2. Design of chiral phase-transfer catalysts with binaphthyl core structures
It is known that about 90 of the top-500 (18%) best-selling drug products in the world utilizes α-amino acids as the starting materials and/or as the intermediates.5) These include, amoxicillin (antibiotic), captopril, enalapril, lisinopril (hypotensive drugs), norvir, amprenavir (anti-HIV drugs) and acyclovir (antiviral drug), etc. (Scheme 2). Therefore, amino acids are indispensable for the preparation of new drugs, and have entered the mainstream of medicinal products. Asymmetric synthesis of α-amino acids by enantioselective phase-transfer alkylation of a prochiral, protected glycine derivative 1 using a chiral phase-transfer catalyst (PTC) has provided an attractive method for the preparation of both natural and unnatural amino acids (Scheme 3).4),6)


However, in the initial stages of the work on asymmetric phase-transfer chemistry, almost all the chiral phase-transfer catalysts that had been obtained were cinchona alkaloid derivatives (Scheme 4). Unfortunately, the starting cinchonine and cinchonidine are not enantiomeric. Further, because of several β-hydrogens to ammonium cation in such catalysts, the Hofmann elimination takes place under ordinary phase-transfer conditions using base. This constituted a major difficulty in rationally designing and fine-tuning of the catalysts to attain sufficient reactivity and selectivity.7) Thus, it is essential to synthesize desirable chiral phase-transfer catalysts from both enantiomeric starting materials, and also without any β-hydrogens in the catalyst structure. It was against this background that the structurally rigid, chiral spiro-ammonium salts of type (R,R)-2 and (S,S)-2 possessing 3,3′-diaryl substituents, which were derived from commercially available (R)- or (S)-binaphthol, respectively, were designed as new C2-symmetric chiral phase-transfer catalysts, and applied to the asymmetric synthesis of ent-3a and 3a, respectively (Scheme 5).8) An initial attempt on the design of efficient chiral phase-transfer catalysts was made on the benzylation of N-(diphenylmethylene)glycine tert-butyl ester 1a with 1 mol % of symmetric (S,S)-2a in 50% aqueous NaOH–benzene (volume ratio = 1:3) at room temperature and the corresponding benzylation product 3a was obtained in 76% yield with 73% ee. It should be noted that the use of (R,S)-2a dramatically lowered both the reactivity and enantioselectivity in the asymmetric phase-transfer benzylation. Introduction of aromatic substituents (Ar) on the 3,3′-position of one binaphthyl subunit of the catalyst afforded a beneficial effect on the enantiofacial discrimination, as the reaction of 1a with (S,S)-2b resulted in the formation of 3a in 43% yield with 81% ee. Moreover, the reaction in toluene as organic solvent under the influence of (S,S)-2b was completed within 30 min at 0 °C with 50% KOH as an aqueous base giving the product 3a in 82% yield with 89% ee. Switching the catalyst to (S,S)-2c and sterically more hindered (S,S)-2d further increased the enantioselectivity to 96% ee and 98% ee, respectively, and virtually complete stereochemical control was achieved using (S,S)-2e as catalyst. In the same way, the similar catalysts (S,S)-4 possessing 4,4′,6,6′-tetraaryl substituents also exhibited excellent enantioselectivity in the enantioselective synthesis of 3a.9) Interestingly, the combination of 0.05 mol % of each of (R,R)-2d and 18-crown-6 at 0 °C for 3 h greatly accelerated the phase-transfer alkylation, giving ent-3a in 90% yield with 98% ee.10) It should be noted that without 18-crown-6 the yield is significantly lowered (only 4%). Although the conformationally rigid, N-spiro structure created by two chiral binaphthyl subunits is a characteristic feature of 2 and related catalyst 4, it also imposes limitations on the catalyst design due to the need to use two different chiral binaphthyl moieties. Accordingly, here developed a new C2-symmetric chiral quaternary ammonium bromide 5 incorporating an achiral, conformationally flexible biphenyl subunit.11) The phase-transfer benzylation of 1a using the catalyst (S)-5a having a β-naphthyl group on the 3,3′-positions of the flexible biphenyl moiety proceeded smoothly at 0 °C to afford 3a in 85% yield with 87% ee after 18 h (Scheme 6). The observed chiral efficiency could be ascribed to the considerable difference in catalytic activity between the rapidly equilibrated, diastereomeric homo- and heterochiral catalysts; namely, homochiral (S,S)-5a is primarily responsible for the efficient asymmetric phase-transfer catalysis to produce 3a with high enantiomeric excess, whereas heterochiral (R,S)-5a displays low reactivity and stereoselectivity. This unique phenomenon provides a powerful strategy for the molecular design of chiral catalysts, and quaternary ammonium bromides possessing a sterically demanding substituent such as (S)-5b and (S)-5c exhibited higher enantioselectivity (92% ee and 95% ee, respectively) in the asymmetric benzylation of 1a.

Further efforts towards simplifying spiro-type phase-transfer catalysts have led to the development of the three-component coupling approach from the key intermediate 7, arylboronic acid and dialkylamine for the design of new, mono(binaphthyl)-based, chiral phase-transfer catalysts of type 6 as illustrated in Scheme 7.12)
The chiral efficiency of such simplified, chiral phase-transfer catalysts (S)-6 was examined by carrying out asymmetric benzylation of glycine derivatives under phase-transfer conditions. Among various readily available arylboronic acids and dialkylamines investigated, 3,4,5-trifluorophenyl-substituted dibutyl- or didecylammonium bromide (S)-6Db∼Dc showed excellent enantioselectivity (97% ee; Table 1).
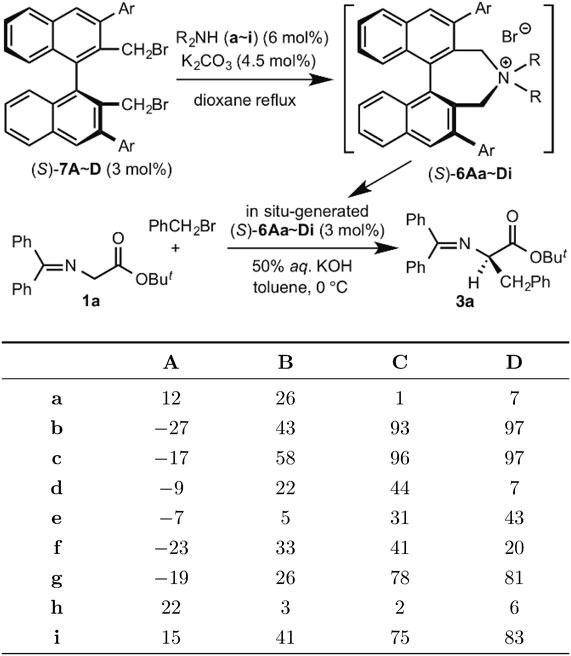
Table 1. Screening of in situ-generated catalysts (S)-6Aa∼Di in the enantioselective phase-transfer benzylation of glycine derivative 1a
These findings led to the conclusion that chiral quaternary ammonium bromide (S)-6Db (now registered as Simplified Maruoka Catalyst®), possessing flexible, straight-chain alkyl groups instead of a rigid binaphthyl moiety, functions as an unusually active chiral phase-transfer catalyst. Most notably, the reaction of 1a with various alkyl halides (R–X) proceeded smoothly under mild phase-transfer conditions in the presence of 0.01–0.05 mol % of (S)-6Db to afford the corresponding alkylation products 3 with excellent enantioselectivities (Scheme 8).12),13)

The synthetic utility of this chiral phase-transfer catalysts was highlighted by the facile synthesis of L-Dopa ester and its analogues, which are usually been prepared by either asymmetric hydrogenation of eneamides or enzymatic processes, and have been tested as potential drug for the treatment of Parkinson’s disease. Catalytic phase-transfer alkylation of protected glycine tert-butyl ester 1a with the requisite benzyl bromide 8 in toluene-50% KOH aqueous solution proceeded smoothly at 0 °C under the influence of (R,R)-2e (1 mol %) to furnish fully protected L-Dopa tert-butyl ester, which was subsequently hydrolyzed with 1 M citric acid in THF at room temperature to afford the corresponding amino ester 9 in 81% yield with 98% ee. Debenzylation of 9 under catalytic hydrogenation conditions produced the desired L-Dopa tert-butyl ester 10 with 94% yield (Scheme 9).8b),14) In a similar manner, L-Azatyrosine can be prepared from 1a and bromide 11 under the influence of chiral phase-transfer catalyst (R,R)-2d and subsequent transformation of 12.15)

In addition to chiral α-monoalkyl-α-amino acids, nonproteinogenic, chiral α,α-dialkyl-α-amino acids possessing stereochemically stable, quaternary carbon centers are also significant synthetic targets, not only because they are often effective enzyme inhibitors, but also because they are indispensable for the elucidation of enzymatic mechanisms. Accordingly, numerous studies have been conducted to develop truly efficient methods for their preparation, and the phase-transfer catalysis has some unique contributions in this process. Since the aldimine Schiff base 1b can be readily prepared from glycine, direct stereoselective introduction of two different side chains to 1b by appropriate chiral phase-transfer catalysis would provide an attractive and powerful strategy for the asymmetric synthesis of structurally diverse α,α-dialkyl-α-amino acids in one pot (Scheme 10).

It should be mentioned that asymmetric mono-alkylation of aldimine Schiff base 14 derived from an α-alkyl-α-amino acid would also provide α,α-dialkyl-α-amino acids as indicated in Scheme 10. This possibility of a one-pot, asymmetric double alkylation of the aldimine Schiff base 1b has been realized by using an N-spiro, chiral phase-transfer catalyst of type 2e, giving α,α-dialkyl-α-amino acid 15 selectively (Scheme 11).16) In addition, asymmetric monoalkylation of α-alkyl-α-amino acid derivatives 14 appears feasible by using (S,S)-2e8b) or (S)-6Db12b) to furnish various types of α,α-dialkyl-α-amino acids 16 and 17, respectively (Scheme 12).

Peptide modification provides a useful strategy for efficient target screening and optimization of lead structures in the application of naturally occurring peptides as pharmaceuticals. The introduction of side chains directly to a peptide backbone represents a powerful method for the preparation of unnatural peptides. An achiral glycine subunit has generally been used for this purpose. However, control of the stereochemical outcome of these transformations in an absolute sense is not easy, especially in the modification of linear peptides, and hence development of an efficient and practical approach to establish sufficient stereoselectivity and general applicability has been crucially important. Accordingly, we examined the chirality transfer in the diastereoselective alkylation of the dipeptide, Gly-L-Phe derivative 18 (Scheme 13).

Firstly, the use of tetrabutylammonium bromide (TBAB) resulted in only low diastereoselectivity (8% de). Interestingly, the reaction with catalyst (S,S)-2c or (R,R)-2c afforded DL-19 (55% de) or LL-19 (20% de), respectively, suggesting (R,R)-2c is a mismatched catalyst for this diastereofacial differentiation of 18. Changing the 3,3′-aromatic substituents (Ar) of 2 dramatically increased the stereoselectivity, and almost complete diastereocontrol (97% de) was realized with (S,S)-2f.17) A similar tendency is also observed in the diastereoselective benzylation of tripeptide 20.
One disadvantage of the asymmetric phase-transfer alkylation of glycine derivative 1 for the synthesis of α-alkyl-α-amino acids is the difficulty of using sterically hindered, unreactive alkyl halides for the preparation of sterically hindered α-alkyl-α-amino acids. In this context, efforts have been intrigued for the development of asymmetric Strecker reactions under phase-transfer conditions. The asymmetric Strecker reaction, catalytic asymmetric cyanation of imines represents one of the most direct and viable methods for the asymmetric synthesis of α-amino acids and their derivatives. Numerous recent efforts in this field have led to the establishment of highly efficient and general protocols, although the use of either alkylmetal cyanide or anhydrous hydrogen cyanide, generally at low temperature, is inevitable. It is worth to mention that this is the first example of a phase-transfer-catalyzed, highly enantioselective Strecker reaction of aldimines using aqueous KCN based on the molecular design of chiral quaternary ammonium salt (R,R,R)-22, having a tetranaphthyl backbone, as a highly efficient organocatalyst (Scheme 14).18) This phase-transfer catalyzed asymmetric Strecker reaction is further elaborated by the use of α-amido sulfone as a precursor of N-arylsulfonyl imine. In this system, the reaction can be carried out with a slight excess of potassium cyanide (1.05 equiv) which leads to completion of the reaction within 2 h.18b)

Although phase-transfer catalyzed enantioselective direct aldol reactions of glycine derivatives with aldehyde acceptors could provide an ideal approach for the simultaneous construction of the primary structure and stereochemical integrity of β-hydroxy-α-amino acids, which is a very important chiral units from the pharmaceutical viewpoint, there are only very limited examples reported till date. Thus, this review reports an efficient, highly diastereo- and enantioselective direct aldol reaction of 1a with a wide range of aliphatic aldehydes under mild phase-transfer conditions.19) Indeed, treatment of 1a with 3-phenylpropanal employing N-spiro chiral quaternary ammonium salt (R,R)-2g as a key catalyst resulted in the formation of the corresponding β-hydroxy-α-amino ester 23 in 76% isolated yield with the anti/syn ratio of 77:23, and the enantiomeric excess of the major anti isomer was determined to be 91% ee. Interestingly, the use of (R,R)-2h possessing 3,5-bis[3,5-bis(trifluoromethyl)phenyl]phenyl substituent as a catalyst enhanced both diastereo- and enantioselectivities (anti/syn = 92:8, 96% ee for anti isomer) (Scheme 15).

The initially developed reaction conditions using 2 equiv of aqueous base (1% NaOH aq) exhibited inexplicably limited general applicability in terms of aldehyde acceptors. For example, reaction of glycine derivative 1a with 4-benzyloxybutanal gave the aldol product with low diastereoselectivity (anti/syn = 58:42; 82% ee for anti isomer). The mechanistic investigation revealed the intervention of an unfavorable yet inevitable retro aldol process involving chiral catalyst 2. Based on this information, a reliable procedure has been established by the use of the catalyst (R,R)-2h (2 mol %) with a catalytic amount of 1% NaOH (15 mol %) and ammonium chloride (10 mol %), which tolerates a wide range of aldehydes to afford the corresponding anti-β-hydroxy-α-amino esters almost exclusively in an essentially optically pure form (Scheme 16).20)

We have developed the diastereo- and enantioselective conjugate addition of nitroalkanes to alkylidenemalonates under mild phase-transfer conditions by the utilization of appropriately designed chiral quaternary ammonium bromide 2h as an efficient catalyst. This new protocol offers a practical entry to optically active γ-amino acid derivatives, from which (R)-Baclofen and (R)-Rolipram can be easily prepared as shown in Scheme 17.21) As an extension of this research, we have also succeeded in the catalytic asymmetric conjugate addition of nitroalkanes to cyclic α,β-unsaturated ketones under phase-transfer condition.22)

An enantioselective Michael addition of β-keto esters to α,β-unsaturated carbonyl compounds is a useful method for the construction of densely functionalized chiral quaternary carbon centers. A characteristic feature of designer chiral phase-transfer catalyst 2g in this type of transformation is that it enables the use of α,β-unsaturated aldehydes as an acceptor, leading to the construction of quaternary stereocenter having three different functionalities of carbonyl origin as demonstrated in the reaction with 2-tert-butoxycarbonylcyclopentanone 24a. It is of interest that the use of fluorenyl ester 24b greatly improved the enantioselectivity. The addition of 24b to MVK was also feasible under similar conditions and the desired Michael adduct was obtained quantitatively with 97% ee (Scheme 18).23)

The combinatorial design approach for the rational design of new phase-transfer catalysts (Scheme 7) is found to be quite useful to develop hitherto difficult asymmetric transformations. For example, asymmetric conjugate addition of α-substituted-α-cyanoacetates 25 to acetylenic esters under phase-transfer condition is quite challenging because of the difficulty to control the stereochemistry of the product. In addition, despite numerous examples of the conjugate additions to alkanoic esters, so far there are no successful asymmetric conjugate additions to acetylenic esters. In this context, we recently developed a new morpholine-derived phase-transfer catalyst (S)-26a and applied it to the asymmetric conjugate additions of α-alkyl-α-cyanoacetates 25 to acetylenic esters. In this asymmetric transformation, an all-carbon quaternary stereocenter can be constructed in a high enantiomeric purity (Scheme 19).24)

With this information in hand, an enantioselective organocatalytic, one-pot synthesis of hexahydropyrrolizine and octahydroindolizine core structures 27 (n = 1 or 2) was considered as the starting from readily available glycine esters 1c in combination with several different organocatalytic reactions (Scheme 20). Our key strategy is based on the development of asymmetric conjugate addition of N-(diphenylmethylene)glycine ester 1c and α,β-enones 30 (n = 1 or 2) by using a chiral phase transfer catalyst of type (S)-26a (Scheme 20). Organocatalytic intramolecular reductive amination of conjugate adduct 29 (n = 1 or 2) with Hantzsch ester would proceed in a stereoselective manner to give pyrrolidine derivative 28 (n = 1 or 2) as an intermediate, which is susceptible to the subsequent cyclization with iminium ion formation and reductively aminated to furnish a hexahydropyrrolizine or octahydroindolizine core structure 27 (n = 1 or 2), respectively.

Here, we firstly examined asymmetric conjugate addition of N-(diphenylmethylene)glycine di(tert-butyl)methyl ester 1c and ethyl vinyl ketone with K2CO3 under the influence of chiral phase-transfer catalyst (S)-26a to furnish conjugate adduct 31 with low enantioselectivity. However, the use of chiral phase-transfer catalyst of type (S)-26b and 10 mol % of CsCl in ether at 0 °C gave conjugate adduct 31 in 84% yield with 94% ee (Scheme 21).25) With the optimal reaction condition for asymmetric conjugate addition in hand, we further carried out intramolecular reductive amination of 31 with Hantzsch ester (2 equiv) and CF3CO2H (1 equiv) in aqueous EtOH at 60 °C to furnish 2,5-disubstituted cis-pyrrolidine 32 stereospecifically in 84% yield.25) Thus, one-pot synthesis of cis-pyrrolidine 32 without isolation of adduct 31 was realized in a reasonable yield.

This information was further used to develop a facile and short asymmetric synthesis of physiologically active (+)-Monomorine in a highly stereoselective manner by the way of key octahydroindolizine core structure 35 (Scheme 22). Accordingly, asymmetric conjugate addition of glycine ester 1b with α,β-enone 33 (2.0 equiv) and K2CO3 (5 equiv) under the influence of chiral phase-transfer catalyst (S)-26b and catalytic CsCl in ether at 0 °C for 8 h was carried out to give conjugate adduct 34 in 86% yield with 93% ee. Intramolecular reductive amination of the adduct 34 and subsequent iminium ion formation followed by second reductive amination were effected with Hantzsch ester (5 equiv) and CF3CO2H in aqueous EtOH at 60 °C for 48 h to furnish octahydroindolizine core structure 35 in 61% yield without loss of enantioselectivity. The absolute configuration of 35 was unambiguously confirmed through X-ray crystallographic analysis after the reduction and subsequent p-bromobenzoate formation. The one-pot reaction of the whole reaction sequence was also realized without any difficulty by sequentially adding different reagents to afford 35 in 48% overall yield. The transformation of the key intermediate 35 to (+)-Monomorine was realized in 3-step sequence as shown in Scheme 22.26)

The development of new, catalytic asymmetric transformations by using a chiral metal-free catalyst in water solvent under neutral conditions with excellent atom economy is one of the most ideal approaches in current asymmetric synthesis in the viewpoint of green and sustainable chemistry. Therefore, this work focuses on the possibility of realizing such an ideal transformation, and hence to develop an environmentally-benign asymmetric conjugate addition to nitroolefins under essentially neutral conditions in order to fulfill four important factors (i.e., metal-free, water, neutral, and atom economy) as described above. Although quaternary ammonium salts as phase-transfer catalysts are generally believed to require base additives for phase-transfer reactions, in this case it is found that even without any base additives the enantioselective phase-transfer conjugate addition of 3-phenyloxindole 36 to β-nitrostyrene proceeded smoothly in the presence of chiral bifunctional ammonium bromide (S)-37 under neutral conditions in water-rich solvent (i.e., H2O/toluene = 10:1) with both high diastereomeric and enantiomeric ratios (Scheme 23).27) This reaction does not work well under ordinary phase-transfer conditions in aqueous basic solutions, such as aqueous KOH, K2CO3, and PhCO2K, and under homogeneous water-free reaction conditions. This finding is very surprising and unusual result in the long-standing phase-transfer chemistry. It is quite desirable in the green and sustainable chemistry. Specifically, both racemic and optically active conjugate adducts derived from 3-aryloxindoles can be readily transformed to valuable natural products and their analogues as exemplified by the transformation of optically active conjugate adduct 38 (90% ee) to the corresponding cyclization product 39, which comprises a similar core structure with many important natural products such as Flustramines and Flustramides.28) These natural product analogues might possess some important biological activity and hence are valuable for the drug industry.

We are also interested in the development of an environmentally-benign asymmetric conjugate amination to nitroolefins under essentially neutral conditions. This transformation allows the efficient asymmetric synthesis of chiral 1,2-diamino compounds as a useful chiral building block in pharmaceutical areas. After various screening of reaction conditions, an asymmetric neutral amination to nitroolefins was found to be catalyzed by chiral bifunctional tetraalkylammonium salts of type 40a with very low catalyst loading (0.05 mol %) in water-rich biphasic solvent (Scheme 24).29) Here, piperidine-derived catalysts (S)-40a are found to be superior to morphorine-derived catalysts (S)-37 in terms of enantioselectivity. It should be noted that asymmetric conjugate amination does not work well under the ordinary phase-transfer reaction conditions using aqueous base solutions, such as aqueous KOH, K2CO3, and PhCOOK solutions, and under the homogeneous reaction conditions without aqueous solution. Hence, the highly enantioselective conjugate amination of nitroolefins was only achieved when the reaction was performed under the base-free neutral phase-transfer conditions in water-rich biphasic solvent. The resulting amination products can be readily transformed by the catalytic hydrogenation with Raney Ni catalyst to the corresponding 1,2-diamines, which are versatile chiral building blocks from synthetic as well as pharmaceutical viewpoints.

The X-ray crystal structure of chiral ammonium amide (S)-43 provides an important structural information. The bond-lengths of amide moiety indicate that the negative charge of amide anion is delocalized on the nitrogen-carbon-oxygen atoms as shown in Scheme 25.29) Importantly, the hydrogen bonding interaction between the hydroxy group in the binaphthyl unit and the oxygen of amide anion is clearly observed in the crystal structure of (S)-43.
A highly diastereo- and enantioselective conjugate addition of α-substituted nitroacetates to maleimides in the presence of (S)-40b under base-free neutral phase-transfer conditions was developed for the synthesis of α,α-disubstituted α-amino acid derivatives (Scheme 26).30)

Although a variety of chiral quaternary ammonium salts have been developed as reliable catalysts for asymmetric phase-transfer reactions, only a few examples of chiral quaternary phosphonium salts as chiral phase-transfer catalysts have been reported with some limitations. In consideration of the wide availability of various types of commercially available chiral phosphine compounds, the present approach for the search of effective chiral quaternary phosphonium salts (S)-45 relies on the use of commercially available chiral phosphine compounds (S)-44 as catalyst precursors. This approach allows facile construction of a catalyst library of chiral quaternary phosphonium salts with various structures, which is successfully applied to asymmetric conjugate additions under base-free phase-transfer conditions with low catalyst loading (up to 0.1 mol %) (Scheme 27).31)

The fluoride-mediated generation of nucleophiles from organosilicon compounds for selective bond-forming reactions is very useful in organic synthesis. This approach implies the possibility of developing an asymmetric version based on the use of a chiral, nonracemic fluoride ion source as represented by chiral quaternary ammonium bifluorides of type 46. Indeed, the asymmetric nitroaldol reaction and conjugate addition of silyl nitronates are found to be highly efficient under the influence of chiral quaternary ammonium bifluorides (S,S)-46a∼b (Scheme 28).32),33)

Tetraalkylammonium salts are recognized as representative organocatalysts and are often used as phase-transfer catalysts for the activation of anionic nucleophiles through the formation of an ion pair with an ammonium cation. Although the structures of tetraalkylammonium salts are commonly expressed as 47a, their actual ionic structure is discussed differently. Namely, the positive charge of ammonium salts is delocalized on the α-hydrogen atoms, which are known to interact with an anionic counterion through hydrogen bonding, as shown in 47b (Scheme 29).34)

Although the hydrogen-bonding ability of the α-hydrogen atoms on tetraalkylammonium salts is often discussed with respect to phase-transfer catalysts, catalysis that utilizes the hydrogen-bond-donor properties of tetraalkylammonium salts remains unknown. This work successfully demonstrates hydrogen-bonding catalysis with the newly designed tetraalkylammonium salt catalysts in Mannich-type reactions. Indeed, the Mannich-type reaction of N-acyl isoquinolines 50 with ester-derived silyl enol ether 51 under the catalysis of ammonium salts 48a (the actual structure is 49) gave rise to the corresponding Mannich product 52 in 38% yield (Scheme 30).35) It should be noted that the use of related catalysts 53 and 54 gave rise to the product 52 in 9% and 6%, respectively. Fortunately, switching the catalyst from 48a to 48b enhanced the chemical yield to 61%, and the use of longer reaction time (6 h) gave the Mannich product 52 in 90% yield. The structure and the hydrogen-bonding ability of the new ammonium salt 48a were investigated by X-ray diffraction analysis and NMR titration studies.







