Reviews
Prostaglandin terminal synthases as novel therapeutic targets
2017 Volume 93 Issue 9 Pages 703-723
Details
2017 Volume 93 Issue 9 Pages 703-723
Non-steroidal anti-inflammatory drugs (NSAIDs) exert their anti-inflammatory and anti-tumor effects by reducing prostaglandin (PG) production via the inhibition of cyclooxygenase (COX). However, the gastrointestinal, renal and cardiovascular side effects associated with the pharmacological inhibition of the COX enzymes have focused renewed attention onto other potential targets for NSAIDs. PGH2, a COX metabolite, is converted to each PG species by species-specific PG terminal synthases. Because of their potential for more selective modulation of PG production, PG terminal synthases are now being investigated as a novel target for NSAIDs. In this review, I summarize the current understanding of PG terminal synthases, with a focus on microsomal PGE synthase-1 (mPGES-1) and PGI synthase (PGIS). mPGES-1 and PGIS cooperatively exacerbate inflammatory reactions but have opposing effects on carcinogenesis. mPGES-1 and PGIS are expected to be attractive alternatives to COX as therapeutic targets for several diseases, including inflammatory diseases and cancer.
Prostanoids are cyclic and oxygenated metabolites comprised of ω-3 and ω-6 20-carbon essential fatty acids, and they have a broad range of biological activities.1) Prostanoids include what are sometimes referred to as the “classical” prostaglandins (PGs), such as PGD, PGE, and PGF (all of which have a prostanoic acid backbone), as well as prostacyclin (PGI2) and thromboxane. Specific prostanoids are designated by a letter that indicates the type of ring structure, followed by a number that indicates the number of double bonds in the hydrocarbon structure. Among the prostanoids, the most abundant are the “2-series” compounds such as PGE2 that are formed from arachidonic acid (AA).
Three types of enzymes — phospholipase A2 (PLA2), cyclooxygenase (COX), and PG terminal synthase — are involved in the biosynthesis of prostanoids (Fig. 1).2)–5) As the first step of this biosynthesis, AA is released from the sn-2 position of glycerophospholipids by the action of PLA2. In mammals, more than 20 PLA2 enzymes have been identified. Among them, cytosolic cPLA2α plays a critical role in the stimulus-dependent liberation of AA.2) As the second step, the liberated AA is supplied to either of the two COX isozymes, COX-1 or COX-2, to be isomerized to PGH2.3),4) Finally, in the third step, the COX metabolite PGH2 is converted to each prostanoid by specific PG terminal synthases.4),5)
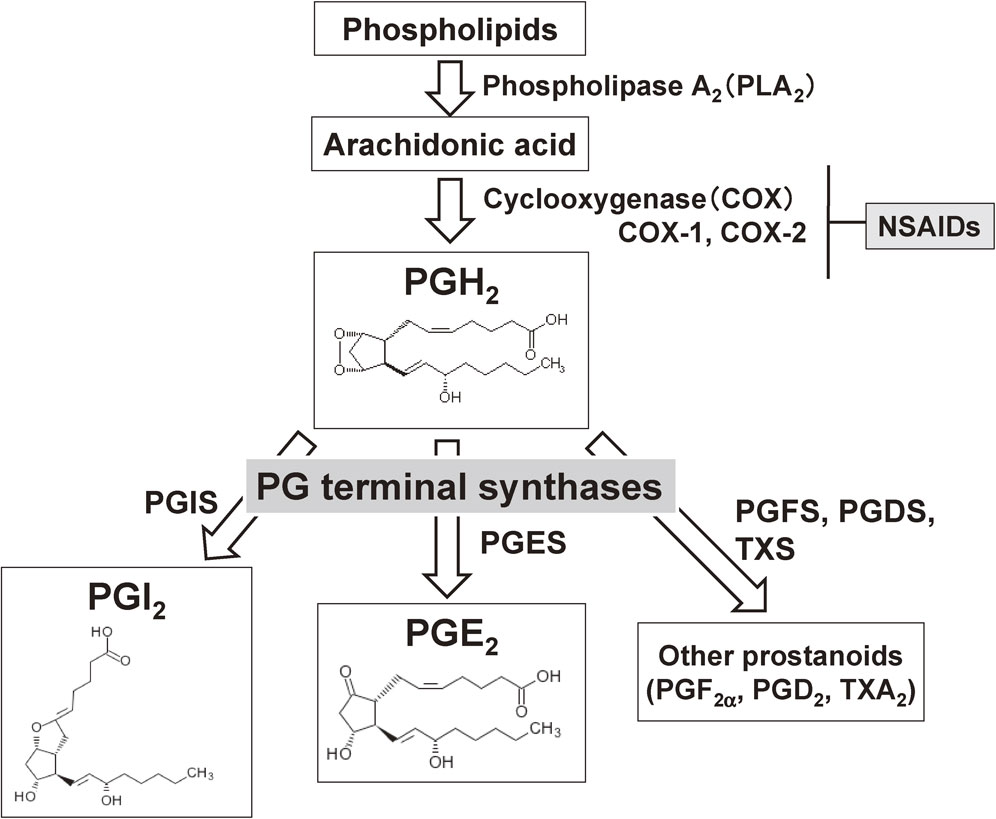
Biosynthetic pathways of prostanoids. Arachidonic acid released from membrane phospholipids by the action of phospholipase A2 (PLA2) is converted to PGH2 by cyclooxygenase (COX), and PGH2 is then isomerized to prostanoids by each PG-specific terminal enzyme. Of the two COX enzymes, COX-1 is expressed constitutively in most tissues, and COX-2 is induced in response to various types of stimuli. NSAIDs exert both beneficial and adverse effects by reducing prostanoid production via the inhibition of COX. PGDS: PGD synthase; PGES: PGE synthase; PGFS: PGF synthase; PGIS: PGI synthase; TXS: thromboxane synthase.
Non-steroidal anti-inflammatory drugs (NSAIDs) exert their anti-inflammatory and anti-tumor effects by reducing prostanoid production via the inhibition of COX.3) As mentioned above, there are two COX isozymes, COX-1 and COX-2. COX-1 is expressed constitutively in most tissues and is generally responsible for the immediate production of prostanoids that control normal physiological functions such as the maintenance of gastrointestinal or renal homeostasis. COX-2 is induced in response to mitogens, cytokines, and cellular transformation, and it mediates the delayed prostanoid generation related to inflammatory reactions and carcinogenesis.6),7)
We investigated mechanism by which COX-2 is induced, and found that certain transcription factors are involved in the induction of COX-2 gene, including NF-IL6 (nuclear factor for interleukin-6) and CREB (cAMP response element-binding protein).8)–10) The involvement of NF-κB (nuclear factor-κB) was also reported by Yamamoto et al.11) NSAIDs exert their effects by inhibiting COX-2, but NSAIDs also inhibit COX-1, and as a result, the long-term use of NSAIDs is associated with severe side effects — mainly gastrointestinal injury and renal irritations. After the discovery of COX-2 in 1991,6),7) several pharmaceutical companies launched the development of COX-2 selective inhibitors as new NSAIDs that would have less adverse effects on the digestive system. These COX-2 selective inhibitors showed reduced gastrointestinal complications as expected, but several clinical trials have indicated a significantly increased cardiovascular risk of these medications.12),13) The specific inhibition of COX-2 alters the balance between platelet-derived thromboxane A2 (TXA2) and endothelium-derived PGI2, leading to increases in the risk of thrombosis due to altered vascular tone. Thus, for the development of novel NSAIDs without adverse side effects, a more selective modulation of prostanoid production appears to be desirable. PG terminal synthases have therefore gained attention as a novel target for NSAIDs.
Multiple PG terminal synthases have been identified (Table 1).4),5) In vitro analyses in which each PG terminal synthase was cotransfected with either COX isozyme into HEK293 cells revealed that each synthase preferentially couples with either COX-1 or COX-2 as its upstream enzyme.14),15) Among the PG terminal synthases, microsomal PGE synthase-1 (mPGES-1) and PGI synthase (PGIS) are the ones that show the greatest preferential coupling with COX-2, suggesting that these two enzymes are involved in COX-2-related diseases. My research group is, therefore, investigating the biochemical properties and the in vivo roles of these two PG terminal synthases. In this review, I summarize the current understanding of mPGES-1 and PGIS, and I discuss their potential as novel therapeutic targets. I also briefly summarize the biochemical and pathophysiological properties of the two other PGE synthases (PGESs) and thromboxane synthase (TXS), which is closely related to PGIS. For the other PG terminal synthases, i.e., the two PGD synthases, please refer to the other reviews.4),5)
| Cytogenetic localization |
No. of amino acid residues |
Molecular weight (kDa) |
Structural characteristics |
Tissue distribution |
Subcellular localization |
COX* coupling |
Phenotypes of KO mice | ||
|---|---|---|---|---|---|---|---|---|---|
| PGES | mPGES-1 | 9q34.11 | 152 | 16 | MAPEG family |
Inflamed tissues, cancerous tissues, kidney |
Perinuclear membrane |
COX-2 > COX-1 |
Impaired inflammatory reactions, suppression of carcinogenesis, etc. |
| mPGES-2 | 9q34.11 | 377 | 33 | Glutaredoxin/ thioredoxin- like |
Almost all tissues |
Cytosol, Golgi |
No preference |
- | |
| cPGES | 12q13.3 | 160 | 23 | - | Almost all tissues |
Cytosol | COX-1 > COX-2 |
Perinatal-lethal | |
| PGDS | H-PGDS | 4q22.3 | 199 | 23 | Glutathione- S-transferase |
Mast cells, microglia, Th2 cells, etc. | Cytosol | COX-1 > COX-2 |
Suppression of neuroinflammation & demyelination, delayed resolution of inflammation, suppression of carcinogenesis, etc. |
| L-PGDS | 9q34.3 | 190 | 26 | Lipocalin | Brain, heart, male genital organs, etc. | Secreted | - | Impaired sleep regulation and pain sensation, atherosclerosis and obesity, etc. | |
| PGIS | PGIS | 20q13.13 | 500 | 56 | Cytochrome P450 |
Vascular endothelial and smooth muscle cells, etc. |
Endoplasmic reticulum |
COX-2 > COX-1 |
High blood pressure, ischemic renal disorders, exacerbation of carcinogenesis, etc. |
| TXS | TXS | 7q34 | 533 | 60 | Cytochrome P450 |
Platelets, bronchial epithelium, etc. | Endoplasmic reticulum |
COX-2 > COX-1 |
Mild hemostatic defect, impaired wound healing |
PGFS enzymes are not included in this table. Despite the long history of research on the physiological and pathological functions of PGF2α, the identity of PGFS enzymes that catalyze PGH2 to PGF2α in vivo is unclear. Some enzymes that belong to the aldo-keto reductase (AKR) superfamily have been shown to exhibit PGFS activity.
*COX coupling indicates which COX isozymes functionally couple with each PG terminal synthase to yield higher amounts of the respective products when each terminal synthase is cotransfected with either of the COX isozymes into HEK293 cells.
PGES, which converts PGH2 to PGE2, is known to comprise a group of at least three structurally and biologically distinct enzymes (Table 1).16) Two of them are membrane-bound and have been designated as mPGES-1 and mPGES-2; the third is a cytosolic enzyme referred to as cytosolic PGES (cPGES). mPGES-1 was identified as the first PGES by Jakobsson et al. in 1999.17) Murakami et al. cloned rat and mouse orthologs of this protein and showed that mPGES-1 is identical to a membrane-associated PGES that had been originally detected in lipopolysaccharide (LPS)-stimulated macrophages (MΦs).14)
mPGES-1 consists of 152–153 amino acids and belongs to the MAPEG (membrane-associated proteins involved in eicosanoid and glutathione metabolism) family. mPGES-1 shows significant homology with other MAPEG superfamily proteins, including MGST-1, MGST-2, MGST-3, 5-lipoxygenase-activating protein, and leukotriene C4 synthase, with the highest homology being found with MGST-1 (38%). A projection structure of mPGES-1 at 10 Å resolution revealed structural properties that were similar to those determined for MGST-1, suggesting that the enzyme is a trimer of four helix bundles, in which the hydrophobic helices traverse the membrane.18) Sjögren et al. determined the crystal structure of human mPGES-1 to a 1.2 Å resolution, and their findings revealed three well-defined active site cavities within the membrane-spanning region in each monomer interface of the trimeric structure (Fig. 2).19) It was also shown that mutation of Asp49, Arg110, and Arg126 in mPGES-1, which is the residue strictly conserved in all MAPEG proteins, abrogates its catalytic function, implying an essential role of this residue.14),20)
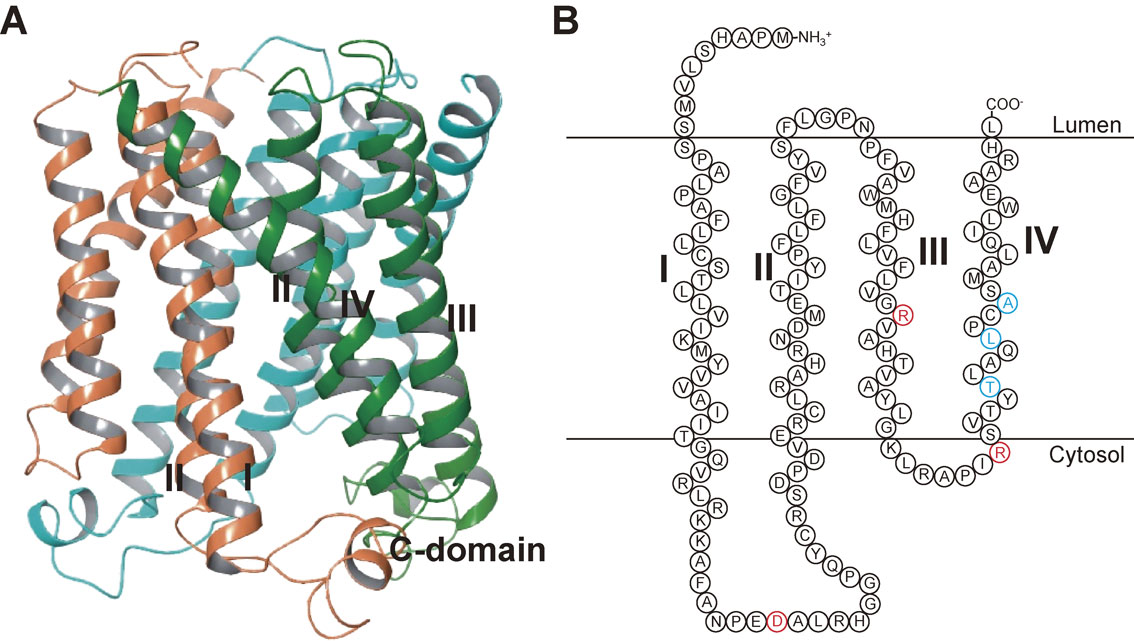
Structure of mPGES-1. (A) Overall structure of the human PGES-1 trimer (PDB4AL0).19) (B) Schematic model structure of the human mPGES-1 monomer. The red letters show the residues critical for mPGES-1 enzymatic activity,14),20) and the blue letters show the residues that account for the species discrepancy in the human-specific mPGES-1 inhibitor.57) I, II, III, and IV indicate transmembrane helix 1, 2, 3, and 4, respectively.
mPGES-1 requires glutathione (GSH) as an essential cofactor for its activity. As shown in Fig. 3, mPGES-1 isomerizes PGH2 to PGE2 in a manner dependent on GSH and two chemical mechanisms have been proposed to explain this activity.20) In addition, purified recombinant mPGES-1 catalyzes a rapid GSH-dependent conversion of PGH2 to PGE2 with a Vmax of 170 µmol/min/mg and a high kcat/Km of 310 min−1 s−1.18) The enzyme also catalyzes a GSH-dependent conversion of PGG2 to 15-hydroperoxy-PGE2. mPGES-1 displays weak GSH-dependent peroxidase activity toward cumene hydroperoxide, 5-hydroperoxyeicosatetraenoic acid, and 15-hydroperoxy-PGE2 and catalyzes a slow but significant conjugation of 1-chloro-2,4-dinitrobenzene to GSH.
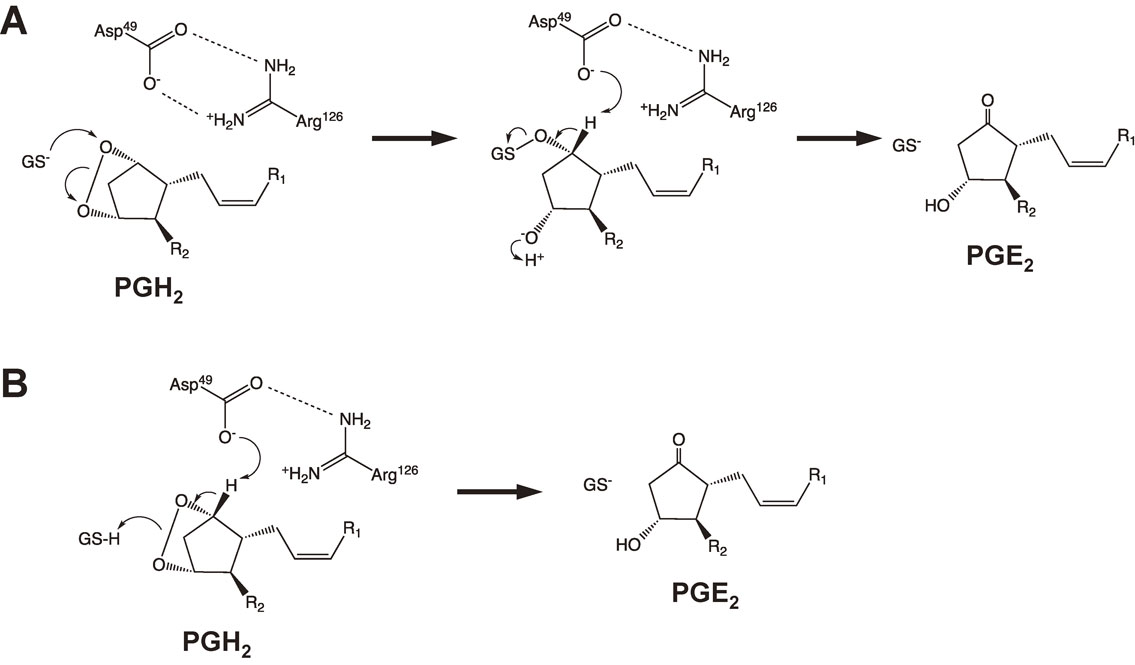
Two suggested chemical mechanisms of GSH-dependent isomerization of PGH2 to PGE2 by mPGES-1.20) (A) The thiolate of GSH could be stabilized by Arg126 and could attack on the endoperoxide oxygen atom at the C-9 carbon of PGH2 to produce an unstable reaction intermediate. The subsequent proton abstraction at C-9 followed by S-O bond cleavages is mediated by Asp49, and then a carbonyl forms and the oxygen sulfur bond is broken to form PGE2. (B) The reaction starts by proton abstraction at C-9 via Asp49, and then a carbonyl forms and the endoperoxide is broken. The thiol of GSH functions as a proton donor to the developing C-11 oxyanion.
Among the PGES enzymes, only mPGES-1 is markedly induced by proinflammatory stimuli, including LPS, tumor necrosis factor α (TNFα) and interleukin (IL)-1β.14),17),21)–23) Although the steady-state expression of mPGES-1 in normal rat tissues is very low, the administration of LPS leads to a dramatic increase in mPGES-1 expression in various tissues.14) Stimulation of various cultured cell lines with proinflammatory or mitogenic stimuli such as 12-O-tetradecanoylphorbol acetate leads to a marked elevation of mPGES-1 expression, often with a concomitant induction of COX-2. The concordant up-regulation of COX-2 and mPGES-1 is accompanied by a marked increase in the production of PGE2.14),21)–23)
mPGES-1 is functionally coupled with COX-2 in marked preference to COX-1. When COX-2 and mPGES-1 are cotransfected into HEK293 cells, considerable amounts of PGE2 are produced from both endogenous and exogenous AA relative to cells transfected with either enzyme alone.14) In comparison, coupling between COX-1 and mPGES-1 is observed only when the supply of AA is increased by explosive activation of cPLA2α. Because both COX-2 and mPGES-1 are present in the perinuclear membrane,14) colocalization of these two enzymes in the same subcellular compartment may allow efficient transfer of the unstable substrate PGH2 between them. Further support for COX-2/mPGES-1 coupling has been provided by studies using antisense oligonucleotides or small interfering RNAs, in which knockdown of mPGES-1 was shown to markedly attenuate COX-2-mediated PGE2 production in cultured cells.14),24) It has also been shown that the coordinate up-regulation of COX-2 and mPGES-1 and the attendant PGE2 generation are downregulated by an anti-inflammatory glucocorticoid.14),21)–23) However, there are some differences in the kinetics of induction between COX-2 and mPGES-1, suggesting that the regulatory mechanisms for their expression are distinct.21)
The promoter of the human mPGES-1 gene is GC-rich but lacks a TATA box, unlike the COX-2 gene promoter. In addition, the 3′ region of the mPGES-1 gene lacks the AUUUA mRNA instability sequences that are found in the COX-2 gene. Naraba et al. found that the tandem GC boxes adjacent to the initiation site in the mPGES-1 promoter play a major role in regulating its inducible transcription.25) Egr-1, an inducible zinc finger protein that recognizes the GC-rich consensus DNA sequence 5′-GCG(T/G)GGGCG-3′, binds to the proximal GC box in the mPGES-1 promoter region and facilitates the inducible transcription of the mPGES-1 gene.
It was very recently reported that hypoxia also induces mPGES-1 gene expression in cancer cells. A single consensus hypoxia-responsive element (HRE) was identified in the mPGES-1 gene promoter. Hypoxia-inducible factor (HIF)-1 and HIF-2 regulate hypoxia-induced mPGES-1 induction via an HRE.26),27)
2.3. The role of mPGES-1 in inflammatory reactions, pain and fever.mPGES-1 knockout (KO) mice were established by Uematsu et al. in 200228) and by Trebino et al. in 2003.29) Uematsu et al. demonstrated that the biosynthesis of PGE2 in wild-type peritoneal MΦs induced by LPS was abolished in mPGES-1-deficient cells.28) It was also shown that the increase in serum PGE2 concentration by the intraperitoneal administration of LPS was suppressed in mPGES-1 KO mice. Following this report, several investigators established mPGES-1 KO mice, and these mice have been used to demonstrate the involvement of mPGES-1 in inflammatory reactions and related diseases.
mPGES-1 KO mice are generally protected against various kinds of inflammatory disease phenotypes, including collagen- or anti-collagen antibody-induced arthritis, LPS-induced bone loss, and antigen-induced edema. Trebino et al. induced arthritis by the subcutaneous administration of chicken collagen type II and then assessed the symptoms of arthritis by clinical evaluation and histopathology.29) Their observations showed that, in this collagen-induced arthritis model, mPGES-1 KO mice developed no or little arthritis. Kamei et al. also revealed that using a collagen antibody-induced arthritis model, in which the influence of lymphocyte-mediated humoral responses could be minimal, the severity of synovial inflammation (including bone destruction and juxta-articular bone loss) were mild in mPGES-1 KO mice compared with wild-type mice, although the incidence of inflammation was unaffected.24) Antigen-induced paw edema was also markedly reduced in the mPGES-1 KO mice compared to the wild-type mice.29) This deficit in edema was accompanied by a marked reduction in the infiltration of white blood cells into the inflamed site.
Likewise, we found that after an intrapleural injection of carrageenan, mPGES-1 KO mice displayed a significantly reduced accumulation of exudate and impaired leukocyte migration into the pleural cavity.16) The migration of MΦs after a peritoneal injection of thioglycollate was also strikingly reduced in mPGES-1 KO mice relative to wild-type mice.24) mPGES-1-derived PGE2 might increase the vascular permeability in inflammatory reactions.
In another study of mPGES-1 KO mice, the formation of inflammatory granulation tissue and attendant angiogenesis in the dorsum induced by the subcutaneous implantation of a cotton thread was also significantly reduced.24) In this model, mPGES-1 deficiency was also associated with a reduced induction of vascular endothelial cell growth factor (VEGF) in the granulation tissue. These results suggested that mPGES-1-derived PGE2, in cooperation with VEGF, may play a critical role in the development of inflammatory granulation and angiogenesis.
mPGES-1 KO mice exhibit reduced pain, fever, and other symptoms associated with inflammatory diseases. Kamei et al. examined the effect of mPGES-1 deficiency on the inflammatory pain response by injecting diluted acetic acid into the peritoneal cavity of mice.24) The pain response, as evaluated based on the amount of writhing, was significantly reduced in mPGES-1 KO mice compared with wild-type mice. This phenotype was particularly evident when the KO mice were primed with LPS, because the peritoneal PGE2 level of these KO mice was far less than that of wild-type mice, which was consistent with the elevated expressions of COX-2 and mPGES-1 in response to LPS. Engblom et al. further observed that in addition to the genetic inactivation of COX-2, mPGES-1 deletion resulted in a reduction of PGE2 levels in the central nervous system in association with the impaired LPS-induced febrile response.30)
2.4. The role of mPGES-1 in neurological diseases.Inflammation is a critical factor in the progression of neurological diseases (including ischemic stroke) and several neurodegenerative diseases, such as multiple sclerosis and Alzheimer’s disease (AD). The role of mPGES-1-derived PGE2 in these diseases has been established in models using mPGES-1 KO mice. In mPGES-1 KO mice, infarction, edema, and apoptotic cell death in the cortex after ischemia were all reduced compared with those in wild-type mice.31) The behavioral neurological dysfunctions observed after ischemia in wild-type mice were also significantly ameliorated in mPGES-1 KO mice. In addition, mPGES-1 KO mice had less-severe symptoms of experimental autoimmune encephalomyelitis.32)
On the other hand, we found that the expression of mPGES-1 is associated with β-amyloid plaques in the cerebral cortex in human AD patients and in Tg2576 mice, a transgenic AD mouse model.33) To investigate whether mPGES-1 contributes to AD-like pathology in vivo, mPGES-1 KO mice were bred with Tg2576 mice, and it was found that mPGES-1 deletion reduced the accumulation of microglia around senile plaques and attenuated learning impairments in the Tg2576 mice. In vitro analysis further showed that mPGES-1 deletion protected primary neuronal cells from the cytotoxic effects of β-amyloid peptide fragment.34)
2.5. The role of mPGES-1 in cancer.Kamei et al. found that, as in COX-2, mPGES-1 levels are increased in a number of human cancers, and they showed the tumorigenic potential of mPGES-1 using cell culture systems.35) The transfection of mPGES-1 in combination with COX-2, but not with COX-1, into HEK293 cells led to cellular transformation with a concomitant increase in PGE2. The COX-2/mPGES-1-cotransfected cells formed a number of large colonies in soft agar culture and were tumorigenic when implanted into nude mice.
Oshima et al. showed that transgenic mice overexpressing both COX-2 and mPGES-1 developed metaplasia, hyperplasia, and tumorous growths in the glandular stomach with heavy MΦ infiltration.36) It was also shown that PGE2 synthesis, cell proliferation, and invasive activity in vitro and xenograft formation in vivo were reduced by mPGES-1 knockdown and conversely enhanced by mPGES-1 overexpression in Lewis lung carcinoma (LLC) cells.37)
In addition to cancer cell-associated mPGES-1, host-associated mPGES-1 also contributes to tumor growth, invasion, and metastasis. LLC tumors grafted subcutaneously into mPGES-1 KO mice grew more slowly than those grafted into wild-type mice, with concomitant decreases in the density of microvascular networks.37) Lung metastasis of intravenously injected LLC cells was also significantly less obvious in mPGES-1 KO mice than in wild-type mice. We further found that the growth of intrasplenically transplanted tumor cells was suppressed in mPGES-1 KO mice.38) The co-culture of tumor cells with bone marrow-derived MΦs (BM-MΦs) isolated from wild-type mice resulted in the induction of mPGES-1 in BM-MΦs and increased the growth of tumor cells in vitro, whereas mPGES-1-null BM-MΦs failed to facilitate tumor growth. The adoptive transfer of wild-type BM-MΦs into mPGES-1 KO mice restored the growth of transplanted tumor cells. These results suggested that mPGES-1 induced in tumor-associated MΦs is important for the growth of adjacent tumor cells.
The effect of mPGES-1 deficiency on intestinal tumorigenesis was also reported. Nakanishi et al. showed that the genetic deletion of mPGES-1 ameliorated both the ApcΔ14-dependent and azoxymethane (AOM)-induced development of intestinal tumors.39),40) We also found that mPGES-1 deletion suppressed AOM-induced colon carcinogenesis with a reduced nuclear translocation of β-catenin and altered the expression profiles of chemokines/cytokines.38) At an early stage of colon carcinogenesis, mPGES-1 deficiency significantly reduced the number of aberrant crypt foci (ACF), whereas its transgenic overexpression increased the number. It is noteworthy that the genetic ablation of mPGES-1 resulted in not only the suppression of carcinogenic PGE2 production but also the reciprocal upregulation of anticarcinogenic PGD2 and PGI2 production in tumor tissues.
In light of the fact that the inhibition of COX-2 suppresses both pro- and anti-tumorigenic PGs, an mPGES-1-specific inhibitor that blunts pro-tumorigenic PGE2 while increasing anti-tumorigenic PGD2 and PGI2 is expected to be a more effective anti-carcinogenic agent than a COX-2-specific inhibitor. Mammary tumor growth and angiogenesis in human epidermal growth factor receptor 2-overexpressing mice were also suppressed by the gene deletion of mPGES-1.41) Very recently, we found that mPGES-1 deletion suppressed chemically induced skin carcinogenesis (manuscript in preparation). An mPGES-1-specific inhibitor is expected to exhibit antitumor effects in various types of tissues.
2.6. Biochemical properties, expression, and in vivo role of mPGES-2.As described above, there are at least three structurally and biologically distinct PGESs (Table 1).16) The second membrane-associated form of PGES, mPGES-2, is a 41 kDa protein consisting of 378–385 amino acids, which is structurally distinct from mPGES-1.42) mPGES-2 has a catalytic glutaredoxin/thioredoxin-like domain and is activated by various thiol reagents. This enzyme is synthesized as a Golgi membrane-associated protein, and proteolytic removal of the N-terminal hydrophobic domain leads to the formation of a mature cytosolic enzyme.42),43) Cotransfection of mPGES-2 with either COX isozyme demonstrated that mPGES-2 could be coupled with both COX-1 and COX-2, leading to PGE2 production.43) Unlike mPGES-1, mPGES-2 expression is rather constitutive in various cells and tissues and is not elevated appreciably during inflammation or tissue damage.
mPGES-2 KO mice were established by Jania et al. in 2009,44) but mPGES-2 KO mice showed no specific phenotype and no alteration in PGE2 levels in several tissues (including liver, kidney, heart, and brain) or in LPS-stimulated MΦs. These results suggested that mPGES-2 is not involved in PGE2 synthesis in the physiological and pathological conditions tested thus far. However, Sun et al. recently showed that genetic deletion of mPGES-2 remarkably enhances liver injury in streptozotocin-treated mice via induction of GLUT2.45) The possibility of tissue-specific or particular pathological roles of mPGES-2 has not yet been ruled out.
2.7. Biochemical properties, expression, and in vivo role of cPGES.The third PGES, cPGES, is a GSH-requiring enzyme that is constitutively expressed in a wide variety of cells and is identical to p23, a co-chaperone of heat shock protein 90 (Hsp90).46) Cotransfection and antisense experiments indicated that cPGES is capable of converting COX-1-, but not COX-2-, derived PGH2 to PGE2 in cells, particularly during the immediate PGE2 biosynthetic response elicited by Ca2+-evoked stimuli.46) Localization of cPGES in the cytosol may allow coupling with proximal COX-1 in the endoplasmic reticulum (ER) in preference to distal COX-2 in the perinuclear envelope, although other regulatory mechanisms could be involved. cPGES is directly associated with and phosphorylated by protein kinase CK2. In activated cells, CK2-directed phosphorylation of cPGES occurs in parallel with increased cPGES enzymatic activity and PGE2 production, and these processes are facilitated by interaction with Hsp90.47)
To elucidate the in vivo function of cPGES, cPGES KO mice were developed, but cPGES KO mice were perinatal-lethal with poor lung development, delayed skin maturation, and growth retardation.48)–50) The lung phenotype was consistent with a defective glucocorticoid response and with p23 acting as a co-chaperone for the glucocorticoid receptor/Hsp90 complex. In fibroblasts and tissues from cPGES KO mice, the expression of glucocorticoid-responsive genes, as well as the glucocorticoid transcriptional activation of reporter plasmids, was reduced.48),49) It was noteworthy that although PGE2 levels were reduced in lung and other tissues in cPGES KO mice, primary fibroblasts from these mice showed increased, rather than decreased, PGE2 production. We found that cPGES-deficient fibroblasts decreased the expression of the PGE2 degrading enzyme, 15-hydroxyprostaglandin dehydrogenase, which catalyzes the inactivating conversion of the PGE2 15-OH to a 15-keto group, compared with that of wild-type fibroblasts.51) These results suggested that the PGE2-inactivating pathway may be controlled by the PGE2 biosynthetic enzyme, cPGES.
2.8. Development of mPGES-1 inhibitors and their potential as novel NSAIDs.The experimental data described above using mPGES-1, mPGES-2, and cPGES KO mice indicated that, among these three PGES enzymes, mPGES-1 may be the primary target in the treatment of inflammatory diseases and cancers. It is also noteworthy that the genetic deletion of mPGES-1 in mice, unlike disruption, deletion, or inhibition of COX-2, failed to alter blood pressure or modulate the response to a thrombogenic stimulus.52),53) These studies suggested the possibility that the pharmacological targeting of mPGES-1 may ultimately prove to be less toxic and perhaps more effective than traditional NSAIDs.
New drug candidates have recently been developed for targeting mPGES-1 (Fig. 4), and some of them have been shown to suppress inflammatory reactions or tumor development in animal models. Xu et al. reported that when tested in guinea pigs and a knock-in mouse expressing human mPGES-1, a novel inhibitor of human mPGES-1 named MF63 inhibited LPS-induced pyresis, hyperalgesia, and iodoacetate-induced osteoarthritic pain, and it did not cause gastrointestinal toxic effects.54) Another mPGES-1 inhibitor, PF-4693627, was recently reported by Arhancet et al. to have high potency and selectivity and to be effective in a carrageenan-induced guinea pig model of inflammation.55) Finetti et al. reported that in A431 squamous carcinoma-derived xenografts in nude mice, AF3485 (a compound with human mPGES-1 inhibitory activity) reduced tumor-associated angiogenesis and tumor growth.56)
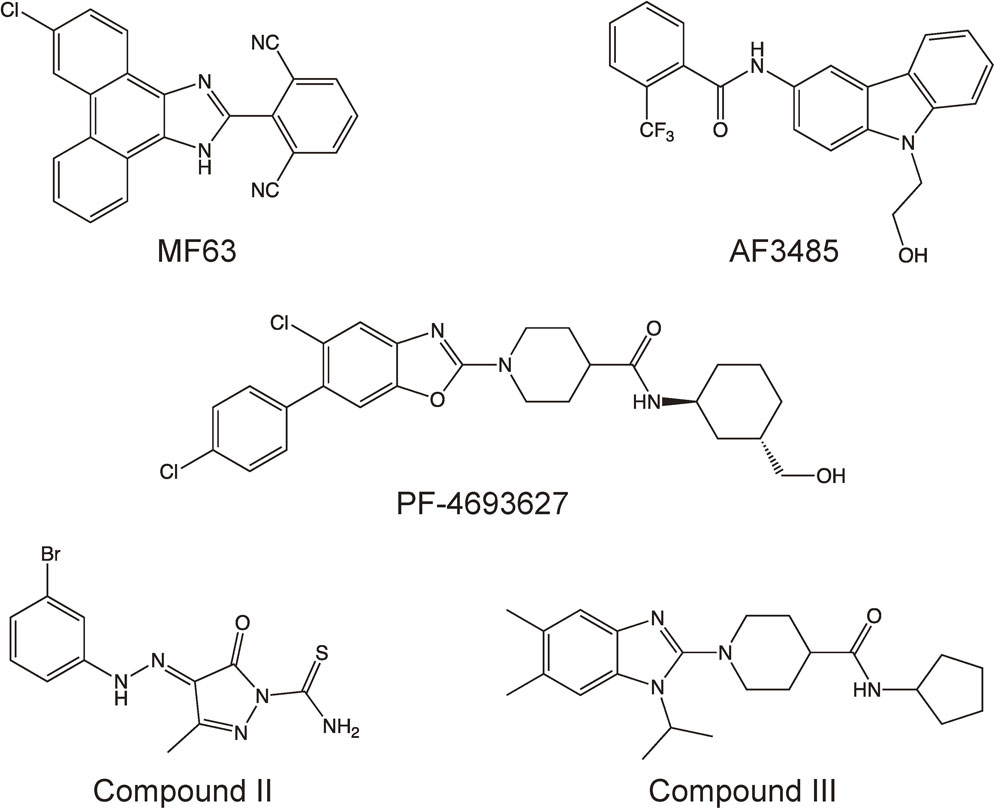
Structural formulae of mPGES-1 inhibitors.
It is noteworthy that these compounds, which are currently being developed by pharmaceutical companies, inhibit human or guinea pig mPGES-1 enzymes but do not have any effect on rat or mouse mPGES-1,54)–56) which complicates animal testing in preclinical studies. The observed interspecies differences (human/guinea pig vs rat/mouse) might lead to a delay in bringing these drugs to the market. Pawelzik et al. demonstrated three individual amino acid residues (131Thr, 135Leu, and 138Ala) that are not conserved between human/guinea pig and rat/mouse mPGES-1 (Fig. 2) and are located close to the active site of mPGES-1 (in transmembrane helix 4), and these accounted for the species discrepancy of human/guinea pig-specific inhibitors.57)
Leclerc et al. developed new mPGES-1 inhibitors, compounds II and III, and they showed that both of these compounds were able to inhibit both human and rat mPGES-1, predominantly through competition with PGH2 for the active site of mPGES-1.58),59) They further reported that compound II attenuated both the acute and delayed inflammatory responses in rat adjuvant-induced arthritis58) and that compound III reduced cell migration into inflamed sites in a mouse air pouch model.59) Compounds II and III are now expected to be used in studies of mPGES-1 inhibition in rodent models of human diseases.
mPGES-1-derived PGE2 acts on four subtypes of PGE2 receptor EPs (EP1, EP2, EP3, and EP4) to mediate inflammatory reactions and carcinogenesis.60) All four EPs are known to be involved in inflammatory diseases and cancers.60)–62) Thus, in addition to mPGES-1 inhibitors, several EP antagonists have been developed as candidates for new anti-inflammatory or anti-tumor drugs. However, although mPGES-1 inhibitors inhibit all receptors downstream of mPGES-1-derived PGE2, subtype-specific EP antagonists target only one pathway. More than one antagonist may be required to suppress and/or treat inflammatory diseases and cancers. It is also noteworthy that, in the human body, PGE2 is constantly produced to physiologically maintain homeostasis, and EP mediates this physiological action of PGE2 as well as the pathological action of mPGES-1-derived PGE2. Attention should be paid to any possible mechanisms by which EP antagonists might block the physiological actions of PGE2.
Although mPGES-1-specific inhibitors may ultimately become novel NSAIDs with less toxicity than COX-2-specific inhibitors and EP antagonists, it should be remembered that the pharmacologic blockade of mPGES-1 could upregulate the production of prostanoids other than PGE2 by shunting PGH2 to the other PG terminal synthases. Peña Silva et al. reported that mPGES-1 deficiency increased the incidence of subarachnoid hemorrhage in a mouse model of intracranial aneurysm.63) They further showed that aspirin, but not PGE2, attenuated the increased mortality in mPGES-1 KO mice. These results suggested that in the absence of mPGES-1, accumulated PGH2 could be converted to harmful prostanoids. Chronic treatment with mPGES-1 inhibitors might induce compensatory responses to affect the balance of production of each prostanoid. Moreover, it is important to remember that mPGES-1-derived PGE2 is crucial for maintaining gastrointestinal mucosal homeostasis. In a mouse model of dextran sodium sulfate-induced colitis, we found that mPGES-1 deficiency induced severe epithelial loss and crypt abscesses in the colon.16) It was also reported that mPGES-1 together with COX-2 was induced in the granulation tissues of acetic acid-treated mouse stomachs, and that the healing of acetic acid-induced ulcers was significantly delayed in mPGES-1 KO mice compared with wild-type mice.64) Therefore, any mPGES-1 inhibitors that are developed will need to be used carefully in patients with inflammatory bowel diseases or gastrointestinal ulcers.
PGIS and TXS catalyze the isomerization of PGH2 to PGI2 and TXA2, respectively. Ullrich and co-workers were the first to provide spectral evidence that both of PGIS and TXS are cytochrome P450s,65),66) but unlike most P450s, neither of them requires NADPH and O2 as cosubstrates.65)–67)
PGIS and TXS act on PGH2 from the opposite direction. After the hemolytic cleavage of the O-O bond of PGH2, PGIS coordinates with the oxygen atom at C-11 and TXS coordinates with the oxygen atom at C-9 (Fig. 5).68) In addition to TXA2, almost the same amounts of malondialdehyde and 12-hydroxyheptadecatrienoic acid (12-HHT) are produced by the TXS reaction.66) 12-HHT was recently found to act on leukotriene B4 type-2 receptor (BLT2)69) and then to promote epidermal wound healing by accelerating keratinocyte migration.70)
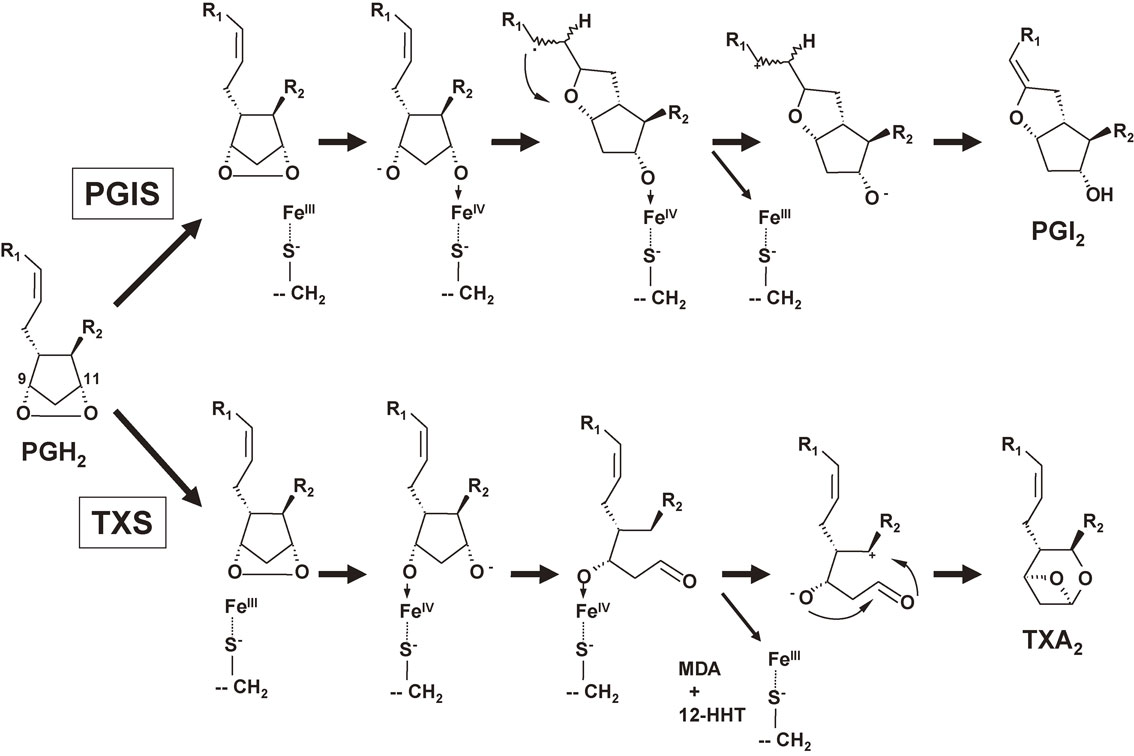
Mechanism of PGI2 and TXA2 biosynthesis by PGIS and TXS, respectively.68) For PGI2 biosynthesis, PGH2 is bound to the active site of PGIS with its C-11 oxygen oriented close to the heme iron. The homolytic scission of the endoperoxide bond followed by the immediate formation of an [S−⋯FeIV⋯O-R] species leads to an alkoxyradical. Subsequent cyclization to the carbon radical, Fe(IV) oxidation to the carbocation, and loss of the C-6 proton yield the vinyl ether, PGI2. In contrast, for TXA2 biosynthesis, PGH2 is bound to the active site of TXS with its C-9 oxygen at the heme iron. In addition to TXA2, almost the same amounts of malondialdehyde (MDA) and 12-HHT are produced by the TXS reaction.
In 1994, we collaborated with Ullrich and colleagues to purify and characterize PGIS from bovine aorta, and the results demonstrated that the purified PGIS catalyzes the conversion of PGH2 to PGI2 with a Vmax of 2.0 µmol/min/mg.71) We further isolated bovine and human cDNAs for PGIS and designated this enzyme as CYP8A1 in the P450 family.71),72) The PGIS cDNA encoded a 56-kDa protein consisting of 500–501 amino acid residues. On the other hand, human TXS cDNA encodes a 60-kDa protein consisting of 533 amino acid residues.73) The homology of PGIS and TXS is very low (only 14%), although both enzymes reacted with the same substrate PGH2. TXS is designated as CYP5A1 in another P450 family. Site-directed mutagenesis of human PGIS revealed that, similar to other P450s, Cys441 in the Cys-pocket and Glu347 and Arg350 of the EXXR motif are important for the expression of the enzymatic activity of PGIS.74)
The crystal structures of ligand-free and substrate analog-bound PGIS have also been determined.75),76) It was revealed that PGIS differs from other P450s in that a conformational change on the proximal side accompanied by heme relocation is observed upon ligand binding. In PGIS, a heme barrier might be evoked to ensure strict binding specificity for PGH2, leading to a radical-mediated isomerization with high product fidelity.
TXS is localized in the ER and perinuclear membranes and could be coupled with both COX-1 and COX-2. The beneficial effect of low-dose aspirin on the incidence of cardiovascular diseases is due to suppression of the synthesis of prothrombotic TXA2 by platelets, in which COX-1/TXS coupling occurs because COX-1 is the dominant COX isozyme in platelets.77) On the other hand, MΦs produce TXA2 via constitutive COX-1 after Ca2+ ionophore stimulation and via inducible COX-2 after LPS stimulation.78) Cotransfection experiments using HEK293 cells revealed that TXS is coupled with COX-2 in preference to COX-1, yet the supply of a high concentration of AA (from high-level cPLA2α activation) allows TXS to be coupled with COX-1.15) These observations suggested that TXS-COX coupling might depend on substrate availability and that COX-1-dependent TXA2 production could occur in cells intrinsically expressing a high level of cPLA2α, such as platelets and MΦs. The observations that PGI2, unlike TXA2, was preferentially produced via COX-2 in MΦs79) and endothelial cells80) and that the serum PGI2 level was blunted after in vivo administration of COX-2-selective inhibitors81) suggested that PGIS is rather strictly coupled with COX-2. Cotransfection of PGIS with either COX isozyme in HEK293 cells confirmed that the preference for COX-2 was more strict in PGIS than that in TXS.15) This could be explained in part by the restricted localization of PGIS in the perinuclear membranes, where COX-2 is located.15)
3.2. Expression and transcriptional regulation of PGIS.Although PGIS is abundantly expressed in vascular endothelial and smooth muscle cells, our in situ hybridization analysis revealed that this enzyme is expressed not only in vascular cells but also in other cells such as fibroblasts in the heart myocardium and lung parenchyma cells.71),72),82) Peritoneal MΦs, dendritic cells, and T cells also express PGIS.83)–85) It has been shown that inflammatory cytokines, including IL-1β, IL-6, and TNF-α, hypoxia, and hemodynamic forces such as cyclic strain and shear stress increased PGIS expression levels in vascular cells, but the increase in PGIS expression in response to these stimuli was smaller than that of COX-2 or mPGES-1.71),72),86),87) PGIS is essentially expressed as a constitutive enzyme, although as described above, PGIS is functionally coupled with inducible COX-2 in marked preference to COX-1, as is the case for mPGES-1.15)
The 5′-flanking region of the human PGIS gene contains both GC-rich and pyrimidine-rich regions but it does not have a typical TATA box.88) Transient transfection experiments using vascular endothelial cells demonstrated that the GC-rich region possessed significant promoter activity. This region contained tandem repeats of the 9-base pair (bp) sequence CCGCCAGCC, including Sp1 (and AP-2) binding sites. Iwai et al. identified a repeat polymorphism that is associated with promoter activity.89) The number of 9-bp repeats varied between 3 and 7 in Japanese subjects. A luciferase reporter analysis indicated that alleles with 3 and 4 repeats had less promoter activity in vascular endothelial cells. It was further shown that this repeat polymorphism might be a risk factor for systolic hypertension and cerebral infarction in the Japanese population.89),90)
3.3. The role of PGIS in vascular homeostasis.PGIS-derived PGI2 is a strong vasodilator and the most potent endogenous inhibitor of platelet aggregation, whereas TXS-derived TXA2 is a potent stimulator of platelet activation and aggregation and vascular constriction. PGI2 also inhibits the growth of vascular smooth muscle cells. We showed that the overexpression of PGIS in vascular smooth muscle cells in vitro increased PGI2 synthesis and then inhibited cell growth.91) Therefore, PGIS has been considered to play an important role in cardiovascular diseases. Yokoyama et al. established PGIS KO mice and demonstrated that PGIS gene deletion impaired vascular homeostasis.92) The blood pressure of PGIS KO mice was significantly higher than that of wild-type mice. PGIS KO mice also developed ischemic renal disorders, including nephrosclerosis and renal infarction.
The in vivo function of PGIS indicated that the gene transfer of PGIS may be useful for gene therapy for cardiovascular diseases. The efficacy of gene transfer of PGIS on cardiovascular disease has already been shown in several animal models. We demonstrated that the gene transfer of PGIS prevents neointimal formation after a carotid balloon injury in rats.93) Monocrotaline- or hypoxia-induced pulmonary hypertension was also ameliorated by PGIS gene transfer.94),95) Koike et al. reported that cotransfection of the PGIS gene with the hepatocyte growth factor (HGF) gene enhanced the effect of HGF gene transfer on angiogenesis and resulted in an improvement of neuropathy in a mouse hindlimb ischemia model.96)
3.4. The role of PGIS in inflammatory reactions.Studies using PGI2 receptor (IP) KO mice revealed that PGI2 is involved in inflammatory and pain responses as well as the regulation of vascular tone.84),97),98) We showed that inflammatory reactions such as exudation of leukocytes and inflammatory pain hypersensitivity were significantly suppressed in PGIS KO mice, and that these reactions tended to be suppressed more effectively in PGIS and mPGES-1 double KO (DKO) mice than in PGIS single KO mice (Fig. 6A).85) These results suggested that PGIS-derived PGI2 acts on IP at inflamed sites and exacerbates inflammation in cooperation with mPGES-1-derived PGE2.
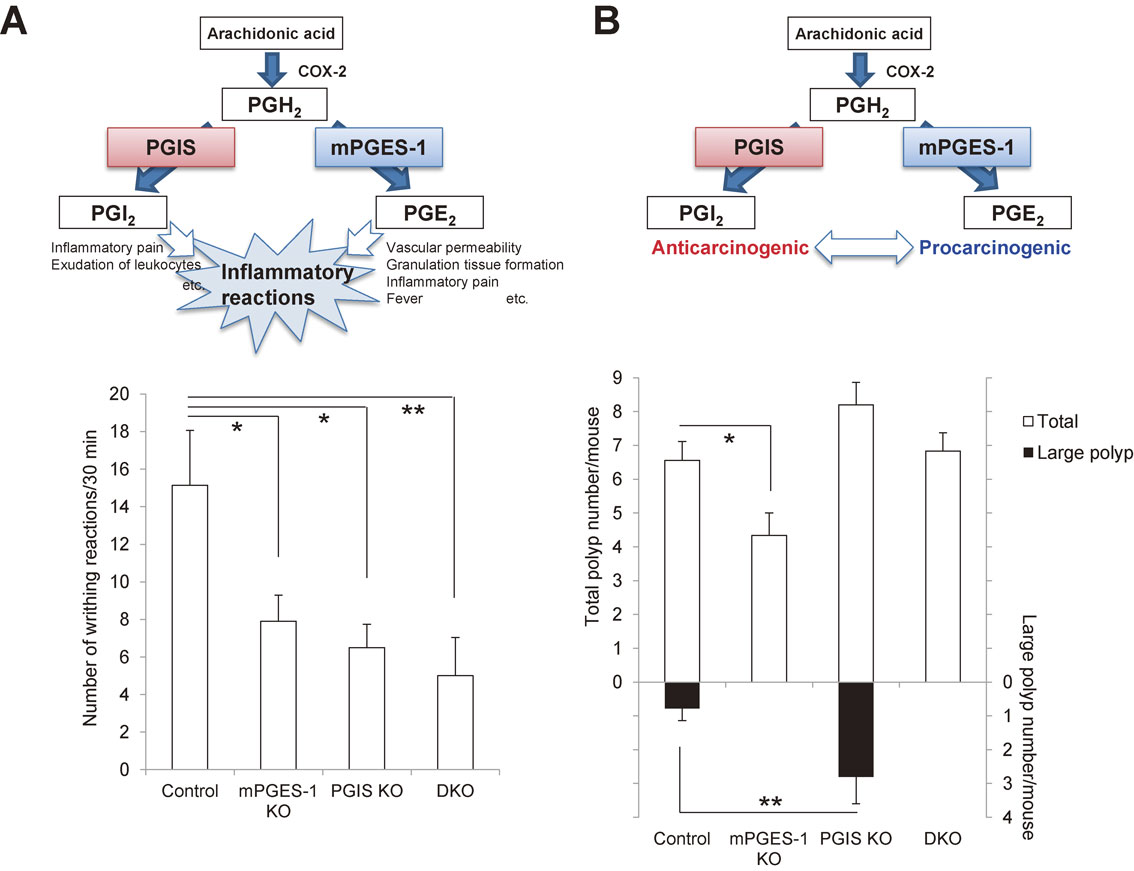
Crosstalk between PGIS-derived PGI2 and mPGES-1-derived PGE2 in inflammatory reactions (A) and carcinogenesis (B). A: Crosstalk between PGIS and mPGES-1 in inflammatory reactions. Both COX-2/PGIS-derived PGI2 and COX-2/mPGES-1-derived PGE2 function as proinflammatory agents. They cooperatively exacerbate inflammatory reactions. As shown in the lower panel, the LPS-primed acetic acid-induced writhing reaction was reduced in both the PGIS KO and mPGES-1 KO mice, and it was suppressed more effectively in the PGIS/mPGES-1 DKO mice compared with the PGIS KO and mPGES-1 KO mice (*p < 0.05 and **p < 0.01 vs control mice).85) B: Crosstalk between PGIS and mPGES-1 in carcinogenesis. COX-2/PGIS-derived PGI2 functions as an anticarcinogenic agent in contrast to COX-2/mPGES-1-derived PGE2 in various tissues. When mPGES-1-derived PGE2 increases and then exceeds the anticarcinogenic actions of PGIS-derived PGI2, a tumor might progress. As shown in the lower panel, in an AOM-induced colon cancer model, mPGES-1 deficiency decreased the number of polyps, whereas PGIS deficiency tended to increase the polyp numbers and significantly increased the number of large polyps (*p < 0.05 and **p < 0.01 vs control mice).85)
A role of PGIS in carcinogenesis has also been suggested. Pulmonary-specific overexpression of PGIS decreased lung tumor multiplicity in chemical- and cigarette-smoke-induced murine lung cancer models.99),100) We also showed that in an AOM-induced colon cancer model, the genetic deletion of PGIS up-regulated both ACF formation in the early stage of carcinogenesis and polyp formation in a later stage (Fig. 6B).85) It was also found that colon-26 cells grafted subcutaneously into PGIS KO mice grew faster than those grafted into littermate control mice.101) These results suggest that PGIS may be chemopreventive in cancer.
Several groups of investigators have examined the role of tandem repeat polymorphism and methylation in the PGIS promoter in colorectal cancer. A large case-control study of adenomatous and hyperplastic colon polyps by Poole et al. showed an increased risk of these polyps in individuals when both PGIS promoter alleles had a variable number of tandem repeat lengths less than six.102) In addition, Frigola et al. reported the hypermethylation of the CpG dinucleotides in the PGIS promoter in colorectal cancer.103) A very marked loss in PGIS mRNA levels was also observed in primary human lung tumor samples compared with matched normal controls.104)
These observations suggested that the balance between COX-2/mPGES-1-derived PGE2 and COX-2/PGIS-derived PGI2 might be critical for carcinogenesis (Fig. 6B). Even when COX-2 expression was induced, if PGIS-derived PGI2 overcomes the pro-carcinogenic actions of mPGES-1-derived PGE2, tumor progression might be suppressed. However, if PGIS is downregulated or mPGES-1 is highly induced, and then mPGES-1-derived PGE2 exceeds the anti-carcinogenic actions of PGIS-derived PGI2, the tumor might progress.
The carcinogen-induced lung tumor incidence in mice was not affected by IP gene deficiency.105) Anti-tumor effects of PGIS overexpression were observed even in IP KO mice. These results suggested that the anti-tumor effects of PGIS-derived PGI2 are not mediated through the activation of IP. PGI2 is known to act on several G-protein-coupled receptors (GPCRs) other than IP.106) Although the possibility that PGIS-derived PGI2 exerts its anti-tumor effects through these GPCRs, orphan GPCRs, or ion channels cannot be excluded, transcriptional factors such as members of the peroxisome proliferator-activated receptor (PPAR) family are strong candidates for targeting PGI2-mediated cancer chemoprevention. In vitro studies showed that PGI2 can activate PPARs,107),108) and the expressions of PPARγ and PPARδ are found to be elevated in several types of cancers, including colorectal cancer.109),110) It was also shown that transgenic mice overexpressing PPARγ developed fewer lung tumors.105) In addition to PPARγ, a PGI2-mediated activation of PPARδ has also been shown to be involved in the negative growth of lung cancer cell lines.111) These findings suggested that PPARγ or PPARδ might be a target for PGI2-mediated cancer chemoprevention.
Several studies of the role of PPARδ in cancer, however, have produced contradictory results, and there is no scientific consensus on whether PPARδ promotes or prevents cancer formation.110) For example, Wang et al. reported that PGI2 activated PPARδ, leading to a subsequent acceleration of intestinal tumor growth in ApcMin/+ mice.112) Furthermore, activation of PPARs by PGI2 itself remains controversial. Forman et al. reported that a significantly higher concentration of PGI2 and its analogues are required for activation of PPARs compared with concentrations that are able to stimulate IP.107) It was also shown that the binding of PGI2 to PPARs is not specific. Further studies are needed to elucidate the mechanism underlying the suppression of carcinogenesis by PGIS-derived PGI2.
3.6. Expression and in vivo role of TXS.TXS-derived TXA2 is a potent inducer of platelet aggregation and of platelet release reactions. TXA2 is also known to cause respiratory tract and vascular smooth muscle contraction,113) and has been implicated as a mediator in diseases such as myocardial infarction, stroke, and bronchial asthma.114) In addition to its expression in platelets,66),73) TXS is expressed in many tissues, including spleen, lung and thymus, and in leukocytes and peritoneal MΦs.83),115),116) Gene deletion of TXS caused a mild hemostatic defect and protected mice against AA-induced shock and death.117) TXS inhibitors were originally considered to be promising antiplatelet agents, but clinical trials of various inhibitors yielded unsatisfactory results compared with low-dose aspirin. Nonetheless, TXS inhibitors have been evaluated for their potential in other TXA2-involving diseases, such as bronchial asthma and pulmonary hypertension.118)
It is also important to remember that 12-HHT is also produced by the TXS reaction as well as TXA2,66) and 12-HHT has the ability to promote epidermal wound healing by accelerating keratinocyte migration via BLT2.69),70) TXS KO mice exhibited impaired re-epithelialization and delayed wound closure after skin punching.
3.7. PGIS as a novel therapeutic target.As described above, the tandem repeat polymorphism and methylation of the gene promoter affect the expression of PGIS, and the downregulation of PGIS is associated with cardiovascular diseases and cancer.89),90),102),103) Several single-nucleotide polymorphisms in the PGIS gene have been identified, and some were shown to affect PGIS enzymatic activity.119) An association between a certain type of single-nucleotide polymorphism and the risk of cancers was also reported.120) Genotypes of the PGIS gene may be useful as a predictive factor for cardiovascular diseases and cancer.
Although PGIS-derived PGI2 is involved in inflammatory reactions, it may be difficult to use a PGIS inhibitor as an anti-inflammatory or analgesic drug, because the inhibition of PGIS might disturb vascular homeostasis. On the other hand, the upregulation of PGIS, such as in gene therapy using the PGIS gene, is very promising as a treatment for cardiovascular diseases. PGI2 itself and several IP agonists such as beraprost and iloprost are now used to treat pulmonary arterial hypertension, chronic arterial occlusion, and other diseases in which the blood vessels are constricted and blood cannot flow to the tissues.121) However, these drugs have short durations and require repeated administration. Gene therapy using the PGIS gene is expected to be more effective and convenient than these drugs. Moreover, the anti-tumor effects of PGIS-derived PGI2 are not mediated through the activation of IP. Unlike the case of IP agonists, PGIS gene therapy may, therefore, have some effects on cancers, as well as cardiovascular diseases.
Indeed, Kawakami et al. reported that PGIS gene transfer using adeno-associated virus (AAV) vector significantly reduced hypoxia-induced pulmonary hypertension in a mouse model.95) However, many problems remain to be solved before PGIS gene therapy can be used in clinical practice. For example, to continuously produce the large amounts of PGI2 necessary for therapy of patients, not only PGIS upregulation but also a sufficient supply of PGH2 to PGIS might be necessary. Ruan et al. engineered an expression vector for a hybrid enzyme linking COX-2 and PGIS.122) This hybrid enzyme, expressed in HEK293 cells and COS-7 cells, could actively convert AA into PGI2. Such approaches might solve the problem, but the matter of highest importance in gene therapy for patients is establishment of the safety and efficacy. Kawakami et al. reported that with PGIS gene transfer using the AAV vector, there was no evidence of inflammatory responses or tumor formation at the vector-injected site in mice.95) As this report indicated, AAV is a relatively safe and nontoxic virus among viruses used for gene therapy; however, it has also been shown that AAV injection less often caused oncogenesis, immune response, and toxicity.123),124) Further improvements in the safety and efficacy of manufacture of gene delivery systems are needed for PGIS gene therapy in clinical use.
It has become apparent that there are multiple PG terminal enzymes in mammalian cells, and distinct PG terminal enzymes may control the different pathophysiological aspects of the spatial and temporal production of prostanoids in particular tissues and cells. Findings from studies by my own research group and others have demonstrated that among the PG terminal synthases, mPGES-1 and PGIS are the most attractive alternatives to COX as therapeutic targets. I expect that future studies will lead to the development of novel NSAIDs that do not have the adverse effect associated with current therapies.

Shuntaro Hara was born in Tokyo in 1962. He graduated from the University of Tokyo in 1986 and received his M.S. and Ph.D. degrees from the Graduate School of Pharmaceutical Sciences, the University of Tokyo in 1988 and 1991, respectively. He started his research career by studying lipid mediators as a postdoctoral fellow in Professor Keizo Inoue’s laboratory at the University of Tokyo. After postdoctoral training, Dr. Hara has worked as a research associate in Dr. Tadashi Tanabe’s laboratory at the National Cardiovascular Center Research Institute (1992–), as a visiting fellow in Professor Shamshad Cockcroft’s laboratory at University College London (1995–), as an assistant professor in Professor Nobumasa Imura’s laboratory at Kitasato University (1997–), and as an associate professor in Professor Ichiro Kudo’s laboratory at Showa University (2003–). Dr. Hara became a professor at the School of Pharmacy, Showa University in 2009. Throughout most of his research career, he has been working on biosynthetic enzymes of lipid mediators (phospholipase A2, acyl CoA synthase, cyclooxygenase, prostaglandin E synthase, and prostacyclin synthase).
I thank Dr. Tadashi Tanabe, the late Dr. Ichiro Kudo and all other co-workers, present and past, for their invaluable contributions to this body of work. This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and from the Japan Society for the Promotion of Science, and by Grants-in-Aid for Comprehensive Research on Aging and Health, and Research on Dementia from the Ministry of Health, Labor, and Welfare of Japan.