Reviews
Lipoquality control by phospholipase A2 enzymes
2017 Volume 93 Issue 9 Pages 677-702
Details
2017 Volume 93 Issue 9 Pages 677-702
The phospholipase A2 (PLA2) family comprises a group of lipolytic enzymes that typically hydrolyze the sn-2 position of glycerophospholipids to give rise to fatty acids and lysophospholipids. The mammalian genome encodes more than 50 PLA2s or related enzymes, which are classified into several subfamilies on the basis of their structures and functions. From a general viewpoint, the PLA2 family has mainly been implicated in signal transduction, producing bioactive lipid mediators derived from fatty acids and lysophospholipids. Recent evidence indicates that PLA2s also contribute to phospholipid remodeling for membrane homeostasis or energy production for fatty acid β-oxidation. Accordingly, PLA2 enzymes can be regarded as one of the key regulators of the quality of lipids, which I herein refer to as lipoquality. Disturbance of PLA2-regulated lipoquality hampers tissue and cellular homeostasis and can be linked to various diseases. Here I overview the current state of understanding of the classification, enzymatic properties, and physiological functions of the PLA2 family.
In terms of signal transduction, the phospholipase A2 (PLA2) reaction, which hydrolyzes the sn-2 position of phospholipids to yield fatty acids and lysophospholipids, has been considered to be of particular importance, since arachidonic acid (AA, C20:4), one of the polyunsaturated fatty acids (PUFAs) released from membrane phospholipids by PLA2, is metabolized by cyclooxygenases (COXs) and lipoxygenases (LOXs) to lipid mediators including prostaglandins (PGs) and leukotrienes (LTs), which are often referred to as eicosanoids (Fig. 1). Lysophospholipids or their metabolites, such as lysophosphatidic acid (LPA) and platelet-activating factor (PAF), are categorized into another class of PLA2-driven lipid mediators (Fig. 2A, B). More recently, a novel class of anti-inflammatory lipid mediators derived from ω3 PUFAs, such as eicosapentaenoic acid (EPA, C20:5) and docosahexaenoic acid (DHA, C22:6), has also been attracting much attention (Fig. 2C). These lipid mediators exert numerous biological actions on target cells mainly by acting on their cognate G protein-coupled receptors. The pathophysiological roles of individual lipid mediators have been summarized in recent reviews.1)–4)
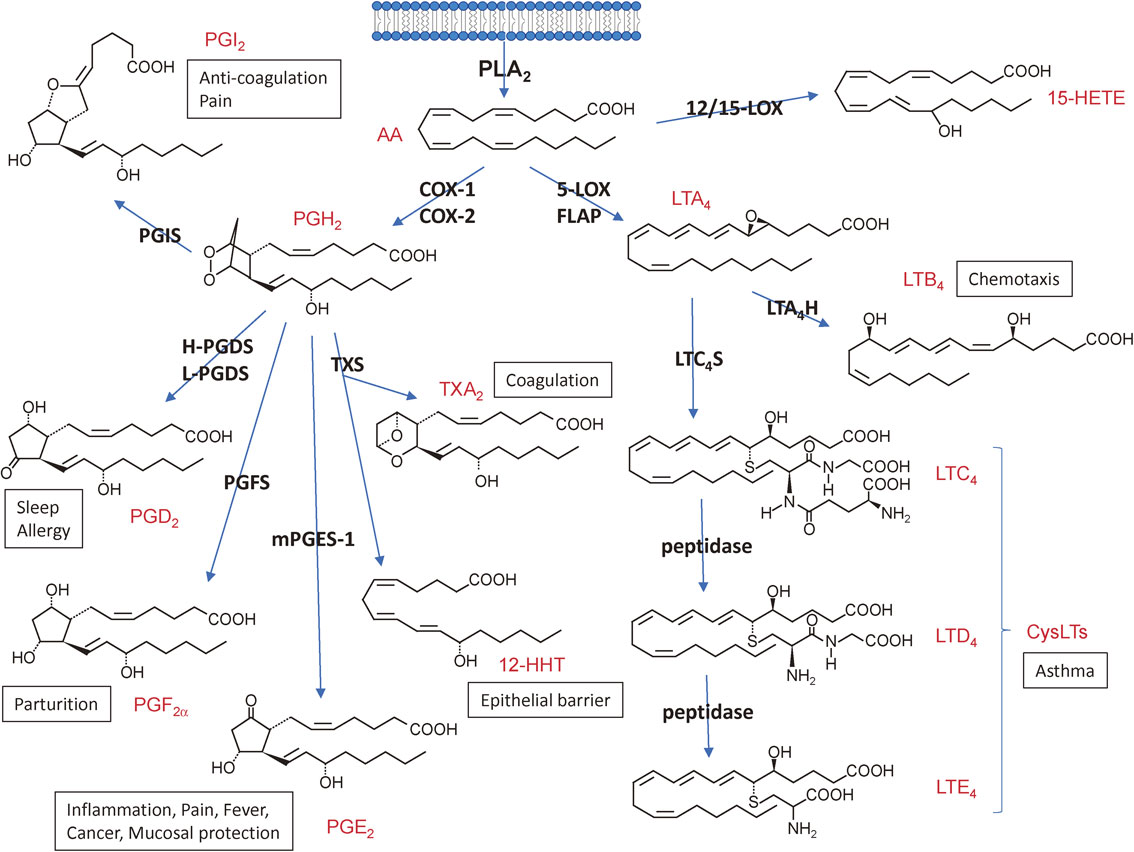
The eicosanoid-biosynthetic pathway (AA metabolism). The AA released by PLA2 from cellular membrane is metabolized to various eicosanoids through the COX and LOX pathways. Structures and representative bioactivities of individual eicosanoids and their biosynthetic enzymes are shown. H- and L-PGDS, hematopoietic and lipocalin-type PGD2 synthases, respectively; PGFS, PGF2α synthase, PGIS, PGI2 synthase; mPGES-1, microsomal PGE2 synthase-1; TXS, TX synthase; 12-HHT, 12-hydroxyheptadecatrenoic acid; 12-HETE, 12-hydroxyeicosatetraenoic acid; FLAP, 5-LOX-activating protein; LTA4H, LTA4 hydrolase; LTC4S, LTC4 synthase.
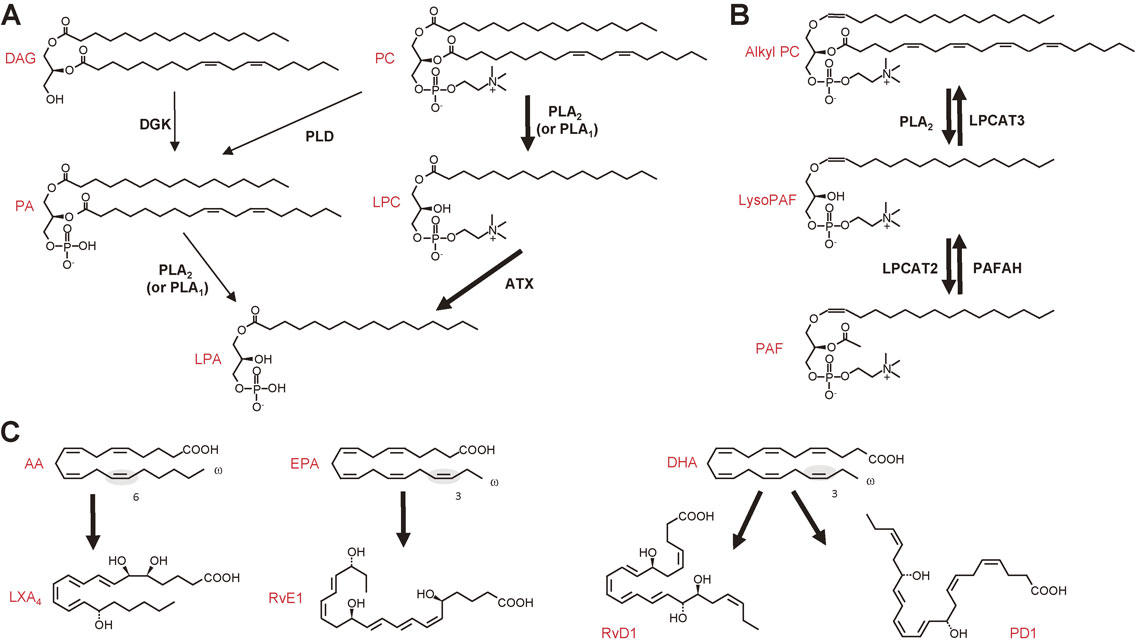
Lysophospholipid-derived lipid mediators (LPA and PAF) and PUFA-derived anti-inflammatory lipid mediators (lipoxin, resolvin and protectin). (A) Two biosynthetic pathways for LPA. LPA is produced by fatty acid deacylation of phosphatidic acid (PA) by PLA2 (or PLA1), or by removal of the polar head group of lysophosphatidylcholine (LPC), which is produced from PC by PLA2 (or PLA1), by a lysophospholipase D termed autotaxin (ATX). In most if not all in vivo situations, the ATX-dependent route is dominant for the production of LPA. DAG, diacylglycerol; DGK, diacylglycerol kinase; PLD, phospholipase D. (B) Biosynthesis and degradation of PAF. Alkyl-PC is converted by PLA2 to alkyl-LPC (LysoPAF), which is then acetylated by LPC acyltransferase 2 (LPCAT2) to give rise to PAF. PAF is deacetylated to LysoPAF by PAFAH, a unique group of PLA2s. LysoPAF is converted back to alkyl-PC by LPCAT3. (C) Anti-inflammatory PUFA metabolites derived from ω6 AA (lipoxin A4; LXA4), ω3 EPA (resolvin E1; RvE1), and ω3 DHA (RvD1 and protectin D1; PD1). The double bond characteristic of the ω3 and ω6 PUFAs is shadowed.
However, this principal concept appears to be insufficient to fully explain the biological aspects and physiological roles of the PLA2 family. Phospholipids comprise numerous molecular species that contain various combinations of fatty acids esterified at the sn-1 and sn-2 positions and several polar head groups at the sn-3 position. Many, if not all, PLA2 enzymes recognize such differences in the fatty acyl and/or head group moieties in their substrate phospholipids. Moreover, several enzymes in the PLA2 family also catalyze the phospholipase A1 (PLA1), lysophospholipase, neutral lipid lipase, or even transacylase/acyltransferase reaction rather than or in addition to the genuine PLA2 reaction. Therefore, the fatty acids and lysophospholipids released by different PLA2s are not always identical; rather, in many situations, specific fatty acids and lysophosholipids can be released by a particular PLA2 in the presence of a given microenvironmental cue. In this context, PLA2 enzymes act as one of the critical regulators of spatiotemporal lipid profiles, namely the quality of lipids (lipoquality). To comprehensively understand the lipoquality regulation by individual PLA2s in various pathophysiological contexts, their precise enzymatic, biochemical and cell biological properties, tissue and cellular distributions, and availability of phospholipid substrates in various pathophysiological settings should be taken into consideration. Herein, I overview current understanding of the biological aspects of various PLA2 enzymes in the context of lipoquality.
Obviously, the substrate specificity of individual PLA2s is the critical determinant of lipoquality. The in vitro enzymatic activity of PLA2s may be influenced by the assay conditions employed, such as the composition of the substrate phospholipids, concentrations of PLA2s and substrates, presence of detergents, and pH. Hence, the enzymatic properties of individual PLA2s determined in different studies may not be entirely identical. Since natural membranes contain numerous phospholipid molecular species, the results obtained using artificial phospholipid vesicles comprising only one or a few phospholipid species may not always reflect the true enzymatic properties of a given PLA2. Addition of an excess amount of recombinant or purified PLA2 to an enzyme assay often results in hydrolysis of bulk phospholipids, which makes precise evaluation of its substrate specificity difficult. The results obtained using a commercially available PLA2 assay kit, in which a synthetic, chromophoric phospholipid is used as a substrate, should be interpreted carefully, since some PLA2s are unable to hydrolyze it efficiently. In this regard, mass spectrometric examination of the in vitro hydrolysis of natural membrane phospholipids extracted from the affected tissues or cells by PLA2, particularly at a low (physiologically relevant) concentration of the enzyme, could provide a valuable clue to the in vivo substrates and products of this enzyme.5)–7) The overall tendency in this in vitro assay using natural membranes is recapitulated in several in vivo systems, often with even more selective patterns of hydrolysis that are relevant to the results of studies using PLA2 knockout and/or transgenic mice (see below). Importantly, the mobilization of distinct lipids by PLA2s in vivo relies not only on their intrinsic enzymatic properties, but also on tissue- or disease-specific contexts such as the lipid composition of target membranes, the spatiotemporal availability of downstream lipid-metabolizing enzymes, or the presence of cofactor(s) that can modulate the enzymatic function, which may account for why distinct PLA2 enzymes even in the same subfamily exert specific functions with different lipid profiles in distinct settings.
Hereafter, I describe the current understanding of various PLA2s in the context of lipoquality. The classification, distributions, properties and functions of individual PLA2s, whose pathophysiological functions have currently been studied using their gene-manipulated mice, are summarized in Table 1.
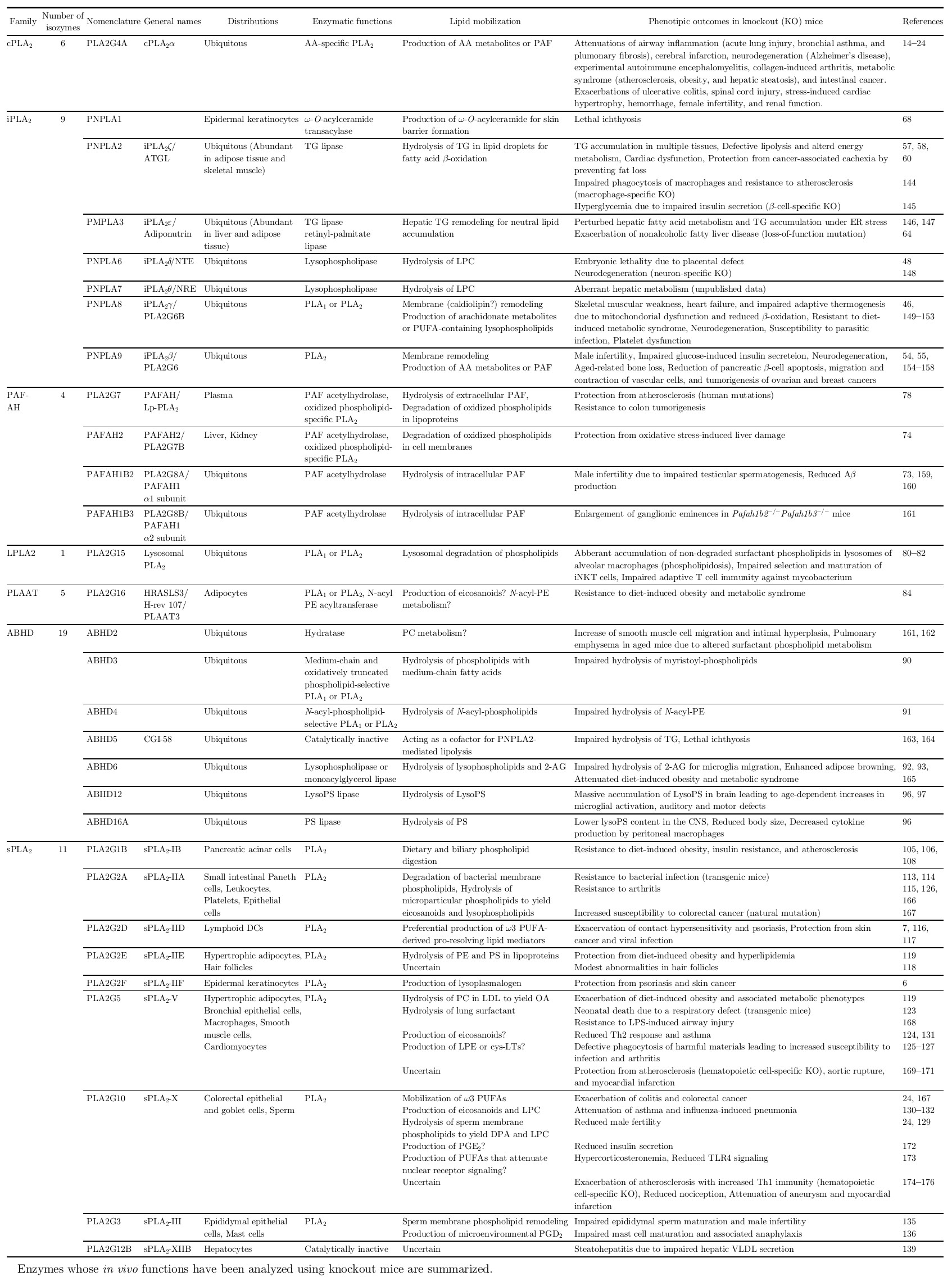
Properties of PLA2 subtypes and their biological roles
The cytosolic PLA2 (cPLA2) family comprises 6 isoforms (α–ζ), among which cPLA2β, δ, ε and ζ map to the same chromosomal locus (Fig. 3A).8) cPLA2α (also known as group IVA PLA2) is undoubtedly the best known PLA2 and its biological roles in association with lipoquality have been well documented.9) cPLA2α is the only PLA2 that shows a striking substrate specificity for AA-containing phospholipids. Strictly speaking, cPLA2α can also hydrolyze phospholipids containing EPA, yet the low abundance of this ω3 PUFA relative to other fatty acids including ω6 AA in cell membranes allows cPLA2α to release AA rather specifically in most situations. Upon cell activation, cPLA2α translocates from the cytosol to the phosphatidylcholine (PC)-rich perinuclear, endoplasmic reticulum (ER) and Golgi membranes (particularly Golgi) in response to an increase in the µM range of cytosolic Ca2+ concentration, and is maximally activated by phosphorylation through mitogen-activated protein kinases (MAPKs) and other kinases.10),11) In addition, the phosphoinositide PIP2 and ceramide-1-phosphate modulate the subcellular localization and activation of cPLA2α.12),13) The AA released by cPLA2α is converted by the sequential action of constitutive COX-1 or inducible COX-2 and terminal PG synthases to PGs or by the sequential action of 5-LOX and terminal LT synthases to LTs (Fig. 3B).
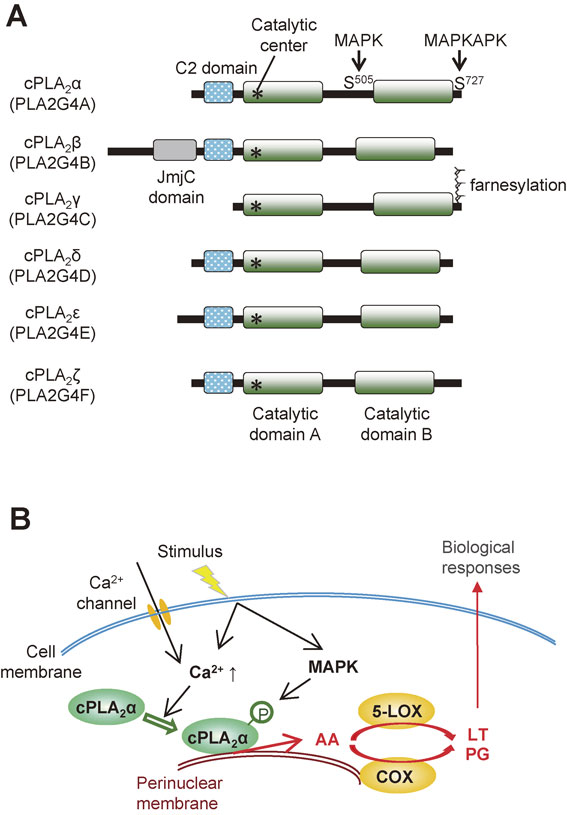
The cPLA2 family. (A) Structures of cPLA2 enzymes (α-ζ). The C2 domain, which is essential for Ca2+-dependent membrane translocation, is conserved in cPLA2 enzymes except for cPLA2γ, whose C-terminal region is farnesylated. (B) A schematic diagram of stimulus-induced cPLA2α activation. For details, see the text.
Mice deficient in cPLA2α display a number of phenotypes that can be explained by reductions of PGs and/or LTs. Under physiological conditions, cPLA2α-deficient mice display a hemorrhagic tendency, impaired female reproduction, gastrointestinal ulcer, and renal malfunction, among others.14)–18) Under pathological conditions, cPLA2α-deficient mice are protected against bronchial asthma, pulmonary fibrosis, cerebral infarction, Alzheimer’s disease, experimental autoimmune encephalomyelitis, collagen-induced arthritis, metabolic diseases, intestinal cancer and so on, whereas they suffer from more severe colitis and spinal cord injury.15),19)–24) Most of these phenotypes are recapitulated in mice lacking one or more of the biosynthetic enzymes or receptors for PGs and LTs, lending strong support to the notion that cPLA2α lies upstream of eicosanoid biosynthesis in many situations. For instance, as is the case for cPLA2α-deficient mice, mice lacking LTC4 synthase (LTC4S), LTD4 receptor (CysLT1), LTB4 receptor (BLT1), or PGD2 receptor (DP1) are protected from asthma,25)–27) revealing the critical role of the cPLA2α-LTB4/LTC4/PGD2 axis in this allergic disease. Likewise, the decrease of PGE2 in cPLA2α-deficient mice can account largely, even if not solely, for the mitigation of arthritis, autoimmune encephalomyelitis, cancer and neurodegeneration as well as the exacerbation of colitis, since these phenotypes are mimicked by mice lacking PGE2 synthase (mPGES-1) or either of the four PGE2 receptors (EP1∼4).28)–32) Furthermore, cPLA2α-triggered release of AA by platelets is coupled not only with biosynthesis of the pro-thrombotic eicosanoid thromboxane A2 (TXA2), but also with β-oxidation-mediated bioenergetics for blood clotting.33) Importantly, inherited human cPLA2α mutations are associated with reduced eicosanoid biosynthesis, platelet dysfunction, and intestinal ulceration,34),35) thus mimicking cPLA2α deletion in mice.
On the other hand, the enzymatic activities and biological functions of cPLA2 isoforms other than cPLA2α have remained largely unknown. Reportedly, cPLA2β (group IVB PLA2), which has a unique JimC domain in the N-terminal region, display PLA1, PLA2 and lysophospholipase activities.36) cPLA2γ (group IVC PLA2), which uniquely lacks the C2 domain characteristic of the cPLA2 family, is C-terminally farnesylated and possesses lysophospholipase and transacylase activities in addition to PLA2 activity.37) cPLA2δ (group IVD PLA2), whose expression is elevated in human psoriatic skin,38) shows PLA1 activity in preference to PLA2 activity.36) cPLA2ε (group IVE PLA2) exhibits a unique transacylase activity that transfers sn-1 fatty acid of PC to an amino residue of phosphatidylethanolamine (PE) to form N-acyl-PE, a precursor of the endocannabinoid lipid mediator N-acylethanolamine.39) cPLA2ζ (group IVF PLA2) displays both PLA1 and PLA2 activities without fatty acid selectivity.40) However, these enzymatic properties of cPLA2β–ζ vary according to the in vitro assays employed, implying that analyses using gene-manipulated mice for these enzymes will be necessary for clarifying their biological roles in the context of lipoquality.
The iPLA2/PNPLA family.The human genome encodes 9 Ca2+-independent PLA2 (iPLA2) enzymes (Fig. 4). These enzymes are now more generally referred to as patatin-like phospholipase domain-containing lipases (PNPLA1∼9), as all members in this family share a patatin domain, which was initially discovered in patatin (iPLA2α), a potato protein.41),42) Mammalian iPLA2/PNPLA isoforms include lipid hydrolases or transacylases with specificities for diverse lipids such as phospholipids, neutral lipids, sphingolipids, and retinol esters. Generally speaking, enzymes bearing a large and unique N-terminal region (PNPLA6∼9) act mainly on phospholipids (phospholipase type), whereas those lacking the N-terminal domain (PNPLA1∼5) act on neutral lipids (lipase type). Analysis of mutant mouse models and clinical symptoms of patients with mutations for these enzymes have provided valuable insights into the physiological roles of the iPLA2/PNPLA family in various forms of homeostatic lipid metabolism that are fundamental for life.
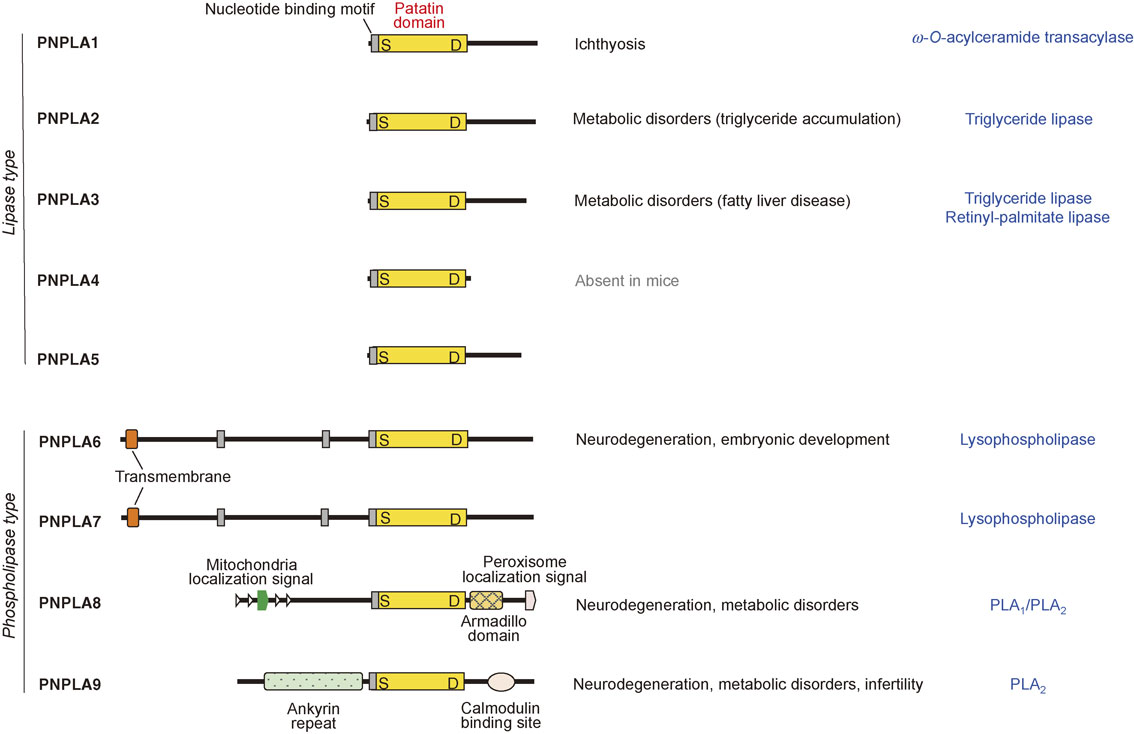
The iPLA2/PNPLA family. Structures of iPLA2/PNPLA enzymes (PNPLA1∼9), which are subdivided into lipase and phospholipase types, are shown. The patatin domain, which is characteristic of this family, is conserved in all of these enzymes. The biological functions and enzymatic properties of the individual enzymes are indicated on the right. For details, see the text.
Among the iPLA2/PNPLA family, PNPLA9 (iPLA2β, also known as group VIA PLA2) is the only isoform that acts primarily as a PLA2 with poor fatty acid selectivity.43),44) Although PNPLA8 (iPLA2γ or group VIB PLA2) displays PLA2 activity, it acts as a PLA1 toward phospholipids bearing sn-2 PUFA.45),46) Accordingly, hydrolysis of PUFA-bearing phospholipids by PNPLA8/iPLA2γ typically gives rise to 2-lysophospholipids (having a PUFA at the sn-2 position) rather than 1-lysophospholipids (having a saturated or monounsaturated fatty acid at the sn-1 position). PNPLA6 (iPLA2δ) and its closest paralog PNPLA7 (iPLA2θ) have lysophospholipase activity that cleaves lysophosphatidylcholine to yield fatty acid and glycerophosphocholine.47),48) Genetic mutations or deletions of these phospholipid-targeting PNPLAs cause various forms of metabolic dysfunction and neurodegeneration.49)–53) In particular, PNPLA9/iPLA2β is also referred to as the parkinsonism-associated protein PARK14, whose mutations impair Ca2+ signaling in dopaminergic neurons.54) Apart from the metabolic and neurodegenerative phenotypes, the lack of PNPLA9/iPLA2β leads to male infertility through an unknown mechanism.55)
PNPLA2 (iPLA2ζ), more generally known as adipose triglyceride lipase (ATGL), is a major lipase that hydrolyzes triglycerides in lipid droplets to release fatty acids as a fuel for β-oxidation-coupled energy production, a process known as lipolysis.56) Genetic deletion or mutation of PNPLA2 leads to massive accumulation of triglycerides in multiple tissues leading to multi-organ failures,57) while protecting from cancer-associated cachexia by preventing fat loss.58) The activity of PNPLA2 is regulated positively by ABHD5 (see below) and negatively by perilipin and G0S2, which modulate the accessibility of PNPLA2 to lipid droplets.59) The fatty acids released from lipid droplets by PNPLA2 act as endogenous ligands for the nuclear receptor PPARα or PPARδ, which accelerates energy consumption.59),60) The regulatory mechanisms and metabolic roles of PNPLA2 have been detailed in other elegant reviews.61),62) Mutations of PNPLA3 (iPLA2ε) are highly associated with non-alcoholic fatty liver disease.63) Although the catalytic activity of PNPLA3 is controversial, it may serve as a triglyceride lipase, since its loss-of function mutation increases cellular triglyceride levels.64) Furthermore, recent evidence suggests that PNPLA3 acts as a retinyl-palmitate lipase in hepatic stellate cells to fine-tune the plasma levels of retinoids. The expressions of PNPLA2 and PNPLA3 are nutritionally regulated in a reciprocal way; PNPLA2 is upregulated, while PNPLA3 is downregulated, upon starvation, and vice versa upon feeding.65) Biochemical and cell biological studies have suggested that PNPLA4 (iPLA2η, which is absent in mice) might be involved in retinol ester metabolism66) and that PNPLA5 might participate in triglyceride lipolysis coupled with autophagosome formation,67) although the in vivo relevance of these in vitro observations is unclear.
Unlike most PNPLA isoforms that are ubiquitously expressed in many tissues, PNPLA1 is localized predominantly in the upper layer of the epidermis. PNPLA1 acts as a unique transacylase, catalyzing the transfer of linoleic acid (LA; C18:2) in triglyceride to the ω-hydroxy residue of ultra-long-chain fatty acid in ceramide to form ω-O-acylceramide, a lipid component essential for skin barrier function.68),69) Accordingly, genetic deletion or mutation of PNPLA1 hampers epidermal ω-O-acylceramide formation, thereby severely impairing skin barrier function and causing ichthyosis. The unique role of PNPLA1 in the acylceramide-metabolic pathway in the epidermis is depicted in Fig. 5.

The role of PNPLA1 in epidermal acylceramide biosynthesis. Structures of the metabolites and enzymes or transporters responsible for individual steps in the acylceramide-biosynthetic pathway are indicated. Mutations or deletions of these enzymes cause ichthyosis in both human and mouse. PNPLA1 catalyzes the transacylation of LA from triglyceride to ω-OH ceramide, leading to the formation of ω-O-acylceramide, which is an essential component of lipid lamellae and the cornified lipid envelope in the uppermost epidermis. For details, see the text. ELOVL6, fatty acid elongase 6; CYP4F22/39, cytochrome P450 family F22 (in human) and F39 (in mouse); CERS3, ceramide synthase 3; ABCA12, ABC transporter 12; UGCG, UDP-glucose ceramide glucosyltransferase; GBA, β-glucocerebrosidase; ALOXE3, epidermal-type lipoxygenase 3; ALOX12B, 12R-lipoxygenase; TGM1, transglutaminase 1.
The PAF-acetylhydrolase (PAFAH) family comprises one extracellular and three intracellular PLA2s that were originally found to have the capacity to deacetylate and thereby inactivate the lysophospholipid-derived lipid mediator PAF.70),71) Type-I PAFAH is a heterotrimer composed of two catalytic subunits, group XIIIA and XIIIB PLA2s, and a regulatory subunit LIS-1, the causative gene for a type of Miller Diecker syndrome.72) Deficiency of type-I PAFAH leads to male infertility through an unknown mechanism.73) Type-II PAFAH (group VIIB PLA2) preferentially hydrolyzes oxidized phospholipids (i.e., phospholipids having an oxygenated fatty acid at the sn-2 position) in cellular membranes, thereby protecting cells from oxidative damage.74) Although plasma-type PAFAH (group VIIA PLA2) is a secreted protein, it is described here as its structure is close to type-II PAFAH. Plasma-type PAFAH is now more generally called lipoprotein-associated PLA2 (Lp-PLA2), existing as a low-density lipoprotein (LDL)-bound form in human plasma.75) A series of studies have revealed the correlation of Lp-PLA2 with atherosclerosis, likely because this enzyme liberates toxic oxidized fatty acids from modified LDL with pro-atherogenic potential.76),77) Furthermore, deficiency of Lp-PLA2 decreases intestinal polyposis and colon tumorigenesis in ApcMin/+ mice,78) suggesting an anti-tumorigenic role for PAF in this setting.
Lysosomal PLA2.Lysosomal PLA2 (LPLA2), also known as group XV PLA2, is homologous with lecithin cholesterol acyltransferase (LCAT) and catalytically active under mildly acidic conditions.79) LPLA2 hydrolyzes both sn-1 and sn-2 fatty acids in phospholipids and contributes to phospholipid degradation in lysosomes. Genetic deletion of LPLA2 results in unusual accumulation of non-degraded lung surfactant phospholipids in lysosomes of alveolar macrophages, leading to phospholipidosis,80) perturbed presentation of endogenous lysophospholipid antigens to CD1d by invariant natural killer T (iNKT) cells,81) and impairment of adaptive T cell immunity against mycobacterium.82)
The PLAAT family.The PLA-acyltransferase (PLAAT) family (3 enzymes in humans and 5 enzymes in mice) is structurally similar to lecithin retinol acyltransferase (LRAT). Members of this family, including group XVI PLA2 (PLA2G16), display PLA1 and PLA2 activities, as well as acyltransferase activity that synthesizes N-acyl-PE, to various degrees.83) PLA2G16 is highly expressed in adipocytes, and PLA2G16-deficient mice are resistant to diet-induced obesity.84) PLA2G16 and its paralogs in this family have also been implicated in tumor invasion and metastasis,85) vitamin A metabolism,86) peroxisome biogenesis,87) and cellular entry and clearance of Picornaviruses.88)
The ABHD family.The α/β hydrolase (ABHD) family is a newly recognized group of lipolytic enzymes, comprising at least 19 enzymes in humans.89) Enzymes in this family typically possess both hydrolase and acyltransferase motifs. Although the functions of many of the ABHD isoforms still remain uncertain, some of them have been demonstrated to act on neutral lipids or phospholipids as lipid hydrolases. ABHD3 selectively hydrolyzes phospholipids with medium-chain fatty acids.90) ABHD4 releases fatty acids from multiple classes of N-acyl-phospholipids to produce N-acyl-lysophospholipids.91) ABHD6 acts as lysophospholipase or monoacylglycerol lipase, the latter being possibly related to the regulation of 2-arachidonoyl glycerol (2-AG) signaling.92),93) 2-AG is an endocannabinoid lipid mediator that plays a role in the retrograde neurotransmission and is considered to be produced mainly by diacylglycerol lipase α.94) Interestingly, in the brain, the AA released from 2-AG by monoacylglycerol lipase, rather than that released from phospholipids by cPLA2α (see above), is linked to the production of a pool of PGE2 that promotes fever.2),95) ABHD12 hydrolyzes lysophosphatidylserine (LysoPS), and is therefore referred to as LysoPS lipase.96) Mutations in the human ABHD12 gene result in accumulation of LysoPS in the brain and cause a disease called PHARC, which is characterized by polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataract.97) ABHD16A acts as a phosphatidylserine (PS)-selective PLA2 (referred to as PS lipase), being located upstream of ABHD12 in the PS-catabolic pathway.96) Although ABHD5 (also called CGI-58) does not have a catalytic activity because of the absence of a serine residue in the catalytic center, it greatly enhances PNPLA2-directed hydrolysis of triglycerides in lipid droplets by acting as an essential lipolytic cofactor.98)
The secreted PLA2 (sPLA2) family contains 10 catalytically active isoforms and one inactive isoform in mammals.42),99) Based on the structural and evolutional relationships, these enzymes are categorized into classical (IB, IIA, IIC, IID, IIE, IIF, V and X) and atypical (III and XII) classes (Fig. 6). The sPLA2 family strictly hydrolyzes the sn-2 position of phospholipids, a feature that differs from intracellular PLA2s that often display PLA1, lysophospholipase, lipase, or transacylase/acyltransferase activity (see above). Individual sPLA2s exhibit unique tissue and cellular distributions, suggesting their distinct biological roles. As sPLA2s are secreted and require Ca2+ in the mM range for their catalytic action, their principal targets are phospholipids in the extracellular space, such as microparticles, surfactant, lipoproteins, and foreign phospholipids in microbe membranes or dietary components. The biochemical properties and pathophysiological functions of sPLA2s have been detailed in several recent reviews.5),100) Here, I describe several key features of lipoquality regulation by the sPLA2 family.

The sPLA2 family. The phylogenetic tree of sPLA2 isoforms, which are subdivided into classical sPLA2s (I/II/V/X branch) and atypical sPLA2s (III and XII branches), is shown. The pathophysiological roles and related types of lipid metabolism (target substrates or products; shown in blue) for the individual isoforms are indicated. For details, see the text.
In terms of the lipoquality, sPLA2s have long been considered to display no apparent selectivity for sn-2 fatty acid species in the substrate phospholipids. This view was based on the fact that sPLA2-IB and -IIA, two prototypic sPLA2s that were initially identified through classical protein purification from the pancreas and sites of inflammation, respectively,101),102) as well as a number of snake venom PLA2s that belong to group I and II sPLA2s, are capable of releasing fatty acids non-selectively. However, recent lipidomics-based evaluation of the substrate specificity of sPLA2s toward natural membranes (see above) has revealed that several sPLA2s can distinguish sn-2 fatty acyl moieties in phospholipids under physiologically relevant conditions. In general terms, sPLA2-IB, -IIA and -IIE do not discriminate fatty acid species, sPLA2-V tends to prefer those with a lower degree of unsaturation such as oleic acid (OA; C18:1), and sPLA2-IID, -IIF, -III and -X tend to prefer PUFAs including AA and DHA. Several sPLA2s can also distinguish differences in the polar head groups of phospholipids. For instance, sPLA2-X is very active on PC, while sPLA2-IIA has much higher affinity for PE than for PC, and this substrate selectivity has been partly ascribed to their crystal structures.103),104) Therefore, in order to comprehensively understand the specific biological roles of this enzyme family, it is important to consider when and where different sPLA2s are expressed, which isoforms are involved in what types of pathophysiology, why they are needed, and how they exhibit their unique functions by driving specific types of lipid metabolism.
Classical sPLA2s.sPLA2-IB, also known as “pancreatic sPLA2”, is synthesized as an inactive zymogen in the pancreas, and its N-terminal propeptide is cleaved by trypsin to yield an active enzyme in the duodenum.101) The main role of sPLA2-IB is to digest dietary and biliary phospholipids in the intestinal lumen. Perturbation of this process by gene disruption or pharmacological inhibition of sPLA2-IB leads to resistance to diet-induced obesity, insulin resistance, and atherosclerosis due to decreased phospholipid digestion and absorption in the gastrointestinal tract.105)–108) The human PLA2G1B gene maps to an obesity-susceptible locus.109)
sPLA2-IIA is often referred to as “inflammatory sPLA2”, since its expression is induced by pro-inflammatory cytokines such as TNFα and IL-1β or by bacterial products such as lipopolysaccharide.110) In mice, however, sPLA2-IIA in mice is distributed only in intestinal Paneth cells (in BALB/c, C3H, NZB and DBA, etc.) or not expressed at all due to a natural frameshift mutation (in C57BL/6, A/J, C58/J, P/J, 129/Sv and B10.RIII, etc.).111),112) The best-known physiological function of sPLA2-IIA is the degradation of bacterial membranes, thereby providing the first line of antimicrobial defense in the host.113),114) Consistent with this, sPLA2-IIA preferentially hydrolyzes PE and phosphatidylglycerol, which are enriched in bacterial membranes. Under sterile conditions, sPLA2-IIA attacks phospholipids in microparticles, particularly those in extracellular mitochondria (an organelle that evolutionally originated from bacteria), which are released from activated platelets or leukocytes at inflamed sites.115) Hydrolysis of microparticular phospholipids by sPLA2-IIA results in production of pro-inflammatory eicosanoids and lysophospholipids as well as in release of mitochondrial DNA as a danger-associated molecular pattern (DAMP). Thus, sPLA2-IIA is primarily involved in host defense by killing bacteria and triggering innate immunity, while over-amplification of the response leads to exacerbation of inflammation.
sPLA2-IIA, -IIC, -IID, -IIE and -IIF are often classified into the group II subfamily (sPLA2-IIC is a pseudogene in human), since they share structural characteristics and map to the same chromosome locus. sPLA2-IID is constitutively expressed in dendritic cells (DCs) in lymphoid organs. sPLA2-IID is an “immunosuppressive sPLA2” that attenuates DC-mediated adaptive immunity by hydrolyzing PE probably in microparticles to mobilize anti-inflammatory ω3 PUFAs and their metabolites such as resolvin D1 (RvD1).7) As such, sPLA2-IID-null mice exhibit more severe contact hypersensitivity and psoriasis, whereas they are protected against infection and cancer because of enhanced anti-viral and anti-tumor immunity.7),116),117) Unlike sPLA2-IIA, which is stimulus-inducible (see above), sPLA2-IID is downregulated by pro-inflammatory stimuli, consistent with its anti-inflammatory role.
In mice, sPLA2-IIE instead of sPLA2-IIA is upregulated in several tissues under inflammatory or other conditions. sPLA2-IIE is expressed in hair follicles in association with the growth phase of the hair cycle118) and induced in adipose tissue in association with obesity in mice.119) sPLA2-IIE hydrolyzes PE without apparent fatty acid selectivity in hair follicles and lipoproteins, and accordingly, sPLA2-IIE-deficient mice display subtle abnormalities in hair follicles118) and are modestly protected from diet-induced obesity and hyperlipidemia.119)
sPLA2-IIF has a long C-terminal extension containing a free cysteine, which might contribute to formation of a homodimer, and is more hydrophobic than other sPLA2s.120) Physiologically, sPLA2-IIF is an “epidermal sPLA2” that is expressed predominantly in the upper epidermis and induced by IL-22, a Th17 cytokine, in psoriatic skin.6) sPLA2-IIF preferentially hydrolyzes PUFA-containing plasmalogen-type PE in keratinocyte-secreted phospholipids to produce plasmalogen-type lysophosphatidylethanolamine (P-LPE; lysoplasmalogen), which in turn promotes epidermal hyperplasia (Fig. 7A–C). Accordingly, sPLA2-IIF-null mice are protected against epidermal-hyperplasic diseases such as psoriasis and skin cancer, while sPLA2-IIF-transgenic mice spontaneously develop psoriasis-like skin.6)
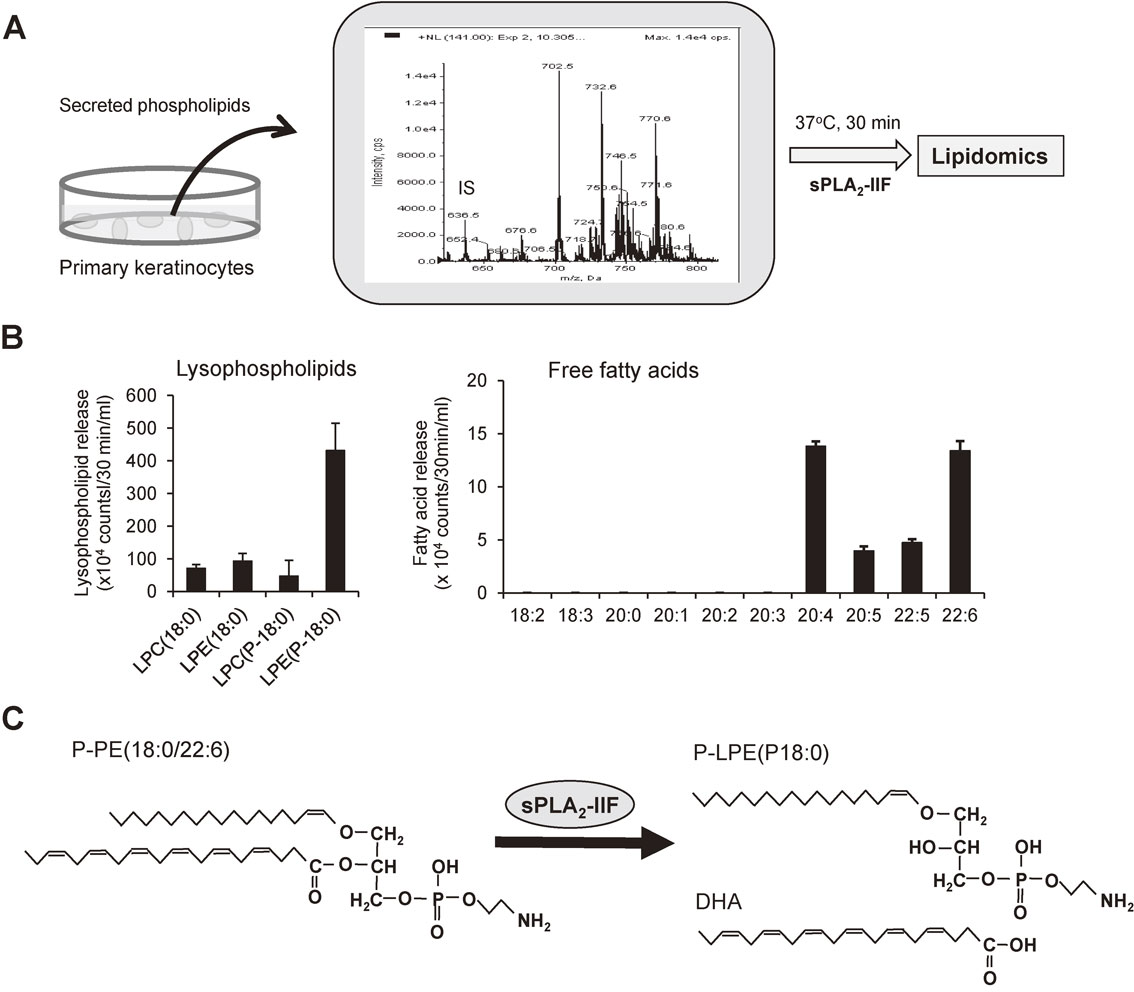
Properties of sPLA2-IIF. (A) A schematic procedure for identification of the lipid metabolism driven by sPLA2-IIF in differentiating keratinocytes. Phospholipids extracted from the culture supernatants of mouse keratinocytes (a representative mass spectrometric profile of phospholipids is shown; IS, internal standard; cps, count per second) were incubated with a physiologically relevant concentration of recombinant sPLA2-IIF and then taken for the lipidomics analysis. (B) In the assay shown in (A), sPLA2-IIF preferentially increased plasmalogen-type (P-) lysophosphatidylethanolamine (LPE) species as well as PUFAs. Values represent AUC (area under the curve; mean ± SEM, n = 4). (C) The results shown in (B), together with in vivo analyses using sPLA2-IIF-transgenic and knockout mice,6) indicate that sPLA2-IIF preferentially hydrolyzes P-PE bearing DHA to liberate P-LPE and DHA under physiological conditions. For more details, please see ref. 6.
Although sPLA2-V was previously thought to be a regulator of AA metabolism,121),122) it is now becoming obvious that this sPLA2 has a preference for phospholipids having fatty acids with a lower degree of unsaturation. sPLA2-V is markedly induced in adipocytes during obesity as a “metabolic sPLA2” and hydrolyzes PC in hyperlipidemic LDL to release OA and to a lesser extent LA, which counteract adipose tissue inflammation and thereby ameliorates obesity-associated metabolic disorders.119) Transgenic overexpression of sPLA2-V, but not other sPLA2s, results in neonatal death due to a respiratory defect, which is attributable to the ability of sPLA2-V to potently hydrolyze PC with palmitic acid (PA, C16:0), a major component of lung surfactant.123) This unique substrate preference of sPLA2-V has also been supported by a recent lipidomics analysis of the spleen (a tissue where sPLA2-V is abundantly expressed), in which the levels of fatty acids with a lower degree of unsaturation (e.g. PA, OA and LA), rather than PUFAs (AA, EPA and DHA), are significantly reduced in sPLA2-V-deficient mice relative to wild-type mice (Fig. 8). This is in contrast to the spleen of sPLA2-IID-deficient mice, in which ω3 PUFAs and their metabolites are selectively diminished,7) revealing distinct lipoquality regulation by different sPLA2s. Another intriguing feature of sPLA2-V is that it is the only “Th2-prone sPLA2” induced in M2 macrophages by the Th2 cytokines IL-4 and IL-13 and promotes Th2-driven pathology such as asthma. Gene ablation of sPLA2-V perturbs proper polarization and function of M2 macrophages in association with decreased Th2 immunity,124) although the underlying lipid metabolism responsible for this event remains obscure. Probably because of this alteration in the macrophage phenotype, sPLA2-V-null macrophages have a reduced ability to phagocytose extracellular materials. Accordingly, sPLA2-V-null mice are more susceptible to fungal infection and arthritis due to defective clearance of hazardous fungi and immune complexes, respectively.125),126) Likewise, sPLA2-V-null mice suffer from more severe lung inflammation caused by bacterial or viral infection,127) which could also be explained by poor clearance of these microbes by alveolar macrophages.
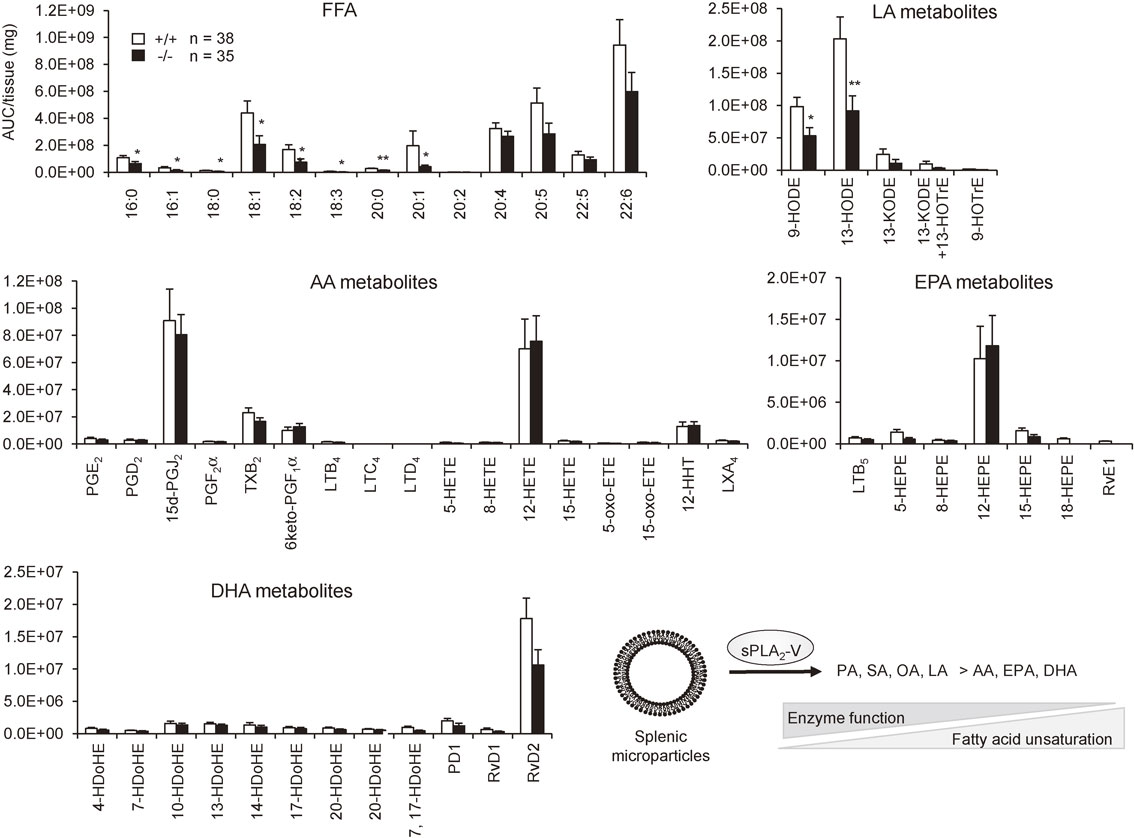
Fatty acid selectivity of sPLA2-V. Lipids extracted from the spleen of 1-year-old sPLA2-V-deficient (−/−) and littermate control (+/+) mice were subjected to mass spectrometric lipidomics analysis (values are mean ± SEM, *P < 0.05 and **P < 0.01). Experiments were performed in accordance with the procedure described previously (5). Y-axis indicate relative abundance (AUC; area under the curve) of each product per mg tissue. Free fatty acid (FFA) species with a lower degree of unsaturation, including PA (16:0), palmitoleic acid (16:1), stearic acid (18:0; SA), OA (18:1), LA (18:2), eicosanoic acid (20:0) and eicosenoic acid (C20:1), but not PUFAs including AA (20:4), EPA (20:5), DPA (22:5) and DHA (22:6), were significantly reduced in sPLA2-V-deficient mice relative to control mice. Accordingly, LA metabolites, including 9- and 13-hydroxyoctadecadienoic acids (HODEs) among others, were substantially decreased in mutant mice relative to control mice, whereas none of the AA, EPA and DHA metabolites differed significantly between the genotypes. These results are consistent with the view that sPLA2-V has a propensity to preferentially hydrolyze phospholipids having sn-2 fatty acids with a lower degree of unsaturation, as illustrated at right bottom.
Among the mammalian sPLA2s, sPLA2-X has the highest affinity for PC leading to release of fatty acids, with an apparent tendency for PUFA preference. sPLA2-X is activated by cleavage of the N-terminal propeptide by furin-type convertases.128) sPLA2-X is expressed abundantly in colorectal epithelial and goblet cells and has a protective role in colitis by mobilizing anti-inflammatory ω3 PUFAs.24) Consistently, sPLA2-X-transgenic mice exhibit global anti-inflammatory phenotypes in association with elevation of systemic ω3 PUFA levels.24) In the process of reproduction, sPLA2-X secreted from the acrosomes of activated spermatozoa hydrolyzes sperm membrane phospholipids to release DHA and docosapentaenioc acid (DPA, C22:5), the latter facilitating fertilization.24),129) Additionally, sPLA2-X-null mice are protected from asthma, accompanied by decreased levels of pulmonary ω6 AA-derived eicosanoids.130) Unlike the situation in sPLA2-V-null mice (see above), however, the Th2 response per se is not affected in the asthma model131) and the lung damage is milder following influenza infection132) in sPLA2-X-null mice, illustrating the distinct actions of different sPLA2s in the same tissue.
Atypical sPLA2s.sPLA2-III is unusual in that it consists of three domains, in which the central sPLA2 domain similar to bee venom group III sPLA2 is flanked by large and unique N- and C-terminal domains.133) The enzyme is processed to the sPLA2 domain-only form that retains full enzymatic activity.134) Although sPLA2-III does not discriminate the polar head groups, it tends to prefer sn-2 PUFAs in the substrate phospholipids. sPLA2-III is expressed in the epididymal epithelium and acts on immature sperm cells passing through the epididymal duct in a paracrine manner to allow sperm membrane phospholipid remodeling, a process that is prerequisite for sperm motility.135) sPLA2-III is also secreted from mast cells and acts on microenvironmental fibroblasts to produce PGD2, which in turn promotes proper maturation of mast cells.136) Accordingly, mice lacking sPLA2-III exhibit male hypofertility and reduced anaphylactic responses.
sPLA2-XIIA is evolutionally far distant from other sPLA2s.137) sPLA2-XIIA is expressed in many tissues at relatively high levels, yet its enzymatic activity is weaker than that of other sPLA2s. The properties and physiological roles of sPLA2-XIIA are currently unclear and await future studies using sPLA2-XIIA-deficient mice. Apart from lipoquality regulation, sPLA2-XIIB is a catalytically inactive protein due to substitution of the catalytic center histidine by leucine.138) sPLA2-XIIB deficiency impairs hepatic lipoprotein secretion,139) although the mechanism is unclear.
sPLA2 receptor.Beyond the lipoquality control by sPLA2s, several sPLA2s binds to sPLA2 receptor (PLA2R1, also known as the C-type lectin Clec13c) with different affinities.140) In mice, PLA2R1 binds to sPLA2-IB, -IIA, -IIE, -IIF and -X with high affinity, sPLA2-V with moderate affinity, and sPLA2-IID, -III and -XIIA with low or no affinity.138) PLA2R1 is homologous to sPLA2-inhibitory proteins present in snake plasma and exists as an integral membrane protein or as a soluble protein resulting from shedding or alternative splicing. PLA2R1 may act as a clearance receptor or endogenous inhibitor that inactivates sPLA2s, as a signaling receptor that transduces sPLA2-dependent signals in a catalytic activity-independent manner, or as a pleiotropic receptor that binds to non-sPLA2 ligands. In support of its clearance role, Pla2r1−/− mice show more severe asthma, likely due to defective clearance of pro-asthmatic sPLA2-X.141) In support of its signaling role, PLA2R1, probably through binding to myocardial sPLA2s or other ways, promotes the migration and growth of myofibroblasts and thereby protects against cardiac rupture in a model of myocardial infarction.142) PLA2R1 has recently attracted attention as a major autoantigen in membranous nephropathy, a severe autoimmune disease leading to podocyte injury and proteinuria,143) although it is not clear whether this role of PLA2R1 is sPLA2-dependent or -independent.
By applying lipidomics approaches to knockout or transgenic mice for various PLA2s, it has become evident that individual enzymes regulate specific forms of lipid metabolism, perturbation of which can be eventually linked to distinct pathophysiological outcomes. Knowledge of lipoquality control by individual PLA2s acquired from studies using animal models should be translated to humans. Current knowledges on the relationship between PLA2 gene mutations and human diseases are summarized in Table 2. Nonetheless, future development of more comprehensive and highly sensitive lipidomics techniques will contribute to the discovery of novel PLA2-driven lipid pathways that could be biomarkers or druggable targets for particular diseases.

Representative PLA2 mutations in human diseases

Makoto Murakami was born in Nagano Prefecture in 1964 and graduated from Faculty of Pharmaceutical Sciences, the University of Tokyo, in 1986. He received a M.S. degree in 1988 and a Ph.D. degree in 1991 from the University of Tokyo. He worked as a postdoctoral fellow at the University of Tokyo under a support of the Japan Society for the Promotion of Science from 1991 to 1993 and then at Harvard Medical School under Professor K. Frank Austen from 1993 to 1995. He then worked as an associate professor at School of Pharmaceutical Sciences, Showa University, from 1995 to 2005 and as a project leader of the Lipid Metabolism project, Tokyo Metropolitan Institute of Medical Science, from 2005 to 2016. He is now working as a professor at Graduate School of Medicine, the University of Tokyo, since 2017. He has authored 180 original articles and 54 review articles (in English) and 100 review articles (in Japanese) on phospholipase A2s and lipid mediators. He is now a committee member of the Japanese Biochemical Society, Japanese Lipid Biochemistry Society, and Japanese Society of Inflammation and Regeneration. He received the Young Investigator Awards for the Pharmaceutical Society of Japan in 1999 and the Japanese Society of Inflammation and Regeneration in 2000, Investigator Awards for the Tokyo Metropolitan Institute of Medical Science in 2008, Award for the Terumo Science Foundation in 2014, and the Bureau of Social Welfare and Public Health at Tokyo Metropolitan Government in 2015.
I sincerely thank my laboratory members at the University of Tokyo and the Tokyo Metropolitan Institute of Medical Sciences who have contributed their expertise to acquire a better understanding of PLA2 biology. In particular, I thank Drs. Kei Yamamoto, Tetsuya Hirabayashi, Yoshitaka Taketomi, Hiroyasu Sato, Yoshimi Miki, Remi Murase, Seiko Masuda and Noriko Ueno among others for collecting the experimental data and information on which this review is based. In the interest of brevity, I have referenced other reviews whenever possible and apologize to the authors of the numerous original papers that were not explicitly cited. This work was supported by AMED-CREST from the Japan Agency for Medical Research and Development and by JSPS KAKENHI Grant Numbers JP15H05905 and JP16H02613.