound AMPPNP, ATP, or ADP, revealing that the conformational change was direct consequence of the nucleotide binding. Our previous RDC experiment on the ADP bound β subunit monomer revealed that this was the open/closed conformational change.39) In contrast, the change of the Tyr307 signal was negligible for MgADP, suggesting that the γ-phosphate of the nucleotide was involved in this conformational change.
Table 1. The dissociation constants (
Kd) and conformational change ability of the wild and mutated TF
1β monomers with MgAMPPNP and the ATPase activity of the reconstituted TF
1α
3β(mutant)
3γ complex
42)
| β subunits |
Kd/mM |
ATPase activity1 |
open/closed
conformational
change of β2 |
| Unit/mg |
% |
| Wild-type |
0.96 |
6.0 |
100 |
yes |
| (D311A) |
0.87 |
4.0 |
67 |
yes |
| (R333A) |
6.1 |
7.7 |
128 |
yes |
| (K164A) |
20.6 |
≤0.01 |
- |
no |
| (T165A) |
2.4 |
0.06 |
1 |
no |
| (D252A) |
1.0 |
0.06 |
1 |
no |
| (R191A) |
0.82 |
0.08 |
1 |
yes |
| (Y307P) |
1.3 |
≤0.01 |
- |
no |
1One unit is the activity that hydrolyzes 1 µmol of ATP/min at 298 K.
2A nucleotide-induced conformational change of the β monomer observed by 1H-NMR.
In the case of β(R191A), β(D311A), and β(R333A), the plots for the Tyr199 and Tyr307 signals were similar to those of the wild-type β, respectively. Although the AMPPNP-binding affinity of β(R333A) was diminished, this mutation scarcely affected the open/closed conformational change induced by nucleotide binding. Therefore, we concluded that the hydrogen bonds of Arg191/phosphate, Ala160/Arg333 and Arg333/Asp311 were not essential for the open/closed conformational change although they might stabilize the closed form. Arg191 would be essential for the catalytic activity (Table 1). In contrast to these mutations, β(K164A), β(T165A), and β(D252A) revealed no chemical shift changes in the nucleotide titration of the Tyr148, Tyr199, and Tyr307 signals. This result indicated that the hydrogen bond networks involving the amino group of Lys164, the hydroxyl group of Thr165 and the carboxyl group of Asp252 were essential for the open/closed conformational change. Loss of any hydrogen bond in this network shut down this conformational change in spite of the presence of a bound nucleotide. The essential roles of these residues were also confirmed in F1 by the ATPase activity of TF1α3β(mutant)3γ. The α3β(mutant)3γ complex with β(K164A), β(T165A) or β(D252A) lost the activity (Table 1).
For detailed analysis of the effects of mutations on the nucleotide-induced conformational change, segmentally isotope-labeled β(K164A), β(T165A), and β(D252A) were examined by NMR.42) 1H-15N TROSY-HSQC spectrum of each mutant protein in the absence of nucleotide was in good agreement with that of the wild-type β. It indicated that the tertiary structure of the nucleotide-free TF1β monomer was not affected by any of the mutations. The spectra in the absence and presence of 20-fold MgATP were measured. The average chemical shift perturbation (Δδave = [ΔδH2 + (ΔδN/5)2/2]1/2) of each NH signal is shown in Fig. 7C–E. Δδ stands for the chemical shift change induced by nucleotide binding. The perturbed residues (Δδave > 0.1 ppm) were 328–344 and 412–426 for β(K164A), 168–174, 328–345 and 412–430 for β(T165A), and 167–174, 330–347 and 410–430 for β(D252A). In the case of β(K164A), the perturbed region was limited to the adenine-binding pocket. For β(T165A) and β(D252A), on the other hands, a part of α helix B (αB), where Lys164 and Thr165 were located, was affected in addition to the adenine-binding pocket. Namely, each mutated β could bind a nucleotide to the adenine-binding pocket but could not induce the conformational change from the open to the closed form.
In the crystal structures of MF1 and TF1,10),31),35) the Lys164 side chain formed a hydrogen bond with the side chain of Asp252 located in the β strand 6 (β6) in the open form (Fig. 8 left), but was attracted to the phosphate group of AMPPNP or ADP and formed hydrogen bonds with the phosphate and the carbonyl group of Gly158 in the P-loop, making the C=O angle of Gly158 suitable for a hydrogen bond with NH of Val308 (Fig. 8 middle) for extension of the β strands 3 and 7. Thus, the movement of the Lys164 side chain from Asp252 to the phosphate group should be the first essential step in the conformational change process on nucleotide binding. Without this, nothing happens except for the adenine binding.

In the cases of β(T165A) and β(D252A), an additional change was observed in the α helix B as mentioned above (Fig. 7D and E), which included Glu170. According to the crystal structure of MF1 and TF1, the carboxyl group of Glu170 forms a hydrogen bond with the amide group of His415 in both open and closed forms. On the other hand, His415 is included in a stretch Phe414–Phe420, forming a part of the adenine-binding pocket, in which a triple ring stacking of Tyr341:adenine:Phe420 stabilizes the adenine binding. A perturbed stretch of Leu410–Val430 in Figs. 5 and 7 connects the α helix B, the adenine binding pocket, and the C-terminal domain (at Leu410 and Val430). Lys164 and Thr165 on the α helix B are directly involved in the nucleotide binding. The fact that the stretch 168–174 was affected by nucleotide binding reveals that the structural perturbation was propagated to the α helix B through the stretch of Leu410–Val430, suggesting a strong structural correlation among the nucleotide binding site, stretch 410–430, and αB. They should work as a coupling device between the catalytic and C-terminal domains in the open/closed conformational change. Actually, a sliding of the α helix B was identified as a major factor in formation of the closed form by free energy simulations.45) The sliding of the helix would accompany a movement of the C-terminal domain through this coupling device. Without Thr165 or Asp252, the sliding of the whole helix could not take place.
The Kd values of β(T165A) and β(D252A) were similar to that of the wild type (Table 1). Nevertheless, the nucleotide binding failed to induce an open/closed conformational change in the absence of either Thr165 or Asp252. The side chain of Thr165 formed a hydrogen bond with Glu201 in the open form and with Asp252 in the closed form in the crystal structures of MF1 and TF1.10),31),35) Namely, formation of the T165/D252 hydrogen bond should be the second essential step to the closed form. Since the side chain of Thr is much shorter than that of Lys, the Thr165/Asp252 hydrogen bonding brings up αB/P-loop together with the C-terminal domain, realizing the closed form (Fig. 8 right). For stabilization of the closed form, formation of the β-sheet structure between Leu156-Gly158 (Leu154-Gly156 for MF1) and Ile306-Val308 (Ile310-Val312) is indispensable, because a secondary-structure breaking mutation, Y307P, shut down the open/closed conformational change and ATPase activity of F1 (Table 1). Extension of the β-sheet should take place in concert with the Thr165/Asp252 hydrogen-bond formation. A model of stepwise mechanism of the conformational conversion from the open to the closed form base on our analysis is schematically presented in Fig. 8. The intermediate conformation in the figure has the features similar to those of the half-closed form of the β subunit in a crystal structure of MF1.46) In the half-closed form bound with ADP·AlF3, Oγ of Thr165 is separated from Oδ of Asp252 by 3.43 Å (2.50 Å for βTP) and from Glu201 by over 10 Å. This mechanism was supported by detailed free energy simulations.45) The open/closed conformational change must be driven by a collective fluctuation of the β monomer, and the interactions mentioned above should thermodynamically stabilize the closed form. Actually, the relative order parameter of the C-terminal domain was found as S = 0.39 in the RCD experiment, suggesting the presence of slow collective motions in the β monomer.47)
It should be noted that the Tyr307 aromatic signal shifted on MgATP or MgAMPPNP binding, but not on MgADP binding. However, the crystal structures reported so far gave no evidence for the involvement of Tyr307 in the interaction with γ-phosphate of MgAMPPNP. It was reported that although a chemical modification of Tyr307 of single β in F1 suppressed ATPase activity for TF1 and EF1,48),49) a simple mutation such as Y307C did not impair the ATPase activity,50) suggesting that the ring itself was not an essential element for the activity. In the crystal structure of MF1 and TF1,10),31),35) the aromatic ring of Tyr307 and the guanidyl group of Arg256 (Arg260 for MF1) are stacked to each other for βTP and βDP. Arg256 is one of the conserved residues. Ahmad and Senior reported that Arg256 (Arg246 for EF1) was critical for formation of transition state in catalysis and for recognition of the cleaved γ-phosphate in the EF1 on the basis of their mutation experiments and reported crystal structures.49) They designated the area including Arg256 and Tyr307 as the Pi-binding pocked. This was also confirmed by a recent crystal structure.51) The chemical shift change of the Tyr307 ring proton described above suggested the interaction of the γ-phosphate of ATP with this pocket in the closed form of the β monomer. This might be explained by the difference in the closed form for the β subunit in F1 and in monomer as presented in Fig. 6. The relative orientation angles were 119° and 110° for the former and latter, respectively. Since the structure of β monomer is more closed than that of β in F1, the γ-phosphate of ATP may come close to the Pi-binding pocket without hydrolysis. Furthermore, the change of Tyr307 aromatic signal turned out to be an indirect effect of nucleotide binding from the Tyr307-vs-Tyr341 plots. Therefore, there should be a local conformational change following the global change, bringing the γ-phosphate of ATP close to the Pi-binding pocket. The final conformation would be similar to a conformation in the catalytic transition state suggested by Ahmad and Senior.
5. Structure and property of β subunit in TF1-ATPase in solution as studied by NMR
The information obtained with TF1β monomer should be examined with TF1. Although this was a challenge for NMR because of its huge molecular mass, this was successfully carried out.52),53) For better resolution, a segmentally isotope-labeled TF1β was used. For better efficiency, reconstitution of the TF1α3β3γ complex was carried out by a denaturation and refolding method in the presence of 10 mM MgAMPPNP and 3 mM MgADP. Furthermore, the C-terminal domain-truncated TF1ε subunit (residues 1–90, εΔC) and 200 mM arginine were added to suppress sample aggregation during NMR measurements. Thus, a segmentally isotope-labeled 360 kDa α3β3γεΔC complex (TF1′) successfully provided well-resolved CRINEPT NMR spectra. The cross-relaxation enhanced polarization transfer (CRINEPT) pulse sequences were developed by Riek, Wüthrich et al. for NMR analysis of large molecular and supramolecular structures.54),55)
The CRINEPT-HMQC-TROSY 15N-1H correlation spectrum of α3β*(391–473)3γεΔC is presented in Fig. 9. Here, β*(391–473) stands for the β subunit segmentally isotope-labeled in the residues 391–473. Signals were unexpectedly well resolved. Most chemical shifts of intense peaks were similar to those for the β*(391–473) monomer in the open form. Therefore, assignment could be performed on the basis of that of the β*(391–473) monomer. The assignment for α3β*(391–473)3γεΔC is shown in Fig. 9. Number of residues with Δδave (average chemical shift difference) between β monomer and TF1′ complex > 0.05 ppm was eleven, which included only two residues (V416 and F420) from the stretch sensitive to the open/closed conformational change (residues 410–428).39),42) This observation provided an important conclusion that at least one of three β subunits in TF1′ took the open form (βE) in solution and its structure was similar to that of the β monomer. The well-resolved spectrum indicated that the C-terminal domain of the β subunit in the open form was dynamically mobile even in TF1′. In contrast, the CRINEPT-HMQC-TROSY 15N-1H correlation spectrum of α3β*(1–124)3γεΔC was poor, suggesting that the N-terminal domain was rigid in TF1′.53)

On closer inspection of the spectrum some residues provided multiple signals, suggesting presence of the closed forms of β in TF1′. Residues showing large chemical shift differences (Δav > 0.1 ppm) between the open and closed forms in the TF1′ complex were F414, V416, E418, Q419, Q423, S426, and G457. Although most of them were located in the global-conformation sensitive stretch 410–428, they might be also sensitive to interactions with the neighboring α and/or γ subunits. However, since F414 and V416 are located inside the protein, they would be barely affected by the interaction with the α or γ subunit. The chemical shifts of their closed-form signals were different from those of TF1β monomer. This strongly suggested that the closed form of the β subunit in the TF1′ complex was different from that of the β monomer. This is in good agreement with the difference in the relative orientation of the N- and C-terminal domains of the nucleotide bound β between the TF1β monomer in solution and MF1 in crystal (Fig. 6).
The mechanism underlying the rotational catalysis has been extensively discussed on the basis of a variety of crystal structures and single molecule analysis.7),14),16),51),56) Major conformational states of the α3β3γ subcomplex along the rotation of the γ subunit have been almost established, although there are still some conflicts. The largest rotation of the γ subunit (80° in TF1) is induced by ATP-binding. The driving force of this rotation is the conformational change from the open to the closed form and the interaction with γ. The dynamic mechanism underlying the open/closed conformational change was elucidated by our work summarized in this review (Fig. 8) and a free energy and molecular dynamics simulations by Ikeguchi’s group.45),57) The conformational change proceeded stepwise. This was also consistent with a series of crystal structures. Representative structures were one in the ground state (GS),10),31) one of transition-state analogue (TS),46) one in pre-nucleotide-release state (PNR),58) and one in pre-phosphate-release state (PPR)51) of the bovine MF1-ATPase (designation by Walker and his colleagues).7) Although the relationship with the rotational substeps is still not yet well established, they include interesting structures of β. GS comprises typical three states of β, namely, βE (empty, open form), βTP (MgATP analogue bound, closed form), and βDP (MgADP bound, closed form). In TS, βE takes a half-closed form with MgADP and phosphate analogue bound. While PNR includes an open form βE with ADP bound, PPR does an open form βE with phosphate analogue bound. This fact suggests that the structure of C-terminal domain of βE is flexible in comparison with the closed structures. It is evident from the NMR spectrum in Fig. 9 that the C-terminal domain of βE is mobile. The RDC experiment revealed that there is a collective fluctuation of the β monomer. Molecular dynamics and free energy simulations also revealed that the β monomer underwent low-frequency motions with propensity for global bending motions.45),57) Presence of a half-closed form is consistent with the stepwise conformational change of TF1β monomer on nucleotide binding shown in Fig. 8. Stabilization of the intermediate structure would be warranted by the interaction with the γ subunit. Even though there are large number of crystal structures of F1, there is no structure which indicates direct interactions between the γ phosphate of ATP analogue and the Pi-binding pocket including Arg256. In view of biochemical results on Arg25649) and our observation on Tyr307, this kind of interaction would take place as an activated state in hydrolysis. It would not be easy to capture the activated state, because of its nature of unstable snapshot.
6. Structural basis of ATP-dependent suppression of ATP hydrolysis by the TF1ε subunit
The ε subunit forms a ternary complex with the γ subunit and the c subunit ring to convert the rotation of the c-ring in Fo to that of γ in F1 or vice versa. It is also known as an endogenous inhibitor of ATP hydrolysis activity of F1 and FoF1 in bacteria and plants,59)–61) although its effect on ATP synthesis by FoF1 is not yet well understood.62) According to crystal63) and solution64) structures, the isolated ε from Escherichia coli (EF1ε) comprised an N-terminal β-sandwich domain and a C-terminal domain (CTD). The two helices of CTD were folded into a hairpin form on top of the N-terminal domain (NTD). This structure was called the “down-state” because whole CTD was located at the bottom of the γ subunit (Fig. 1). However, cross-linking experiments found that CTD interacted with multiple subunits of F1.65),66) This could not be explained by just the down-state structure. Furthermore, cross-linking67) and fluorescence resonance energy transfer (FRET)68) studies on TF1 provided evidence for full extension of the CTD helices along the γ subunit under the conditions of ATPase-inhibition. Thus, the conformation responsible for the inhibition of ATP hydrolysis was called the “up-state”. The crystal structure of EF1 γ′ε complex (γ′: truncated-γ) hinted such an ε conformation.69) The efficiency of the conversion to the down-state was regulated by ATP concentration and membrane potential.67) Actually, specific ATP binding was observed for the isolated ε subunits of TF170) and Bacillus subtilis F1,71) although it was not clear for EF1ε. Since ATP-binding was assumed to be a critical factor regulating inhibitory function of TF1ε, the role of ATP in the conformational conversion of TF1ε was investigated by X-ray crystallography and NMR.72)
TF1ε was tried to be crystallized in the presence and absence of ATP.72) Crystals could be obtained only in the presence of ATP. The crystal structure of TF1ε presented in Fig. 10 (blue) carried an ATP, which was the most important distinction from the crystal structure of the folded EF1ε.63) No Mg2+ was found. Its overall backbone structure was similar to that of the folded EF1ε. TF1ε was composed of two domains, namely, an N-terminal β sandwich (residues 1–84) and a C-terminal α-helical domain (90–133). A 310 helix (85–87) was inserted in the short loop linking NTD and CTD. NTD was composed of ten β strands. Five strands (β1: 4–10, β2: 13–20, β5: 41–45, β8: 66–71, and β9: 74–79) formed one antiparallel β sheet except for a parallel pair, β1 and β9, and the other five strands (β3: 22–27, β4: 30–34, β6: 47–54, β7: 57–64, and β10: 82–84) formed another antiparallel β sheet. CTD comprised two α-helices (α1: 90–104 and α2: 113–130), and a short loop linking these α-helices. The hydrophobic contact surface between two helices was formed by an ‘alanine zipper’-like structure, as in the case of EF1ε. The relative orientation of NTD and CTD was slightly different for TF1ε and EF1ε. If TF1ε was superimposed on NTD of EF1ε, the CTD showed deviation by 8°. When NTD and CTD of two ε subunits were superimposed independently, they fitted well to each other except for the loop in CTD. A well-defined ATP-binding site was found in CTD. The ATP in TF1ε took on an unusual structure, in which the triphosphate chain bended at the β-phosphate. This ATP-specific binding site involved at least four residues. The carbonyl and amide groups of Asp89 specifically recognized the adenine ring through Watson-Crick type hydrogen bonds. The π electron system of the guanidyl group of Arg92 stacked on the adenine ring. Arg92 also formed hydrogen bonds with the ribose and γ-phosphate. These two residues should play key roles in specific recognition of ATP. Furthermore, Ile88 and Ala93 were conserved in the same bacterial ε subunits, and contributed to the adenine-binding pocket of TF1ε. Therefore, I(L)DXXRA (X, any amino acid residue) would be a new ATP-binding motif. Crystallization of ATP-free TF1ε was unsuccessful.
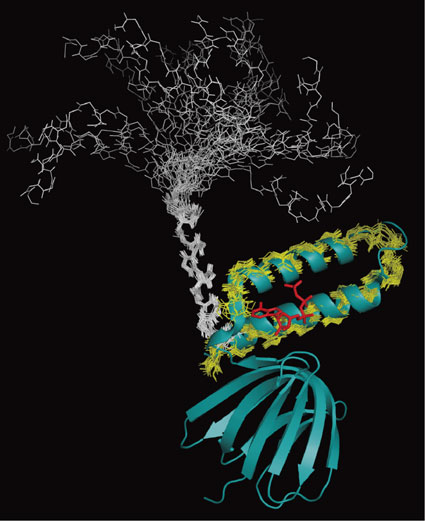
The interaction of TF1ε with ATP in solution was examined by NMR. 2D 1H-15N HSQC spectra of uniformly 15N labeled TF1ε were measured in the presence and absence of ATP. The two spectra were significantly different. In the presence of ATP, the 1H-15N HSQC spectrum was well resolved. All NH resonances except for those of Pro and the N-terminal two residues could be assigned. In the absence of ATP, however, the peak dispersion was worse than that in the presence of ATP. Nevertheless, the NH resonances could be completely assigned except for Pro, the C-terminal six and the N-terminal two residues, which were not observed. Then, the solution structures of residues 88–133 and 88–127 of whole protein in the presence and absence of ATP were determined, respectively. Since NTD and CTD were structurally independent and the ATP-binding site was found in CTD in the crystal structure, we focused our attention on CTD. According to the crystal structure, CTD of TF1ε can be defined as residues 90–133. The conformations of the CTD backbone in the presence of ATP were well converged and very similar to the crystal structure (Fig. 10, yellow lines superimposed with its crystal structure). The backbone RMSD of the 20 best structures (residues 90–131) was 0.50 Å. The Asp89 NH signal was observed at 11 ppm, which could be ascribed to hydrogen bonding with the adenine of ATP as in the crystal structure. In contrast, the best 20 out of 100 structures in the absence of ATP did not converge with the backbone RMSD > 4 Å (Fig. 10, white lines on top of the NTD β-sandwich starting from the linker). Nevertheless, the conformation was basically helical with two well defined helices (residues 90–102 and 113–117). When each helical region was superimposed, they were well defined with backbone RMSD of 0.39 Å and 0.28 Å, respectively (see Fig. 10 for the former). Inter-helix NOEs were not observed. On the basis of the structural analysis, it can be concluded that although the ATP-bound hairpin structure of TF1ε CTD held in solution, the helices were not folded anymore in the absence of ATP. Analysis of relaxation parameters (NOE, T1 and T2) of TF1ε also supported this conclusion. In the case of the ATP-bound TF1ε, NTD and CTD formed a rigid body through strong interdomain interactions mediated by ATP. In contrast, the two domains revealed completely different dynamic properties in the absence of ATP. While NTD was a rigid body, CTD got more and more flexible with approaching to the C terminus. The conformational change induced by ATP binding can be inferred from Fig. 10.
We also examined ATP binding to the isolated EF1ε by NMR. ATP titration experiments revealed that Asp90, Arg93, and Ala94 (corresponding to Asp89, Arg92, and Ala93 for TF1ε) were included in the major affected residues. Especially, the NH signal of Asp90 disappeared at the molar ratio of ATP/ε = 1. This result strongly suggested that the ATP binding site of EF1ε was the same as that of TF1ε, although the affinity is significantly lower (Kd = 22 ± 1 mM at 25 °C). Kd of the TF1ε/ATP complex was 1.4 µM and 0.67 mM at 36 and 65 °C,67) respectively. The latter is the physiological temperature for TF1ε.
The structure of ε in the up-state was determined later for EF1α3β3γε73) and TF1α3β3γε35) by X-ray crystallography. As can be seen in Fig. 11, εCTD was basically extended along the γ subunit in both structures. While two helices of TF1ε CTD lined up almost in a straight manner, the first helix of EF1ε CTD shifted to the β barrel of γ, forming a hook structure. In both cases, the second helix of ε CTD was inserted into the central cavity of F1, interacting with αDP, αE, βDP, βTP, and γ. An important consequence of these interactions was non-closed conformations of βDP (Fig. 11). It took on the half-closed and open conformations in EF1 and TF1, respectively. This would cause the inhibition of ATP hydrolysis in F1. Why the structures of EF1ε CTD and TF1ε CTD are different? It is not clear so far. However, this may be ascribed to the difference in the role of ε inhibition in two bacteria. There is a clear difference in ATP-binding affinity. In the case of TF1ε, ATP binding converts the extended CTD (the up-state) to the folded one (the down-state). We also found the ATP binding site in EF1ε CTD as mentioned above. Actually, FRET experiments suggested that 1 mM MgATP or 1 mM MgAMPPNP induced generation of the folded conformation to some extent, although the extended conformation was the dominant population in the absence of nucleotides and in the presence of ADP and Pi.74) The dissociation constants of the ε/ATP complex under physiological conditions were 22 and 0.67 mM for EF1ε and TF1ε, respectively. Namely, efficiency of ATP in conformational conversion of εCTD depends on the bacterial species presumably because of different survival strategies. In the case of E. coli, the extended form of εCTD turned out to inhibit not only ATP-hydrolysis but also ATP-synthesis.62) Nevertheless, the major population of εCTD is supposed to take the extended conformation.62),74) Thus, it is possible for EF1 to synthesize ATP in the presence of the extended εCTD under the physiological conditions. The hook structure of EF1ε would be more flexible to cope with effects of the rotation driven by the proton motive force (pmf) during the ATP synthesis than the structure of TF1ε. The intermediate structure proposed on the basis of EF1α3β3γε crystal structure63) may be included in this flexibility as well. In the case of TF1, εCTD would mainly take the folded conformation in the presence of physiological concentration of ATP, although the extended conformation was reported to inhibit the ATP synthesis to some extent as well.75) There is no ATP-binding motif in the sequences of ε subunits of chloroplasts and cyanobacteria.63),76) Their ε subunits revealed very strong inhibition of ATP hydrolysis in vitro, suggesting that most εCTDs took on the extended structure under the physiological conditions.76) Since the structure of an isolated ε subunit from a cyanobacterium, Thermosynecoccus elongatus, determined by NMR was found in a folded structure,76) some driving forces including pmf should work for the conformational conversion.

7. Concluding remark
In this review, I have tried to convince the readers that solution NMR provided unique information for understanding the mechanism of the rotational catalysis in F1-ATPase with the help of crystal structures. Using a variety of isotope labeling methods, we could get reasonable spectra even for 360 kDa TF1′. Segmental isotope-labeling of TF1β enabled us to get the information from most residues of TF1β. It turned out that the conformational change from the open to the closed form of the β subunit on nucleotide binding was its intrinsic property and could work as a driving force of the rotational catalysis. The conformational change does not need energy. The conformational change was initiated by nucleotide binding and driven in a stepwise manner by switching of the hydrogen bond networks involving Walker A and B motifs. Segmentally labeled TF1 provided a well resolved NMR spectra, revealing while the open form of β was identical for the TF1β monomer and TF1, its closed form could be different. The activated state in the hydrolysis reaction was suggested in connection with the closed form of the β monomer. ATP-binding was also found to be a critical factor in the regulation of TF1 activity by the ε subunit. The structures of the ATP-bound and ATP-free TF1ε were determined by X-ray crystallography and NMR, which revealed a structural conversion of TF1ε depending on ATP concentration. Thus, the works reviewed here contributed to elucidation of the basic mechanisms underlying important elements of rotational ATP hydrolysis by F1.
Profile

Hideo Akutsu was born in 1944 in Tokyo. He graduated from the University of Tokyo in 1967 and received Doctor of Science degree in 1973. He was appointed as an assistant professor at Institute for Protein Research, Osaka University in 1972 to work with Prof. Y. Kyogoku, then moved to Faculty of Engineering, Yokohama National University as an associate professor in 1985. Meanwhile, he joined the group of Prof. J. Seelig at University of Basel as a research fellow of Japan Society for the Promotion of Science (JSPS) and EMBO from 1978 to 1980 to work on membrane structure by solid-state NMR. He became a professor in 1991 at the same university and moved back to Institute for Protein Research in 2000. He served as director of the institute from 2004 to 2006. He was appointed to be a WCU professor at Seoul National University from 2009 to 2013, and to be director of the JSPS Stockholm Office from 2014 to 2016. His research has been focusing on NMR structural biology of lipids and proteins involved in energy transduction. He elucidated electron transport mechanisms in a variety of biological processes involving tetraheme cytochromes, and the mechanism underlying the nucleotide-induced conformational change of the β subunit, driving the rotational catalysis of F1-ATPase. In order to analyze huge biomolecular complexes such as FoF1-ATP synthase, he has developed biological solid-state NMR, which was used for the investigation of Foc-subunit ring in membranes to elucidate the mechanism underlying proton translocation across the membrane.
Acknowledgments
The author would like to thank Drs. Hiromasa Yagi and Kaeko Tozawa for their excellent works, and Profs. Masasuke Yoshida and Tomitake Tsukihara for the fruitful collaborations with them.
References
- 1) Boyer, P.D. (1997) The ATP synthase — a splendid molecular machine. Annu. Rev. Biochem. 66, 717–749.
- 2) Yoshida, M., Muneyuki, E. and Hisabori, T. (2001) ATP synthase — a marvellous rotary engine of the cell. Nat. Rev. Mol. Cell Biol. 2, 669–677.
- 3) Senior, A.E., Nadanaciva, S. and Weber, J. (2002) The molecular mechanism of ATP synthesis by F1Fo-ATP synthase. Biochim. Biophys. Acta 1553, 188–211.
- 4) Futai, M. (2006) Our research on proton pumping ATPases over three decades: their biochemistry, molecular biology and cell biology. Proc. Jpn. Acad., Ser. B 82, 416–438.
- 5) Dimroth, P., Von Ballmoos, C. and Meier, T. (2006) Catalytic and mechanical cycles in F-ATP synthase. EMBO Rep. 7, 276–282.
- 6) Kagawa, Y. (2010) ATP synthase: from single molecule to human bioenergetics. Proc. Jpn. Acad., Ser. B 86, 667–693.
- 7) Walker, J.E. (2013) The ATP synthase: the understood, the uncertain and the unknown. Biochem. Soc. Trans. 41, 1–16.
- 8) Hisabori, T., Sunamura, E., Kim, Y. and Konno, H. (2013) The chloroplast ATP synthase features the characteristic redox regulation machinery. Antioxid. Redox Signal. 19, 1846–1854.
- 9) Nakanishi-Matsui, M., Sekiya, M. and Futai, M. (2016) ATPase from Escherichia coli: Mechanism of rotational catalysis, and inhibition with the ε subunit and phytopolyphenols. Biochim. Biophys. Acta 1857, 129–140.
- 10) Abrahams, J.P., Leslie, A.G.W., Lutter, R. and Walker, J.E. (1994) Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621–628.
- 11) Noji, H., Yasuda, R., Yoshida, M. and Kinosita, K. Jr. (1997) Direct observation of the rotation of F1-ATPase. Nature 386, 299–302.
- 12) Yasuda, R., Noji, H., Yoshida, M., Kinosita, K. Jr. and Itoh, H. (2001) Resolution of distinct rotational substeps by submillisecond kinetic analysis of F1-ATPase. Nature 410, 898–904.
- 13) Shimabukuro, K., Yasuda, R., Muneyuki, E., Hara, K.Y., Kinoshita, K. Jr. and Yoshida, M. (2003) Catalysis and rotation of F1 motor: cleavage of ATP at the catalytic site occurs in 1 ms before 40° substep rotation. Proc. Natl. Acad. Sci. U.S.A. 100, 14731–14736.
- 14) Adachi, K., Oiwa, K., Nishizawa, T., Furuike, S., Noji, H., Itoh, H., Yoshida, M. and Kinoshita, K. Jr. (2007) Coupling of rotation and catalysis in F1-ATPase revealed by single-molecule imaging and manipulation. Cell 130, 309–321.
- 15) Watanabe, R., Iino, R. and Noji, H. (2010) Phosphate release in F1-ATPase catalytic cycle follows ADP release. Nat. Chem. Biol. 6, 814–820.
- 16) Suzuki, T., Tanaka, K., Wakabayashi, C., Saita, E. and Yoshida, M. (2014) Chemomechanical coupling of human mitochondrial F1-ATPase motor. Nat. Chem. Biol. 10, 930–936.
- 17) Fillingame, R.H., Angevine, C.M. and Dmitriev, O.Y. (2003) Mechanics of coupling proton movements to c-ring rotation in ATP synthase. FEBS Lett. 555, 29–34.
- 18) Stock, D., Leslie, A.G. and Walker, J.E. (1999) Molecular architecture of the rotary motor in ATP synthase. Science 286, 1700–1705.
- 19) Seelert, H., Poetsch, A., Dencher, N.A., Engel, A., Stahlberg, H. and Muller, D.J. (2000) Proton-powered turbine of a plant motor. Nature 405, 418–419.
- 20) Mitome, N., Suzuki, T., Hayashi, S. and Yoshida, M. (2004) Thermophilic ATP synthase has a decamer c-ring: indication of noninteger 10:3 H+/ATP ratio and permissive elastic coupling. Proc. Natl. Acad. Sci. U.S.A. 101, 12159–12164.
- 21) Meier, T., Polzer, P., Diederichs, K., Welte, W. and Dimroth, P. (2005) Structure of the rotor ring of F-type Na+-ATPase from Ilyobacter tartaricus. Science 308, 659–662.
- 22) Matthies, D., Preiss, L., Klyszejko, A.L., Muller, D.J., Cook, G.M., Vonck, J. and Meier, T. (2009) The c13 ring from a thermoalkaliphilic ATP synthase reveals an extended diameter due to a special structural region. J. Mol. Biol. 388, 611–618.
- 23) Watt, I.N., Montgomery, M.G., Runswick, M.J., Leslie, A.G. and Walker, J.E. (2010) Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc. Natl. Acad. Sci. U.S.A. 107, 16823–16827.
- 24) Nesci, S., Trombetti, F., Ventrella, V. and Pagrialani, A. (2016) The c-ring of the F1Fo-ATP synthase: facts and perspective. J. Membr. Biol. 249, 11–21.
- 25) Miwa, K. and Yoshida, M. (1989) The α3β3 complex, the catalytic core of F1-ATPase. Proc. Natl. Acad. Sci. U.S.A. 86, 6484–6487.
- 26) Yoshida, M., Poser, J.W., Allison, W.S. and Esch, F.S. (1981) Identification of an essential glutamic acid residue in the β subunit of the adenosine triphosphatase from the thermophilic bacterium PS3. J. Biol. Chem. 256, 148–153.
- 27) Ohtsubo, M., Yoshida, M., Ohta, S., Kagawa, Y., Yohda, M. and Date, T. (1987) In vitro mutated β subunits from the F1-ATPase of the thermophilic bacterium, PS3, containing glutamine in place of glutamic acid in positions 190 or 201 assembles with the α and γ subunits to produce inactive complexes. Biochem. Biophys. Res. Commun. 146, 705–710.
- 28) Tozawa, K., Ohbuchi, H., Yagi, H., Amano, T., Matsui, T., Yoshida, M. and Akutsu, H. (1996) Unusual pKa of the carboxylate at the putative catalytic position of the thermophilic F1-ATPase β subunit determined by 13C-NMR. FEBS Lett. 397, 122–126.
- 29) Kohda, D., Sawada, T. and Inagaki, F. (1991) Characterization of pH titration shifts for all the nonlabile proton resonances in a protein by two-dimensional NMR: The case of mouse epidermal growth factor. Biochemistry 30, 4896–4900.
- 30) Amano, T., Tozawa, K., Yoshida, M. and Murakami, H. (1994) Spatial precision of a catalytic carboxylate of F1-ATPase β subunit probed by introducing different carboxylate-containing side chains. FEBS Lett. 348, 93–98.
- 31) Bowler, M.W., Montgomery, M.G., Leslie, A.G.W. and Walker, J.E. (2007) Ground state structure of F1-ATPase from bovine heart mitochondria at 1.9 Å resolution. J. Biol. Chem. 282, 14238–14242.
- 32) Beke-Somfai, T., Lincoln, P. and Nordén, B. (2011) Double-lock ratchet mechanism revealing the role of αSER-344 in FoF1 ATP synthase. Proc. Natl. Acad. Sci. U.S.A. 108, 4828–4833.
- 33) Hayashi, S., Ueno, H., Shaikh, A.R., Umemura, M., Kamiya, M., Ito, Y., Ikeguchi, M., Komoriya, Y., Iino, R. and Noji, H. (2012) Molecular mechanism of ATP hydrolysis in F1-ATPase revealed by molecular simulations and single-molecule observations. J. Am. Chem. Soc. 134, 8447–8454.
- 34) Shirakihara, Y., Leslie, A.G.W., Abrahams, J.P., Walker, J.E., Ueda, T., Sekimoto, Y., Kambara, M., Saika, K., Kagawa, Y. and Yoshida, M. (1997) The crystal structure of the nucleotide-free α3β3 oligomer of F1-ATPase from the thermophilic Bacillus PS3 is a symmetric trimer. Structure 5, 825–836.
- 35) Shirakihara, Y., Shiratori, A., Tanikawa, H., Nakasako, M., Yoshida, M. and Suzuki, T. (2015) Structure of a thermophilic F1-ATPase inhibited by an ε-subunit: deeper insight into the ε-inhibition mechanism. FEBS J. 282, 2895–2913.
- 36) Tozawa, K., Sekino, N., Soga, M., Yagi, H., Yoshida, M. and Akutsu, H. (1995) Conformational dynamics monitored by His-179 and His-200 of isolated thermophilic F1-ATPase β subunit which reside at the entrance of the “conical tunnel” in Holoenzyme. FEBS Lett. 376, 190–194.
- 37) Yagi, H., Tozawa, K., Sekino, N., Iwabuchi, T., Yoshida, M. and Akutsu, H. (1999) Functional conformational changes in the TF1 ATPase β subunit probed by twelve tyrosine residues. Biophys. J. 77, 2175–2183.
- 38) Tozawa, K., Yagi, H., Hisamatsu, K., Ozawa, K., Yoshida, M. and Akutsu, H. (2001) Functions and ATP-binding responses of the twelve histidine residues in the TF1-ATPase β subunit. J. Biochem. 130, 527–533.
- 39) Yagi, H., Tsujimoto, T., Yamazaki, T., Yoshida, M. and Akutsu, H. (2004) A conformational change of H+-ATPase β monomer revealed on segmental isotope labeling NMR spectroscopy. J. Am. Chem. Soc. 126, 16632–16638.
- 40) Yamazaki, T., Otomo, T., Oda, N., Kyogoku, Y., Uegaki, K., Ito, N., Ishino, Y. and Nakamura, H. (1998) Segmental isotope labeling for protein NMR using peptide splicing. J. Am. Chem. Soc. 120, 5591–5592.
- 41) Xu, R., Ayers, B., Cowburn, D. and Muir, T.W. (1999) Chemical ligation of folded recombinant proteins: Segmental isotopic labeling of domains for NMR studies. Proc. Natl. Acad. Sci. U.S.A. 96, 388–393.
- 42) Yagi, H., Kajiwara, N., Iwabuchi, T., Izumi, K., Yoshida, M. and Akutsu, H. (2009) Stepwise propagation of the ATP-induced conformational change of the F1-ATPase β subunit revealed by NMR. J. Biol. Chem. 284, 2374–2382.
- 43) Walker, J.E., Saraste, M., Runswick, M.J. and Gay, N.J. (1982) Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1, 945–951.
- 44) Löbau, S., Weber, J., Wilke-Mounts, S. and Senior, A.E. (1997) F1-ATPase, roles of three catalytic site residues. J. Biol. Chem. 272, 3648–3656.
- 45) Ito, Y., Oroguchi, T. and Ikeguchi, M. (2011) Mechanism of the conformational change of the F1-ATPase β subunit revealed by free energy simulations. J. Am. Chem. Soc. 133, 3372–3380.
- 46) Menz, R.I., Walker, J.E. and Leslie, A.G.W. (2001) Structure of Bovine Mitochondrial F1-ATPase with nucleotide bound to all three catalytic sites: Implications for the mechanism of rotary catalysis. Cell 106, 331–341.
- 47) Tolman, J.R., Flanagan, J.M., Kennedy, M.A. and Prestegard, J.H. (1997) NMR evidence for slow collective motions in cyanometmyoglobin. Nat. Struct. Biol. 4, 292–297.
- 48) Yoshida, M. and Allison, W.S. (1990) The ATPase activity of the α3β3 complex of the F1-ATPase of the thermophilic bacterium PS3 is inactivated on modification of tyrosine 307 in a single β subunit by 7-chloro-4-nitorobenzofuran. J. Biol. Chem. 265, 2483–2487.
- 49) Ahmad, Z. and Senior, A.E. (2004) Mutagenesis of residue βArg-246 in the phosphate-binding subdomain of catalytic sites of Escherichia coli F1-ATPase. J. Biol. Chem. 279, 31505–31513.
- 50) Odaka, M., Kiribuchi, K., Allison, W.S. and Yoshida, M. (1993) In vivo affinity label of a protein expressed in Escherichia coli coenzyme A occupied the AT(D)P binding site of the mutant F1-ATPase β subunit (Y307C) through a disulfide bond. FEBS Lett. 336, 231–235.
- 51) Bason, J.V., Montgomery, M.G., Leslie, A.G.W. and Walker, J.E. (2015) How release of phosphate from mammalian F1-ATPase generates a rotary substep. Proc. Natl. Acad. Sci. U.S.A. 112, 6009–6014.
- 52) Kobayashi, M., Yagi, H., Yamazaki, T., Yoshida, M. and Akutsu, H. (2008) Dynamic inter-subunit interactions in thermophilic F1-ATPase subcomplexes studied by cross-correlated relaxation-enhanced polarization transfer NMR. J. Biomol. NMR 40, 165–174.
- 53) Kobayashi, M., Akutsu, H., Suzuki, T., Yoshida, M. and Yagi, H. (2010) Analysis of the open and closed conformations of the β subunits in thermophilic F1-ATPase by solution NMR. J. Mol. Biol. 398, 189–199.
- 54) Riek, R., Wider, G., Pervushin, K. and Wüthrich, K. (1999) Polarization transfer by cross-correlated relaxation in solution NMR with very large molecules. Proc. Natl. Acad. Sci. U.S.A. 96, 4918–4923.
- 55) Riek, R., Fiaux, J., Bertelsen, E.B., Horwich, A.L. and Wüthrich, K. (2002) Solution NMR techniques for large molecular and supramolecular structures. J. Am. Chem. Soc. 124, 12144–12153.
- 56) Watanabe, R. and Noji, H. (2014) Timing of inorganic phosphate release modulates the catalytic activity of ATP-driven rotary motor protein. Nat. Commun. 5, 3486.
- 57) Ito, Y. and Ikeguchi, M. (2010) Molecular dynamics simulations of the isolated β subunit of F1-ATPase. Chem. Phys. Lett. 490, 80–83.
- 58) Rees, D.M., Montgomery, M.G., Leslie, A.G.W. and Walker, J.E. (2012) Structural evidence of a new catalytic intermediate in the pathway of ATP hydrolysis by F1-ATPase from bovine heart mitochondria. Proc. Natl. Acad. Sci. U.S.A. 109, 11139–11143.
- 59) Laget, P.P. and Smith, J.B. (1979) Inhibitory properties of endogenous subunit ε in the Escherichia coli F1 ATPase. Arch. Biochem. Biophys. 197, 83–89.
- 60) Sternweis, P.C. and Smith, J.B. (1980) Characterization of the inhibitory (ε) subunit of the proton-translocating adenosine triphosphatase from Escherichia coli. Biochemistry 19, 526–531.
- 61) Kato-Yamada, Y., Bald, D., Koike, M., Motohashi, K., Hisabori, T. and Yoshida, M. (1999) ε Subunit, an endogenous inhibitor of bacterial F1-ATPase, also inhibits FoF1-ATPase. J. Biol. Chem. 274, 33991–33994.
- 62) Iino, R., Hasegawa, R., Tabata, K.V. and Noji, H. (2009) Mechanism of inhibition by C-terminal α-helices of the ε subunit of Escherichia coli FoF1-ATP synthase. J. Biol. Chem. 284, 17457–17464.
- 63) Uhlin, U., Cox, G.B. and Guss, J.M. (1997) Crystal structure of the ε subunit of the proton-translocating ATP synthase from Escherichia coli. Structure 5, 1219–1230.
- 64) Wilkens, S., Dahlquist, F.W., McIntosh, L.P., Donaldson, L.W. and Capaldi, R.A. (1995) Structural features of the ε subunit of the Escherichia coli ATP synthase determined by NMR spectroscopy. Nat. Struct. Biol. 2, 961–967.
- 65) Dallmann, H.G., Flynn, T.G. and Dunn, S.D. (1992) Determination of the 1-ethyl-3-[(3-dimethylamino)propyl]-carbodiimide-induced cross-link between the β and ε subunits of Escherichia coli F1-ATPase. J. Biol. Chem. 267, 18953–18960.
- 66) Tsunoda, S.P., Rodgers, A.J.W., Aggeler, R., Wilce, M.C.J., Yoshida, M. and Capaldi, R.A. (2001) Large conformational changes of the ε subunit in the bacterial F1Fo-ATP synthase provide a ratchet action to regulate this rotary motor enzyme. Proc. Natl. Acad. Sci. U.S.A. 98, 6560–6564.
- 67) Suzuki, T., Murakami, T., Iino, R., Suzuki, J., Ono, S., Shirakihara, Y. and Yoshida, M. (2003) FoF1-ATPase/synthase is geared to the synthesis mode by conformational rearrangement of ε subunit in response to proton motive force and ADP/ATP balance. J. Biol. Chem. 278, 46840–46846.
- 68) Iino, R., Murakami, T., Iizuka, S., Kato-Yamada, Y., Suzuki, T. and Yoshida, M. (2005) Real-time monitoring of conformational dynamics of the ε subunit in F1-ATPase. J. Biol. Chem. 280, 40130–40134.
- 69) Rodgers, A.J.W. and Wilce, M.C.J. (2000) Structure of the γ-ε complex of ATP synthase. Nat. Struct. Biol. 7, 1051–1054.
- 70) Kato-Yamada, Y. and Yoshida, M. (2003) Isolated ε subunit of thermophilic F1-ATPase binds ATP. J. Biol. Chem. 278, 36013–36016.
- 71) Kato-Yamada, Y. (2005) Isolated ε subunit of Bacillus subtilis binds ATP. FEBS Lett. 579, 6875–6878.
- 72) Yagi, H., Kajiwara, N., Tanaka, H., Tsukihara, T., Kato-Yamada, Y., Yoshida, M. and Akutsu, H. (2007) Structures of the thermophilic F1-ATPase ε subunit suggesting ATP-regulated arm motion of its C-terminal domain in F1. Proc. Natl. Acad. Sci. U.S.A. 104, 11233–11238.
- 73) Cingolani, G. and Duncan, T.M. (2011) Structure of the ATP synthase catalytic complex (F1) from Escherichia coli in an autoinhibited conformation. Nat. Struct. Mol. Biol. 18, 701–708.
- 74) Borsch, M. and Duncan, T.M. (2013) Spotlighting motors and controls of single FoF1-ATP synthase. Biochem. Soc. Trans. 41, 1219–1226.
- 75) Masaike, T., Suzuki, T., Tsunoda, S.P., Konno, H. and Yoshida, M. (2006) Probing conformations of the β subunit of FoF1-ATP synthase in catalysis. Biochem. Biophys. Res. Commun. 342, 800–807.
- 76) Yagi, H., Konno, H., Murakami-Fuse, T., Isu, A., Oroguchi, T., Akutsu, H., Ikeguchi, M. and Hisabori, T. (2010) Structural and functional analysis of the intrinsic inhibitor subunit ε of F1-ATPase from photosynthetic organisms. Biochem. J. 425, 85–94.