Reviews
Glutamate as intracellular and extracellular signals in pancreatic islet functions
2019 年 95 巻 6 号 p. 246-260
詳細
2019 年 95 巻 6 号 p. 246-260
l-Glutamate is one of the most abundant amino acids in the body and is a constituent of proteins and a substrate in metabolism. It is well known that glutamate serves as a primary excitatory neurotransmitter and a critical neuromodulator in the brain. Recent studies have shown that in addition to its pivotal role in neural functions, glutamate plays many important roles in a variety of cellular functions, including those as intracellular and extracellular signals. In pancreatic islets, glutamate is now known to be required for the normal regulation of insulin secretion, such as incretin-induced insulin secretion. In this review, we primarily discuss the physiological and pathophysiological roles of glutamate as intracellular and extracellular signals in the functions of pancreatic islets.
Communicated by Hiroo IMURA, M.J.A.
l-Glutamate (hereafter referred to as glutamate) is a naturally occurring amino acid (i.e., classified as a non-essential amino acid) and is one of the most abundant amino acids in the body. Glutamate is synthesized in most tissues, including brain, liver, kidney, skeletal muscle, and pancreatic islets. Besides synthesis in the body, glutamate is contained in many different types of food, and dietary glutamate (as either free form or monosodium form or both) is also a source of glutamate in the body. In addition to being a constituent of a variety of proteins and a key substrate in mammalian metabolism, glutamate has multiple functions as a signal transduction molecule. In the brain, glutamate serves as a principal excitatory neurotransmitter and a critical neuromodulator, which regulate synaptic plasticity, learning, memory, and neural development.1) These effects are mediated by glutamate receptors.2),3) Although glutamate in the blood is present in substantial concentrations (30–100 µM), it is only present in low concentrations in the extracellular brain fluid (0.5–2 µM).4),5) In most regions of the brain, the uptake of glutamate from circulation is limited by the blood-brain-barrier; therefore, free glutamate is primarily derived from de novo synthesis from l-glutamine (hereafter referred to as glutamine) in local regions.
Dietary glutamate exerts various effects, including in umami taste, digestion, nutrient absorption, and metabolism via brain activation.6) These effects may be mediated through glutamate sensors, e.g., the umami taste receptor, which is functionally linked to the afferent sensory nerves.7)
In pancreatic islets, glutamate is present in and released from α-cells.8),9) Glutamate at pharmacological concentrations potentiates insulin secretion from perfused rat pancreas and pancreatic islets.10),11) However, a recent study has shown that inhibition of the N-methyl-D-aspartate (NMDA) receptor in pancreatic β-cells enhances insulin secretion, suggesting that endogenous glutamate might tonically inhibit insulin secretion in pancreatic islets, which are exposed to relatively high concentrations of extracellular glutamate.12)
In contrast to the functions of extracellular glutamate, little is known about the role of intracellular glutamate in cellular functions. We showed recently that glutamate in pancreatic β-cells acts as a critical signal in incretin-induced insulin secretion (IIIS).13) In this review, we discuss the roles of glutamate as an intracellular and extracellular signaling molecule in pancreatic islet functions.
Glutamate is formed primarily from glutamine and α-ketoglutarate (α-KG) in distinct metabolic pathways in normal conditions (Fig. 1). In the mitochondrion, glutamate is formed by the reductive amination of the Krebs cycle intermediate α-KG by glutamate dehydrogenase (GDH1 and 2). In the cytosol, glutamate is formed by an aspartate aminotransferase 1 (AST1)-catalyzed reaction between aspartate and α-KG to produce oxaloacetate as part of the malate-aspartate (MA) shuttle. Glutamate is also formed from glutamine by glutaminase (GLS) in some tissues such as the kidney, liver, and brain. GLS has tissue-specific isozymes: GLS1 referred to as kidney-type and GLS2 as liver-type. In addition, glutamate is formed in a reaction catalyzed by glutamine:fructose 6-phosphate aminotransferase between fructose 6-phosphate and glutamine in the hexosamine biosynthesis pathway branching from glycolysis. Relative contributions of the above pathways to glutamate biosynthesis may differ in cell-types and pathological states.

Biosynthesis of glutamate. Glutamate is formed from α-KG by GDH in mitochondria and by AST1 in the cytosol. Glutamate produced in mitochondria is transported to the cytosol through mitochondrial glutamate carrier GC1. Glutamate is also formed from glutamine by GLS located in the mitochondrial inner membrane. In the hexosamine biosynthesis pathway, glutamate is formed from glutamine in a reaction catalyzed by GFAT. α-KG, α-ketoglutarate; Aralar1, aspartate-glutamate carrier; AST1, aspartate aminotransferase 1; BPGA, 1,3-bisphosphoglycerate; F6P, fructose 6-phosphate; G6P, glucose 6-phosphate; GAP, glyceraldehyde 3-phosphate; GC1, glutamate carrier; GDH, glutamate dehydrogenase; GFAT, glutamine:fructose 6-phosphate aminotransferase; GlcN6P, glucosamine 6-phosphate; GLS, glutaminase; MAL, malate; OAA, oxaloacetate; OGC, malate-α-ketoglutarate carrier; UDP-GlcNAc, uridine diphosphate N-acetylglucosamine.
Glutamate is converted to α-KG by several enzymes including glutamate dehydrogenase (GDH1 and 2) and aspartate aminotransferase 2 (AST2, also known as glutamic-oxaloacetic transaminase 2) and alanine aminotransferase 1 (ALT1, also known as glutamic-pyruvic transaminase 1; GPT1) in the mitochondria. In the brain, glutamate is converted to glutamine by glutamine synthetase in the presence of ammonia.14),15) Glutamate is also converted to γ-aminobutyric acid by glutamate decarboxylase (GAD1 and GAD2) in the brain and pancreatic β-cells.
Extracellular glutamate exerts various effects on cellular responses through cell surface receptors, i.e., glutamate receptors. Glutamate receptors are classified into two glutamate receptor families: ionotropic glutamate receptors (iGluRs) and metabotropic glutamate receptors (mGluRs) (Fig. 2).2),3),16),17) iGluRs are ligand-gated ion channels that produce glutamate-evoked currents. There are three subfamilies of iGluRs: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), kainate, and NMDA receptors. iGluRs function as a tetramer of subunits. NMDA receptors comprise two GluN1 subunits and either GluN2 or combination of GluN2 and GluN3 subunits. AMPA receptors assemble as a homotetramer or heterotetramer of GluA1 to GluA4. Subunits of kainate receptors (GluK1–3) form a homo- or heterotetramer, but GluK4 and GluK5 require GluK1–3 to form functional receptors.17)

Glutamate receptors. Ionotropic glutamate receptors (iGluRs) assemble as tetrameric complexes of subunits. There are three main groups of iGluRs: N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate. Metabotropic glutamate receptors (mGluRs) exist as homo- or heterodimers.3) Subunit types and isoforms identified in pancreatic islets, α-, β-, or δ-cells are indicated in blue. For details, see the text.
On the other hand, mGluRs are G-protein coupled receptors that control cellular processes via G-protein signaling. mGluRs are classified into three groups: Groups I, II, and III.3),18) Group I includes mGluR1 and mGluR5, which mediate Gq signaling. Group II includes mGluR2 and mGluR3, which mediate Gi/o signaling. Group III includes mGluR4, mGluR6, mGluR7, and mGluR8, which also mediate Gi/o signaling. mGluRs are different from other G-protein coupled receptors in that they have a large extracellular domain that contains a ligand binding site and they exist as a dimer.
Glutamate is transported across the membrane through a family of glutamate transporters, which is composed of two subfamilies: the excitatory amino acid transporter (EAAT) family19),20) and the vesicular glutamate transporter (VGLUT) family.21),22)
The plasma membrane glutamate transport is mediated by EAAT1–5, whereas vesicular membrane glutamate transport is mediated by VGLUT1–3. In the brain, EAATs transport glutamate from the synaptic cleft into glial cells and neurons, and VGLUTs transport glutamate from the cytoplasm into synaptic vesicles.
The role of glutamate in glucose-induced insulin secretion (GIIS) has been discussed for many years. In the 1990s, glutamate dimethyl ester, a membrane permeable glutamate precursor, was shown to enhance insulin secretion in rat islets.23) It was reported that mitochondrial activation directly triggered insulin secretion in pancreatic β-cells;24) subsequent studies proposed that glutamate derived from mitochondria acts as a messenger in GIIS.25),26) Glutamate produced by GDH in mitochondria was suggested to be transported to the cytosol through the glutamate carrier (GC1),27) then transported into insulin secretory granules to augment insulin secretion, although how glutamate is transported into the granules was not shown in these studies. Knockdown of GC1 in INS-1E β-cells was found to partially reduce GIIS (by ∼30%). GDH has been considered to be a key enzyme in controlling GIIS.28) While overexpression of GDH in rat islets amplified GIIS,29) GIIS in the islets from GDH-deficient mice was reduced by 50% compared with that from control islets.30),31) Hyperinsulinemia and hyperammonemia in infants are caused by activating mutations of GDH, which is associated with increased activity of GDH due to reduced GTP-mediated inhibition or higher sensitivity to the allosteric activator ADP.32)–34) However, the role of mitochondrial glutamate in GIIS has remained controversial. It was reported that the intracellular glutamate level was not altered by glucose stimulation in pancreatic islets and β-cell lines and that there was no correlation between the elevation of intracellular glutamate content and the amount of insulin secretion.35),36) In addition, mitochondrial glutamate was found to occupy a relatively small fraction of total cellular glutamate content in MIN6-K8 β-cells.13) Thus, although the activity of GDH in mitochondria is important for the regulation of GIIS, the physiological role of mitochondrial glutamate produced by GDH in GIIS still remains to be clarified.
Role of β-cell glutamate as a signal in incretin-induced insulin secretion.Incretins, such as glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide, are released from gut enteroendocrine cells upon meal ingestion, and they potentiate GIIS by binding to specific receptors on β-cells and activating cAMP signaling.37),38) The glucose-dependent effect of incretins on insulin secretion has led to the development of incretin-related drugs, dipeptidyl peptidase-4 inhibitors and GLP-1 receptor agonists, which are currently used worldwide for the treatment of type 2 diabetes.39),40) However, the relationship between glucose metabolism and incretin signaling in pancreatic β-cells was not known. A recent study using two insulin-secreting β-cell lines, MIN6-K8 and MIN6-K20 β-cells, has clarified that glutamate produced through the MA shuttle is a key signal linking glucose metabolism to incretin/cAMP action to amplify insulin secretion.13) MIN6-K8 and MIN6-K20 β-cells were cloned from the IT6 mouse harboring insulinoma. Although both MIN6-K8 and MIN6-K20 β-cells exhibit GIIS, they differ in incretin responsiveness; MIN6-K8 β-cells exhibit IIIS but MIN6-K20 β-cells do not.41) These cell lines provided a clue to clarify the mechanism of IIIS. Metabolome analysis showed that MIN6-K8 and MIN6-K20 β-cells have distinct metabolic profiles; the contents of metabolites from glucose metabolism including glucose 6-phosphate, fructose 6-phosphate, fructose 1,6-bisphosphate, nicotinamide adenine dinucleotide, glutamate, and aspartate are higher in MIN6-K8 cells than those in MIN6-K20 cells, indicating that both glycolysis and the MA shuttle are enhanced in MIN6-K8 cells. Pharmacological or genetic inhibition of the MA shuttle revealed the shuttle to be required for IIIS but not for GIIS. Mitochondrial glutamate was proposed previously to be a signal for GIIS; however, this notion has remained controversial, as described above. Therefore, whether or not glutamate produced through glucose metabolism acts as a signal in IIIS was explored. To measure cytosolic glutamate produced from glucose metabolism, metabolic flux analysis using stable isotope-labeled [U-13C]-glucose was utilized (Fig. 3). Of the six glutamate isotopomers, M (no substitution with 13C) and M+1 to M+5 (one to five substitutions with 13C, respectively), M and M+1 isotopomers are naturally occurring glutamate in cells, whereas the M+2 to M+5 isotopomers are glutamate derived from metabolism of [U-13C]-glucose. M+2 to M+5 glutamate isotopomers increased in the cytosol when MIN6-K8 β-cells were stimulated with [U-13C]-glucose. Inhibition of the MA shuttle by treatment with aminooxyacetate (AOA), an inhibitor of the MA shuttle, abolished the increase in the levels of M+2 to M+5 isotopomers in the cytosol. In addition, knockout of AST1, an enzyme that produces cytosolic glutamate through the MA shuttle, in clonal β-cells generated using the CRISPR/Cas9 system, almost completely blocked the increase in the levels of M+2 to M+5 glutamate isotopomers and markedly decreased IIIS (Fig. 4).42) Thus, glutamate produced through the MA shuttle is required for IIIS.
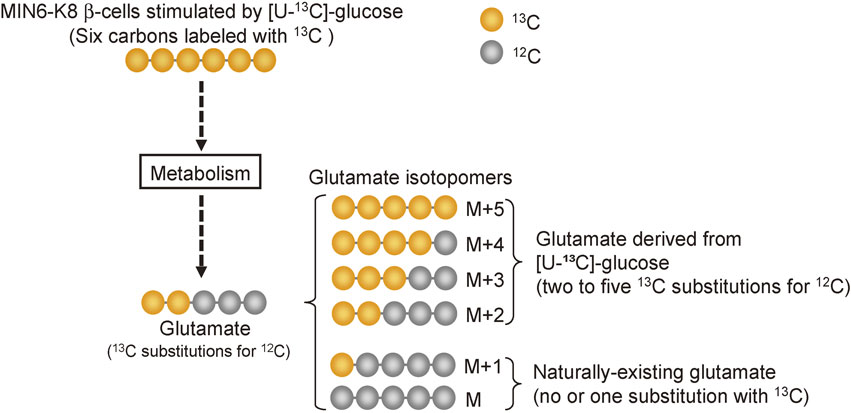
Metabolic flux analysis. Uniformly-labeled [U-13C]-glucose was used to determine the amount of glutamate produced through glucose metabolism. Each glutamate isotopomer is quantified by mass spectrometry. For details, see the text.

Effects of AST1 knockout (KO) on cytosolic glutamate production and incretin-induced insulin secretion (modified from ref. 42). (A) Cytosolic glutamate contents in MIN6-K8 (WT) cells and AST1 KO cells determined by metabolic flux analysis using [U-13C]-glucose. Cells were stimulated with 2.8 or 16.7 mM [U-13C]-glucose, and glutamate isotopomers derived from glucose metabolism (M+2 to M+5) were quantified. (B) Insulin secretion from WT and AST1 KO cells stimulated with 16.7 mM glucose and GLP-1 at concentrations indicated. Response to GLP-1 is presented as fold-change relative to the amount of insulin secretion at 16.7 mM glucose. *P < 0.001; n.s., not significant.
Application of glutamate into primary mouse β-cells dose-dependently stimulated exocytosis in the presence of cAMP, as assessed using capacitance measurements.13) Total internal reflection fluorescence microscopy analysis showed that glutamate dimethyl ester amplifies both the first and the second phases of glucose-induced insulin granule exocytosis, mimicking the incretin effect on potentiation of insulin secretion.13) These findings indicated that cytosolic glutamate acts as a critical amplifying signal in IIIS.
In neurons, glutamate is transported into secretory vesicles through VGLUTs.21),22) Pharmacological inhibition of glutamate transport using Evans Blue or knockdown of VGLUT1 did not affect GIIS, but reduced IIIS. Insulin secretion induced by GLP-1 was abolished, but glutamate dimethyl ester restored insulin secretion in pancreatic islets from VGLUT1 knockout mice, suggesting that glutamate transport into insulin granules through VGLUT1 is required for IIIS. To confirm this, clonal β-cell lines deficient in VGLUT1 were initially established, but IIIS was only partially reduced, possibly due to the compensatory expressions of VGLUT2, VGLUT3, or both in VGLUT1 knockout β-cell lines. Accordingly, VGLUT1, VGLUT2, and VGLUT3 triple knockout (TKO) cell lines were generated using CRISPR/Cas9 system.42) TKO β-cell lines showed a marked impairment in IIIS (Fig. 5). These findings provided direct evidence that glutamate transport into insulin granules through VGLUTs is essential for IIIS. Based on the findings above, it is suggested that the amplification of insulin secretion by incretin/cAMP signaling requires two essential steps: cytosolic glutamate production through the MA shuttle of glucose metabolism and the transport of glutamate into insulin granules by cAMP signaling (Fig. 6).13) However, the precise mechanisms by which glutamate is transported into insulin granules and glutamate in the granules triggers insulin exocytosis are still unknown. It was found that partial suppression of vacuolar H+ ATPase (V-ATPase) by bafilomycin and knockdown of a subunit of V-ATPase reduced IIIS,13) suggesting that granule acidification by V-ATPase is involved in glutamate action in insulin exocytosis. V-ATPase is known to be composed of two domains: an ATP-hydrolytic cytosolic domain (V1) and a proton-translocation domain (V0) located in the vesicle membrane.43) Assembly and disassembly of the V0 and V1 domains have been shown to be regulated by intravesicular pH.44) Furthermore, VAMP2, a component of the exocytotic machinery, has been reported to associate with a subunit of the dissociated V0 domain of V-ATPase and to promote exocytosis of synaptic vesicles.45) These findings suggested a possible mechanism by which glutamate signaling regulates the exocytosis of insulin: glutamate transport into insulin granules further decreases granule pH, which induces disassembly of the V0 and V1 domains of V-ATPase, and association of VAMPs and the V0 domain, thereby promoting exocytosis of insulin granules. Further investigation will be required for elucidation of the mechanism.

Effects of VGLUT knockout on incretin-induced insulin secretion (modified from ref. 42). MIN6-K8 (WT) and triple VGLUTs KO (TKO) cells were stimulated with 16.7 mM glucose and GLP-1 at the concentrations indicated. Response to GLP-1 is presented as fold-change relative to the amount of insulin secretion at 16.7 mM glucose. *P < 0.001.

Glutamate signaling in insulin secretion. In pancreatic β-cells, an increase in ATP production by glucose metabolism closes ATP-sensitive potassium (KATP) channels, depolarizing the β-cell membrane, and opening the voltage-dependent Ca2+ channels (VDCC), which results in the influx of Ca2+. The rise in intracellular Ca2+ level triggers insulin secretion. Incretins such as GLP-1 and GIP increase the intracellular cAMP level and amplify GIIS through the protein kinase A (PKA) and Epac2A (also referred to as cAMP-GEFII) pathways. In β-cells, cytosolic glutamate derived from glucose metabolism is thought to act as a signal to amplify insulin secretion. See also Figs. 1 and 7. AST1, aspartate aminotransferase 1; MA shuttle, malate-aspartate shuttle; PPase, protein phosphatase.
It has been reported that glutamate inhibits protein phosphatases (PPases) in a β-cell line, INS-1E.46) Glutamate inhibits PPase activity in INS-1E cell homogenates with an IC50 of ∼400 µM, suggesting that PPase activity is almost completely inhibited when cells are stimulated by a high concentration of glucose that increases cellular glutamate contents to the mM range. The elevated protein phosphorylation level by inhibition of PPase activities may also contribute to the potential of glutamate to amplify insulin exocytosis (Fig. 6).47)
Glutamate transport into secretory vesicles through VGLUT.VGLUT was initially identified as a brain-specific Na+-dependent inorganic phosphate co-transporter.48) It was later clarified that this transporter was indeed VGLUT1.49),50) Similarly, differentiation-associated Na+-dependent inorganic phosphate co-transporter, another member of the Na+-dependent inorganic phosphate transporters family,51) was identified as a vesicular glutamate transporter and renamed VGLUT2.52)–56) VGLUT3, the third subtype of VGLUT, discovered in 2002, is similar to VGLUT1 and VGLUT2 in its structure and function.57)–60)
The electrochemical proton gradient (ΔµH+) across the synaptic vesicle membrane, which is composed of an electrical gradient (ΔΨ) and a pH gradient (ΔpH) provided by V-ATPase, is a driving force of glutamate transportation through VGLUTs.61) It is well known that glutamate transport through VGLUT is regulated by the Cl− concentration.62)–64) A low millimolar concentration of Cl− stimulates glutamate transport; a high concentration of Cl− inhibits glutamate transport. This is attributed to the dependency of ΔΨ and ΔpH on the intra- and extravesicular concentration of Cl−.65) Several models have been proposed for the mechanism by which a low concentration of Cl− regulates glutamate transport. For example, in one model, Cl− influx into synaptic vesicles through VGLUT or other chloride channels, such as ClC-3, leads to the generation of ΔpH by a decrease in vesicular pH, thereby promoting glutamate transport through VGLUT.66)–68) In another model, Cl− regulates the activity of VGLUT by binding to the allosteric site of VGLUT.69),70)
VGLUT1, VGLUT2, and VGLUT3 were reported to be expressed in rodent islets and β-cell lines.13),71),72) It is possible that the fundamental mechanism of glutamate transport into insulin granules is similar to that of synaptic vesicles, and the ΔµH+ formed by V-ATPase is a driving force for glutamate transport into insulin granules. The influx of glutamate through VGLUT itself also contributes to the formation of ΔΨ and ΔpH.72) Chloride channels (e.g., ClC-3) in insulin granules have also been shown to be involved in insulin exocytosis by regulating pH in insulin granules,73),74) suggesting that the influx of Cl− through ClC-3 contributes to the generation of ΔµH+, which promotes glutamate transport into insulin granules. EAAT2 is expressed in insulin granules in mouse β-cells and mediates glutamate efflux from granules to the cytoplasm, thereby maintaining a certain level of intragranular glutamate.72) Perturbation of the cycle of glutamate influx and efflux by knockdown of EAAT2 or overexpression of EAAT2 or VGLUT reduced the rate of granule exocytosis, as assessed using an indirect assay of human growth hormone release.72) These findings suggested that VGLUTs and EAAT2 modulate insulin exocytosis by regulating the influx and efflux of glutamate, pH, and membrane potential of insulin granules (Fig. 7). Thus, pH in insulin granules, which is regulated by H+, glutamate, and Cl− through V-ATPase, VGLUT, EAAT, and ClC-3 is thought to be critical for potentiation of insulin exocytosis by incretins.
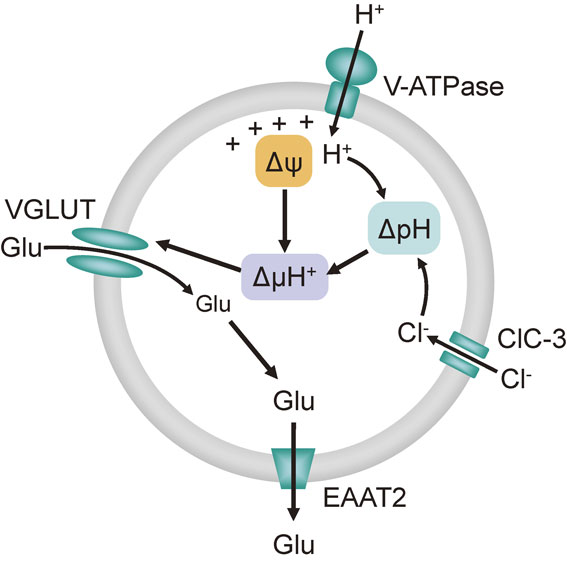
Model of glutamate transport into insulin granules through VGLUTs. The electrochemical proton gradient (ΔµH+) comprised of membrane potential (ΔΨ) and pH gradient (ΔpH) is the driving force of glutamate (Glu) transport into insulin granules. Proton influx through V-ATPase generates ΔΨ and ΔpH. Chloride ion (Cl−) influx through chloride channels (ClC-3) also contributes to the generation of ΔpH. An excitatory amino acid transporter 2 (EAAT2) mediates glutamate efflux, thereby maintaining glutamate at certain levels in insulin granules.
Extracellular glutamate evokes various intracellular signals by acting on its receptors at the cell surface. Ionotropic and metabotropic glutamate receptors are expressed in pancreatic islets and β-cells. GluR1, GluR2, GluR3, and GluR4 of AMPA receptors, KA-1, KA-2, GluR5, GluR6, and GluR7 of kainate receptors, and NR1, NR2A, NR2C, and NR2D of NMDA receptors are expressed in mouse or rat islets and β-cell lines such as MIN6 and RINm5F cells (Fig. 2).11),12),75)–79) Stimulation of glutamate receptors by NMDA, AMPA, kainate, or glutamate depolarizes the plasma membrane and elevates intracellular calcium levels in MIN6 cells and mouse and rat β-cells.11),76),77) AMPA, kainate, and glutamate promote insulin secretion in MIN6 cells and mouse and rat islets, whereas NMDA promotes insulin secretion in MIN6 cells but not in rat β-cells.76),77) Activation of AMPA receptors stimulates cGMP production, which inhibits the ATP sensitive K+ channel, resulting in membrane depolarization and triggering of insulin exocytosis in mouse β-cells.11) Metabotropic glutamate receptors, mGluR5, mGluR2, mGluR3, mGluR4, and mGluR8, are expressed in β-cell lines, an α-cell line, and rat islets.75),79),80) Activation of mGluR5, a G-protein coupled receptor, activates phospholipase C through Gqα and increases inositol 1,4,5-triphosphate (IP3). Elevation of IP3 promotes insulin secretion through mobilization of Ca2+ from intracellular Ca2+ stores. mGluR5 is localized on insulin granules as well as the plasma membrane, suggesting that glutamate transported into insulin granules activates mGluR5 and stimulates vesicle-associated phospholipase C, which generates calcium transients around the vesicles and triggers insulin exocytosis.79) The role of NMDA receptors in insulin secretion from pancreatic islets has been controversial. Pharmacological stimulation of NMDA receptors increased insulin secretion or showed no effect.76)–78) The application of NMDA receptor antagonists also exhibited inconsistent effects on insulin secretion and blood glucose.81)–83) A recent study using genetic and pharmacological approaches has shown that inhibition of NMDA receptors in mice and human islets enhances GIIS without affecting the basal secretion of insulin.12) Considering that the plasma glutamate level is 50–100 µM,84) NMDA receptors in pancreatic islets are fully saturated with glutamate. Upon membrane depolarization, NMDA receptors are activated due to removal of the Mg2+ block. AMPA and kainate receptors are activated only when the intraislet glutamate concentration is elevated; the NMDA receptor and AMPA/kainate receptors have different affinities for glutamate, the former having higher affinity than the latter.17) Inhibition of NMDA receptors results in a prolonged membrane potential burst in β-cells induced by glucose and a longer plateau fraction of glucose-induced Ca2+ oscillations,12) which may contribute to the enhancement of insulin secretion. Based on these findings, the following model is proposed for the regulation of insulin secretion by extracellular glutamate (Fig. 8).12),85) At low glucose levels, NMDA receptors on β-cells are saturated by glutamate, but they are not activated because the β-cell membrane is not depolarized (Fig. 8A). In contrast, high glucose depolarizes the β-cell membrane, thereby activating NMDA receptors due to the relief of Mg2+ blockade. The activation of NMDA receptors has an inhibitory effect on depolarization of the β-cell membrane by increasing outward K+ currents. However, this effect is masked by strong depolarization induced by glucose (Fig. 8B). Inhibition of NMDA receptors by their antagonists such as dextrorphan induces further depolarization of the β-cell membrane by suppressing the polarization by NMDA. As a result, more potentiation of insulin secretion occurs (Fig. 8C). It has been found that plasma glutamate levels were elevated in diabetic mice and human subjects, and prolonged activation of NMDA receptors induced apoptosis as well as dysfunction of β-cells such as an impairment in insulin secretion in clonal β-cell lines.86),87) Treatment of mouse and human islets with NMDA receptor antagonists exerted a protective effect against cell death induced by cytokines.12) Inhibition of NMDA receptors attenuated dysfunctions such as impaired insulin secretion and apoptosis in the clonal β-cell line (MIN6) exposed to a high concentration of glucose.12),86) Administration of NMDA receptor antagonists in vivo improved glucose tolerance in both diabetic mice and patients with type 2 diabetes.12),86) Thus, inhibition of the NMDA receptor-mediated signal in β-cells may have beneficial effects on both β-cell function and β-cell mass.
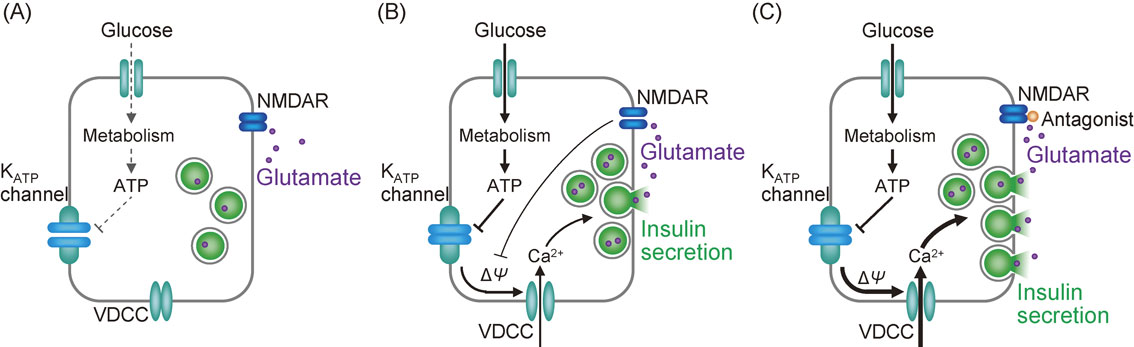
Model of the regulation of insulin secretion by extracellular glutamate through the NMDA receptor. (A) At a low glucose level, extracellular glutamate saturates but does not stimulate NMDA receptors (NMDARs) on β-cells, because the β-cell membrane is not depolarized at a low glucose level. (B) High glucose depolarizes the β-cell membrane, thereby activating NMDARs. The activation of NMDARs inhibits β-cell membrane depolarization, but this effect is masked by the strong depolarization induced by glucose. (C) In the presence of the antagonists of NMDA receptors at high glucose concentration, the β-cells secrete more insulin, compared with that by high glucose alone, because the NMDAR-mediated inhibitory effect on membrane depolarization is canceled. See text for detail and refs. 12 and 85. KATP channel, ATP-sensitive K+ channel; VDCC, voltage-dependent Ca2+ channel.
It has been shown that extracellular glutamate regulates glucagon secretion through activation of its receptor. Both stimulatory and inhibitory effects of glutamate on glucagon secretion have been reported: the former through activation of AMPA receptors was found in rat pancreas and mouse islets,88),89) the latter through activation of a metabotropic receptor, mGluR5, was observed in rat islets.90) In human and monkey islets, functional AMPA/kainate receptors are predominantly present in α-cells, whereas mGluRs are rarely expressed.91) The agonist of the ionotropic glutamate receptors, kainate and AMPA, stimulated glucagon secretion; glutamate-induced glucagon secretion was blocked by iGluR antagonists, whereas both agonists and antagonists of metabotropic glutamate receptors did not affect glucagon release in either human or mouse islets. The injection of glutamate and AMPA into mice increased plasma glucagon levels but did not affect plasma insulin levels. In mice treated with iGluR antagonist, glucagon secretion in response to insulin injection was reduced and insulin-induced hypoglycemia was exacerbated. These findings suggested that the activation of iGluR plays a major role in the regulation of glucagon secretion by glutamate both in vivo and in vitro. The AMPA/kainate glutamate receptors are Na+-permeable non-selective cation channels, and their activation induces influx of Na+ and Ca2+, which leads to the opening of voltage-dependent Ca2+ channels, thereby triggering glucagon release.
VGLUT2 is expressed on glucagon secretory granules in α-cells, and glutamate transported into secretory granules is co-secreted with glucagon.8),9),71) The AMPA/kainate receptors, which have a relatively low affinity for glutamate compared with NMDA receptors, may be activated when glutamate is released from α-cells with glucagon, suggesting that glutamate acts as an autocrine signal in the regulation of glucagon secretion.17),92) δ-cells express the AMPA-type receptor (GluR4), and somatostatin secretion is suggested to be stimulated by glutamate at low glucose concentrations.93)
Studies of animal models provide useful information on the pathogenesis and pathophysiology of diabetes. The Goto-Kakizaki (GK) rat is a model of non-obese type 2 diabetes with defective insulin secretion associated with impaired glucose metabolism in pancreatic β-cells.94),95) The Zucker fatty (ZF) rat is a model of obesity with a mutation in the leptin receptor gene.96) In GK rats, although GIIS is markedly decreased compared with that in control Wistar rats, amplification by incretin is somewhat retained.13) The islets of ZF rats show a higher basal insulin secretion compared with controls and significant GIIS, but not amplification of insulin secretion in response to incretin. Metabolic flux analysis of islets using [U-13C]-glucose showed glucose-stimulated glutamate production in GK rat islets but not in ZF rat islets. These findings indicated that cellular glutamate production is associated with incretin responsiveness (Table 1). Unresponsiveness to endogenous incretins or exogenously administered incretin-related drugs are found in patients with obese type 2 diabetes, so called incretin non-responders.97) Thus, impaired glutamate signaling in β-cells may underlie unresponsiveness to incretins in diabetic patients, especially those with obesity. Therefore, β-cell glutamate signaling might be an effective target for the treatment of patients with obese type 2 diabetes.
| Strain | Impairment in incretin-induced insulin secretion | Impairment in glutamate production by glucose |
|---|---|---|
| Wistar (control) | − | − |
| GK | + | + |
| ZF | +++ | +++ |
−: no impairment +: moderate impairment +++: severe impairment
The extracellular glutamate signal plays important roles in the pathogenesis and pathophysiology of diabetes. In hyperglycemic conditions, β-cell NMDA receptors are thought to be continuously activated due to elevated plasma glutamate levels and depolarization of the β-cell membrane.86) The persistent activation of NMDA receptors may induce β-cell death as well as suppression of insulin secretion. Because the inhibition of NMDA receptors improves glucose tolerance and GIIS in diabetic patients and suppresses β-cell death,12) glutamate receptor signaling may also be a novel therapeutic target to improve islet function by controlling β-cell mass.92)
It is now known that glutamate in β-cells functions as a critical signal in IIIS by linking glucose metabolism to cAMP action in insulin exocytosis, and that extracellular glutamate in the islets participates in the regulation of insulin secretion and glucagon secretion. However, there are several issues yet to be clarified: 1) how does glutamate in insulin granules promote exocytosis?; 2) how is glutamate transport into insulin granules regulated?; 3) how is glutamate produced in α-cells?; 4) how is glutamate transported into glucagon granules?; and 5) how are these mechanisms affected in altered metabolic states such as diabetes and obesity? Answers to these questions should further enhance our understanding of the physiological and pathophysiological roles of intracellular and extracellular glutamate in pancreatic islet functions.
We are grateful to the members of our laboratory who were involved in the studies cited in this review. The studies by our laboratory were supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan. Our laboratory is also supported by MSD K.K, Novo Nordisk Pharma Ltd., Kowa Pharmaceutical Co. Ltd. and Taisho Pharmaceutical Holdings.
Harumi Takahashi graduated from Kyoto University Faculty of Engineering in 1994. She received her Master’s degree in engineering from Kyoto University Graduate School of Engineering in 1996. She graduated from Kyoto Pharmaceutical University and obtained a license for pharmacy in 2000. She was engaged in diabetes care as a certified diabetes educator as well as in general pharmaceutical practices in Rakuwakai Healthcare System until 2005. In 2005, she joined Professor Susumu Seino’s laboratory as a doctorate course student at Kobe University Graduate School of Medicine and started research in diabetes. Her research was centered on cAMP signaling in insulin granule exocytosis. Under the guidance of Professor Seino, she developed an imaging system for analyzing insulin granule dynamics in a living β-cell. After receiving her Ph.D. in medical science in 2009, she has continued research at the Division of Molecular and Metabolic Medicine. She is currently serving as an Assistant Professor. She has been engaged in projects on the molecular mechanisms of the regulation of insulin secretion by cAMP and glutamate signaling, as well as screening small molecules that promote insulin secretion to develop novel anti-diabetic drugs by combining molecular biology, biochemistry, and cell biology.
Norihide Yokoi graduated from Kyoto University Faculty of Agriculture in 1992. He received his Master’s degree at Kyoto University Graduate School of Agriculture in 1994 and his Ph.D. at Kyoto University Graduate School of Medicine in 1998. He joined Chiba University School of Medicine as a Research Associate in 1998. His research focused on the molecular genetics of diabetes. He identified a nonsense mutation in the Cblb gene as a major susceptibility gene in an animal model of type 1 diabetes. In 2003, he moved to Kobe University Graduate School of Medicine as an Associate Professor in the Division of Molecular and Metabolic Medicine, where he has been devoting himself to studies of the pathogenesis and pathophysiology of type 2 diabetes, the molecular mechanism of incretin-induced insulin secretion, and the aging mechanism of pancreatic islets using omics approaches including transcriptomics, proteomics, and metabolomics.

Susumu Seino graduated from Kobe University School of Medicine in 1974. After years of clinical practice at hospitals, he joined the Department of Internal Medicine, Kyoto University School of Medicine as a fellow in 1978, where he began his study of pancreatic hormones and received his D. M. Sci. under the mentorship of Professor Hiroo Imura, M.J.A. After working for 10 years in the U.S.A., where he and his colleagues succeeded in cloning many genes related to glucose metabolism, he was offered a Professorship at Chiba University, Japan, in 1991. In 2003, he moved to Kobe University Graduate School of Medicine, his alma mater, where he is currently appointed Endowed Professor of the Division of Molecular and Metabolic Medicine. His research centers on molecular mechanisms of cell signaling in insulin secretion. He has received many national and international awards. He was also awarded the Medal with Purple Ribbon (Shiju-ho-sho) in 2011 and the Japan Academy Prize in 2018. (see also SS profile in Proc. Jpn. Acad., Ser. B 86:576, 2010)