Reviews
Physiological significance of ghrelin in the cardiovascular system
2019 Volume 95 Issue 8 Pages 459-467
Details
2019 Volume 95 Issue 8 Pages 459-467
Ghrelin, a growth hormone-releasing peptide first discovered in rat stomach in 1999, is a ligand for the growth hormone secretagogue receptor. It participates in the regulation of diverse processes, including energy balance and body weight maintenance, and appears to be beneficial for the treatment of cardiovascular diseases. In animal models of chronic heart failure, ghrelin improves cardiac function and remodeling; these findings have been recapitulated in human patients. In other animal models, ghrelin effectively diminishes pulmonary hypertension. Moreover, ghrelin administration early after myocardial infarction decreased the frequency of fatal arrhythmia and improved survival rate. In ghrelin-deficient mice, endogenous ghrelin protects against fatal arrhythmia and promotes remodeling after myocardial infarction. Although the mechanisms underlying the effects of ghrelin on the cardiovascular system have not been fully elucidated, its beneficial effects appear to be mediated through regulation of the autonomic nervous system. Ghrelin is a promising therapeutic agent for cardiac diseases.
Edited by Hiroo IMURA, M.J.A.
Abbreviations: AgRP: agouti-related protein; CHF: chronic heart failure; CI: cardiac index; COPD: chronic obstructive pulmonary disease; CSNA: cardiac sympathetic nervous activity; ECG: electrocardiogram; GH: growth hormone; GHS-R: growth hormone secretagogue receptor; GOAT: ghrelin O-acyltransferase; HR: heart rate; KO: knockout; MAP: mean arterial pressure; MI: myocardial infarction; NPY: neuropeptide Y; NTS: nucleus of the solitary tract; OMI: old myocardial infarction; RSNA: renal sympathetic nerve activity; SVI: stroke volume index; TAC: transverse aortic constriction; WT: wild-type.
Ghrelin, an acylated 28-amino acid peptide hormone that was originally purified from rat stomach in 1999 (Fig. 1), is the endogenous ligand for the growth hormone secretagogue receptor (GHS-R).1) In the 1999 study, a cultured cell line expressing GHS-R was established and used to identify tissue extracts that can stimulate GHS-R, as monitored by increases in intracellular Ca2+ levels. After several rat tissue extracts were screened, very strong activity was found in stomach extracts.1) The ligand was purified from rat stomach through four steps of chromatography and found to be a 28-amino acid peptide, which was named ghrelin (ghre is the Proto-Indo-European root of the word “grow”).1) Human ghrelin is also a 28-amino acid peptide. Human and rat ghrelins differ in two amino acid residues.1) The molecular weight of human ghrelin is 3370.9 and that of rat ghrelin is 3314.8. Purification of ghrelin from the stomach revealed that the release of growth hormone (GH) from the pituitary is regulated by both hypothalamic GH-releasing hormone and a peripherally derived hormone. A distinguishing structural feature of ghrelin is acyl-modification at its third serine residue (Fig. 1), which is necessary for receptor binding and induction of its biological activity.1) Acyl-modification of ghrelin is mediated by a membrane-bound enzyme called ghrelin O-acyltransferase (GOAT).2) GOAT mRNA level are highest in the stomach, but it is also detectable in the small intestine and colon.2) Subsequent studies revealed that intracerebroventricular administration of ghrelin potently stimulates appetite and increases body weight gain.3) After central administration of ghrelin, Fos, a marker of neuronal activation, appears in regions containing neurons that produce neuropeptide Y (NPY) and agouti-related protein (AgRP); conversely, antibodies and antagonists of NPY and AgRP abolish ghrelin-induced feeding.3) Ghrelin also augments NPY gene expression and blocks leptin-induced feeding reduction, implying that there is a competitive interaction between ghrelin and leptin in feeding regulation.3) Ghrelin-related functions have also been reported in the cardiovascular, gastrointestinal, and immune systems, as well as in bone. In the present review, however, we will focus on the characteristics of ghrelin and its therapeutic potential in cardiovascular diseases.
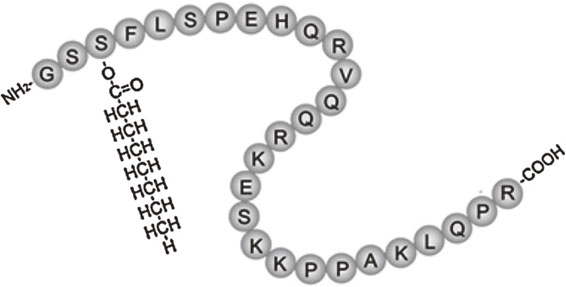
Structure of human ghrelin. Ghrelin is a 28-amino acid peptide discovered in the stomach. One distinguishing structural feature of ghrelin is its n-octanoylation on the third serine residue, which is necessary for its receptor binding and function.
As described previously, the stomach is a major site of ghrelin production (Fig. 2), and an in situ analysis revealed that ghrelin and its mRNA are mainly localized in the submucosal layer.1) There are four types of endocrine cells in the oxyntic mucosa of the stomach — ECL, D, enterochromaffin, and X/A-like cells — and ghrelin is present predominantly in X/A-like cells.4) Rat ghrelin was present in round, compact, electron-dense granules compatible with those X/A-like cells whose hormonal product and physiological functions have not previously been clarified.4) Ghrelin is secreted from the submucosal layer of the stomach into the bloodstream.4) In totally gastrectomized patients, the plasma ghrelin level is reduced to 35% of that in normal controls.5) Plasma ghrelin levels increased by 31% after 12 hours of fasting, but decreased by 22% immediately after feeding.5) In patients with anorexia nervosa, plasma ghrelin levels are markedly higher than those in normal controls, and are negatively correlated with body mass indices.5)
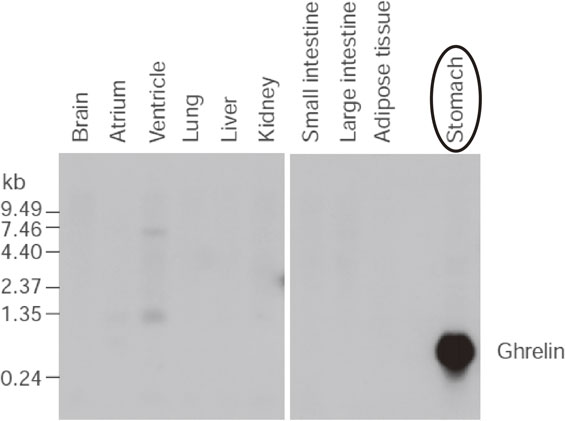
Northern blotting analysis of ghrelin mRNA in rat tissue showing that prepro-ghrelin mRNA occurs in the stomach. Adapted from Kojima et al.1)
GHS-R was identified by expression cloning in 1996.6) It acts as a receptor for ghrelin, ghrelin-induced GH release, and appetite promotion.7) Two transcript variants have been identified in multiple tissues in several species. GHS-R1a encodes a functionally active protein that induces GH release, whereas GHS-R1b is produced by alternative splicing and is functionally inactive.6) Therefore, GHS-R1a is thought to be the primary ghrelin receptor. GHS-R1a cDNA encodes a receptor comprising 366 amino acids with 7 transmembrane domains, whereas the GHS-R1b cDNA encodes a shorter form of the receptor, consisting of 289 amino acids and only 5 transmembrane domains.6) In humans, expression of GHS-R mRNA is predominantly detected in the pituitary, hypothalamus, and hippocampus.8) Expression of GHS-R in the cardiovascular system remains controversial, with some studies reporting expression in the heart and vasculature.9)–11) However, Callaghan et al. did not detect GHS-R expression by immunohistochemistry in the aorta, mesenteric artery, cerebral artery, coronary artery, or myocardial tissue of GHS-R reporter mice, nor did they detect GHS-R1a mRNA by RT-PCR in the aorta or mesenteric vessels of rats.12) Accordingly, further investigations are needed to determine the precise localization of GHS-R within the cardiovascular system.
Regulation of plasma ghrelin concentration.As described previously, ghrelin is primarily produced in the stomach and secreted into the bloodstream. In healthy individuals, ghrelin is regulated by both circadian rhythms and the fasting/fed state, with levels peaking at customary meal times and during fasting.13) Plasma ghrelin levels do not significantly differ between patients with chronic heart failure (CHF) and controls, although the plasma ghrelin level is significantly higher in CHF patients with cachexia than in those without cachexia, and correlates negatively with body mass index in CHF patients.14) Similarly, plasma ghrelin levels are elevated in underweight patients with chronic obstructive pulmonary disease (COPD) relative to those in normal-weight patients and healthy control subjects.15) These data suggest that in patients with CHF of COPD, an increase in ghrelin plasma concentrations plays a compensatory role in the underlying metabolic imbalance.
Peripheral ghrelin administration to healthy male volunteers has been shown to cause hemodynamic changes.16),17) For example, in a randomized, crossover study, an intravenous bolus injection of ghrelin (10 µg/kg) significantly decreased mean arterial pressure (MAP; −12 mmHg) versus vehicle administration without a significant change in heart rate (HR; Fig. 3 left panel).18) Ghrelin also significantly increased cardiac index (CI; 116%) and stroke volume index (SVI; 122%) (Fig. 3 left panel).16) In another study, an intravenous bolus injection of ghrelin (10 µg/kg) significantly decreased both blood pressure and HR.17) In that study, ghrelin administration suppressed sympathetic nerve activity and stimulated parasympathetic nerve activity, as evaluated by analyzing HR variability.18) Matsumura et al. explored the hemodynamic effect of intracerebroventricular injection of ghrelin into conscious rabbits.18) They found that injection of 1 nmol ghrelin decreased arterial pressure, HR, and renal sympathetic nerve activity (RSNA; Fig. 3 right panel).18) In addition, they showed that microinjections of ghrelin (20 pmol) into the nucleus of the solitary tract (NTS) of anesthetized rats decreased arterial blood pressure and renal sympathetic nerve activity.19) Together, these results demonstrate that peripherally and centrally administered ghrelin exerts hypotensive effects without accompanying reflex tachycardia and suggest that the hypotensive effect of ghrelin is not mediated via a direct vasodilative effect but by autonomic nervous regulation.
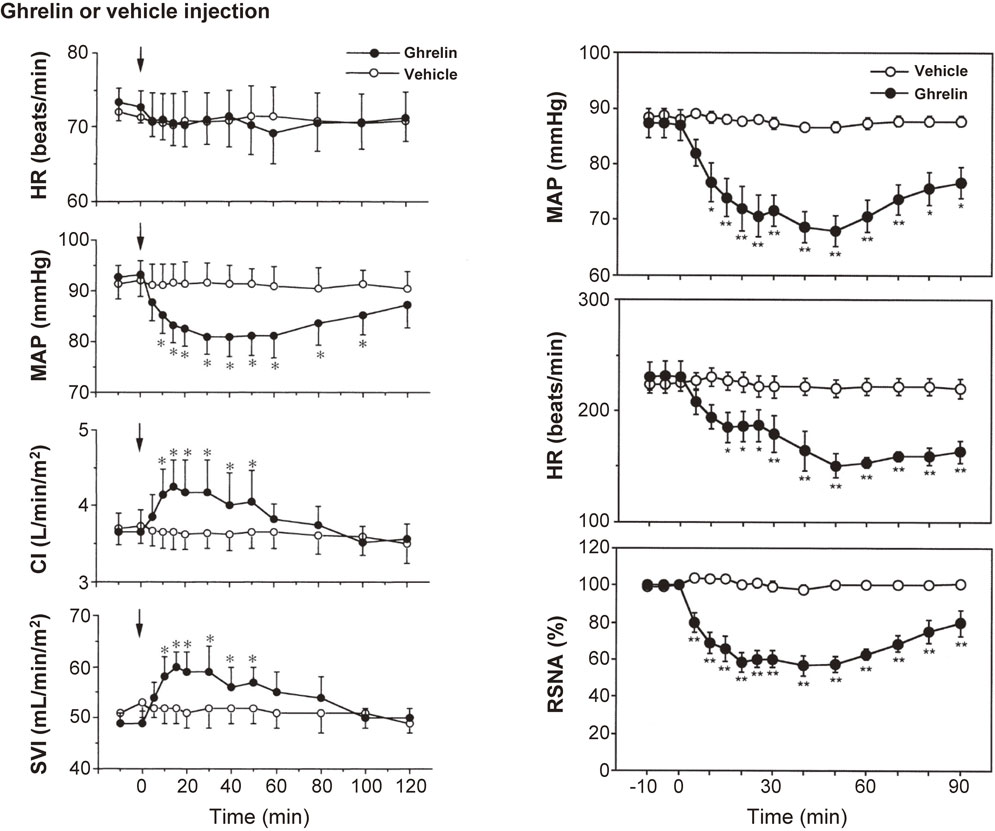
Left panel: Effect of ghrelin on the heart rate (HR), mean arterial pressure (MAP), cardiac index (CI), and stroke volume index (SVI). Data are means ± SE. *P < 0.05 vs. vehicle group. Adapted from Nagaya et al.16) Right panel: Time course of MAP, HR, and renal sympathetic nerve activity (RSNA) elicited by intracerebroventricular injection of 1 nmol of ghrelin or vehicle (n = 6 for each). Values are mean ± SE. *P < 0.05, **P < 0.01 vs. control period. Adapted from Matsumura et al.18)
Our group evaluated the effect of peripherally administered ghrelin on cardiac sympathetic nervous activity (CSNA) in animal models of myocardial infarction (MI).20) Platinum recording electrodes were placed on the portion of the cardiac sympathetic nerve that branches from the stellate ganglion. MI induces a maximal 110% increase in CSNA, which was prevented in rats that were subcutaneously injected with ghrelin (150 µg/kg) 1 minute after infarct. When ghrelin was injected 2 hours after MI (CSNA had increased by about 85%), CSNA then decreased to pre-MI levels (Fig. 4).20) Shirai et al. also demonstrated that subcutaneous or intracerebroventricular ghrelin administration inhibited CSNA and improved mortality and arrhythmic episodes in a rat model of acute MI.21) These results demonstrated that ghrelin can inhibit excessive activation of CSNA in an MI model. By contrast, ghrelin can also modulate parasympathetic nerve activity. Shimizu et al. reported that centrally administered ghrelin activated the cardiac vagal nerve in anesthetized rabbits.22) In their study, they measured norepinephrine and acetylcholine concentrations in dialysate samples after injection of ghrelin (1 nmol) into the lateral cerebral ventricle. Although ghrelin injection did not alter dialysate norepinephrine concentrations, it significantly increased dialysate acetylcholine concentrations (Fig. 5).22)
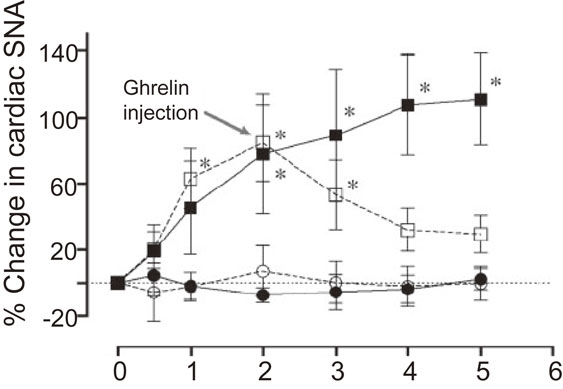
The sympathetic inhibitory effect of ghrelin after myocardial infarction (MI). Transient responses in cardiac sympathetic nervous activity (SNA) in sham rats (closed circles) and three groups of MI rats: untreated (closed squares), ghrelin-treated immediately after MI (open circles), and ghrelin-treated 2 hours after MI (open squares). Immediately after MI or 2 hours after MI, ghrelin treatment effectively reduced the up-regulated SNA. *Significantly different from before MI (time 0) (P < 0.05). Adapted from Schwenke et al.20)
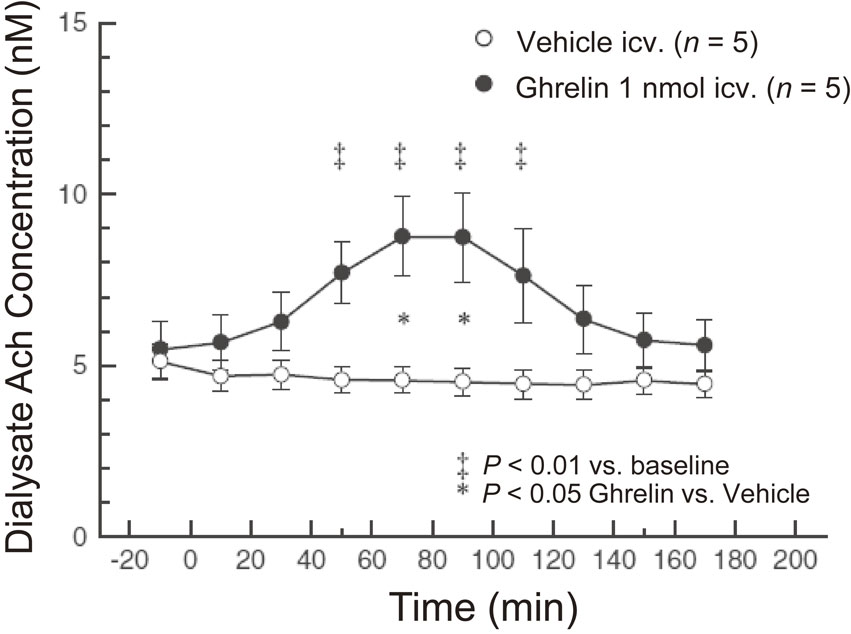
Time courses of dialysate acetylcholine (Ach) concentrations elicited by intracerebroventricular injection of ghrelin (●) or vehicle (○) (n = 5 in each group). Data are concentrations in dialysate samples collected over 20-minute durations. Values are mean ± SE. ‡P < 0.01 vs. baseline before injection. *P < 0.05, ghrelin vs. vehicle. Adapted from Shimizu et al.22)
Given that cardiac vagal afferent nerves send projections to the NTS, ghrelin might decrease cardiac sympathetic nerve activity through the vagal afferent nerve and the NTS (Fig. 6). In fact, we recently showed in an animal study that ghrelin prevents malignant arrhythmia after acute MI through the vagal afferent nerves.23) In that study, wild-type (WT) and ghrelin-knockout (KO) mice were anesthetized with urethane-chloralose, and baseline electrocardiogram (ECG) signals were recorded for 15 minutes. Thereafter, the MI procedure was performed and ECG signals were recorded for 30 minutes. Autonomic nervous function was examined by analyzing HR variability. After establishing MI, we observed a decrease in parasympathetic nerve activity in ghrelin-KO mice relative to WT mice, which was accompanied by an increased percentage of arrhythmic beats. In ghrelin-KO mice, subcutaneous ghrelin injection (150 µg/kg) significantly increased parasympathetic nerve activity after MI and significantly decreased the percentage of arrhythmic beats within 30 minutes after MI. Notably, treatment of the cervical vagal trunk with capsaicin abolished the effects of ghrelin in ghrelin-KO mice.24) Taken together, these findings demonstrated that ghrelin can stimulate cardiac parasympathetic activity.
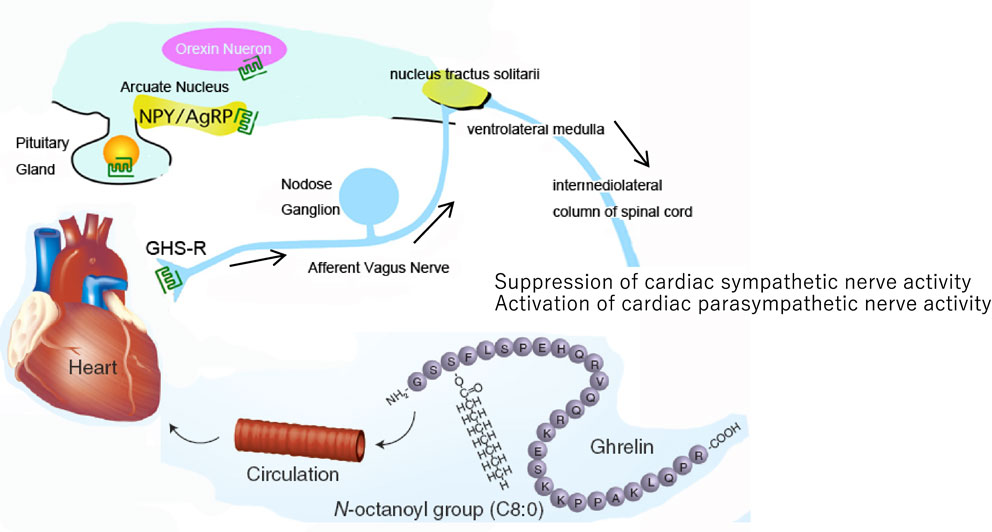
(Color online) Schematic representation of the potential mechanism of ghrelin-mediated regulation of the cardiac autonomic nervous system. By acting on the cardiac vagal afferent nerve terminals, which send signals to the vasomotor center of the medulla through the nucleus of the solitary tract (NTS; shown by arrows), peripheral ghrelin inhibits cardiac sympathetic nerve activity and activates cardiac parasympathetic nerve activity in cardiac disease, which may protect the heart from excessive damage. Adapted from Tokudome et al.38) AgRP: agouti-related peptide, NPY: neuropeptide Y, GHS-R: growth hormone secretagogue receptor.
Left ventricular remodeling after MI is often associated with subsequent heart failure. Previously, we investigated whether peripheral ghrelin administration may attenuate left ventricular dysfunction and remodeling in a rat MI model.24) Two weeks of subcutaneous administration of ghrelin (100 µg/kg, twice a day) significantly improved left ventricular enlargement induced by MI. Furthermore, ghrelin attenuated an increase in the collagen volume fraction of the non-infarct region (Fig. 7).24) We next examined whether subcutaneous injection of ghrelin (150 µg/kg) 1 minute after infarction had a beneficial effect.25) Surprisingly, an early one-shot bolus injection of ghrelin significantly inhibited cardiac remodeling and attenuated the impairment of left ventricular function.25) In another study, we demonstrated that endogenous ghrelin plays crucial roles in protecting heart function and reducing mortality after MI, and that these effects were, in part, the result of sympathetic inhibition.26) Thus, our data demonstrated that ghrelin exerts beneficial effects on cardiac remodeling after MI.
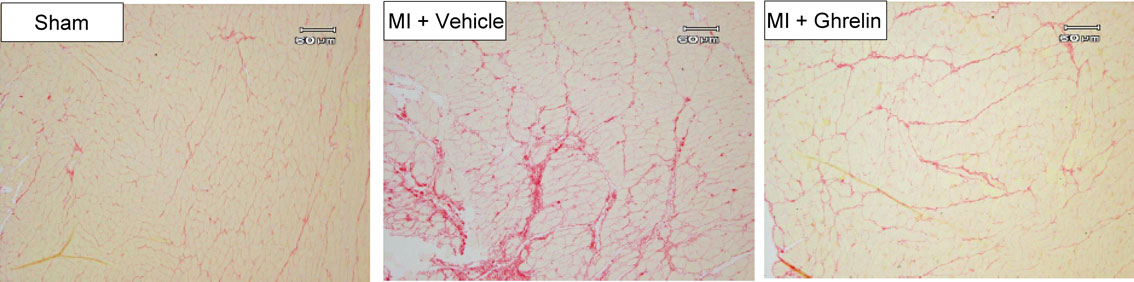
(Color online) Effects of ghrelin treatment on collagen volume in a non-infarcted left ventricular region. After administration of ghrelin for 2 weeks in rats with myocardial infarction, the left ventricular sections were stained with Sirius red. Representative photomicrographs of collagen volume are shown. Adapted from Soeki et al.24)
Malignant ventricular arrhythmia, which can occur during acute MI, is frequently life-threatening, and vagal nerve stimulation exerts an antiarrhythmic effect.27) Soeki et al. reported that intravenous administration of ghrelin (100 µg/kg) 30 minutes after ligation of the left coronary artery decreased the incidence of ventricular tachyarrhythmia through an increase in parasympathetic activity.28) This finding demonstrated that ghrelin also has antiarrhythmic activity.
Nagaya et al. demonstrated that ghrelin treatment improved cardiac performance and attenuated the development of cardiac cachexia in rats with heart failure.29) Four weeks after ligation of the left coronary artery (old myocardial infarction [OMI] model), 100 µg/kg of ghrelin was administered subcutaneously for 3 weeks. In rats with OMI, ghrelin treatment significantly increased serum GH levels and body weight, and also significantly enhanced cardiac output relative to that of rats treated with vehicle. Furthermore, ghrelin administration inhibited left ventricular enlargement in OMI rats.29) The same authors also evaluated the acute and chronic hemodynamic effects of ghrelin in CHF patients.30),31) In 12 patients with CHF who received intravenous infusions of ghrelin (0.1 µg/kg/min) for 60 minutes, ghrelin significantly decreased MAP without significantly increasing HR, significantly increased CI and SVI, and decreased systemic vascular resistance.30) They also evaluated the chronic effects of intravenous ghrelin administration (2 µg/kg, twice a day) in 10 patients with CHF after 3 weeks of treatment.31) Chronic administration of ghrelin significantly decreased plasma norepinephrine and epinephrine concentrations, and increased ejection fraction (27 ± 2% to 31 ± 2%, P < 0.05), in association with an increase in left ventricular mass and a decrease in left ventricular end-systolic volume. In addition, ghrelin administration increased peak workload and peak oxygen consumption during exercise.31) Furthermore, treatment with ghrelin significantly decreased the plasma brain natriuretic peptide level in these CHF patients (238 ± 59 pg/mL to 190 ± 60 pg/mL, P < 0.05).31) Taken together, these results strongly support the potential of ghrelin for the treatment of CHF.
Sustained elevation of pulmonary arterial pressure increases the workload of the heart, which can lead to congestive heart failure, as well as increased morbidity and mortality. Although the exact mechanism responsible for the pathogenesis of pulmonary arterial hypertension has yet to be fully elucidated, numerous studies have reported the disruption of pathways mediated by several vascular vasoactive substances. Our group investigated whether ghrelin administration can impede the pathogenesis of pulmonary arterial hypertension during chronic hypoxia.32) In the study, conscious rats were housed in a hypoxic chamber (10% oxygen); during this hypoxic period, the rats received a daily subcutaneous injection of either saline or ghrelin (150 µg/kg). In the saline-treated rats, chronic hypoxia led to significantly elevated pulmonary arterial pressure, increased the wall thickness of the peripheral pulmonary arteries (Fig. 8), and consequently induced right ventricular hypertrophy. By contrast, in the ghrelin-treated animals, hypoxia-induced pulmonary arterial hypertension, pulmonary vascular remodeling (Fig. 8), and right ventricular hypertrophy were all significantly attenuated.32)
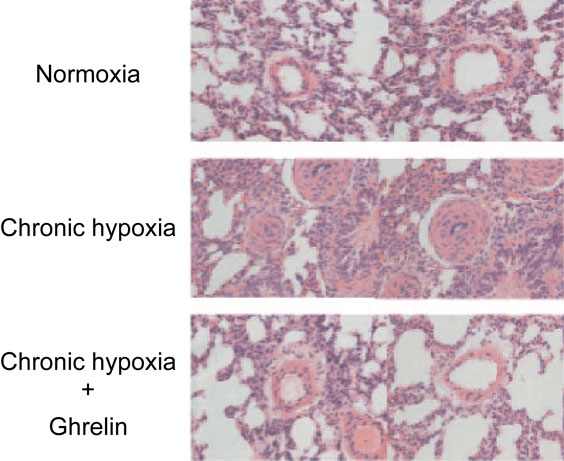
(Color online) Representative histological photomicrographs of peripheral pulmonary arteries from normoxic rats and chronic hypoxic rats treated with either saline or ghrelin. Adapted from Schwenke et al.32)
Patients with COPD have sustained alveolar hypoxia that leads to irreversible remodeling of the pulmonary artery. Furthermore, patients with COPD often exhibit a degree of cachexia, which is an independent risk factor for mortality in CHF.33) In an open-label pilot study, repeat administration of ghrelin improved body composition, muscle wasting, and functional capacity in seven cachectic patients with COPD.34) Collectively, these findings suggest that ghrelin may be beneficial for the treatment of pulmonary hypertension.
Cardiac hypertrophy is an adaptive response of the heart to persistent increases in hemodynamic workload, such as those caused by hypertension. Sustained cardiac hypertrophy significantly increases the risk of heart failure and sudden death. Inflammatory cytokines play important roles in the progression of cardiac hypertrophy.35) Stimulation of the vagus nerve by nicotine, a specific α7-nicotine acetylcholine receptor agonist, decreased the production of these cytokines.36) Ghrelin stimulates cardiac parasympathetic nerve activity, which led us to hypothesize that ghrelin can attenuate cardiac hypertrophy. Indeed, we demonstrated that endogenous ghrelin plays a crucial role in the progression of pressure overload-induced cardiac hypertrophy via a mechanism that involves activation of the cholinergic anti-inflammatory pathway.37) In that study, cardiac hypertrophy was induced by transverse aortic constriction (TAC) in ghrelin-KO mice and their WT littermates. After 12 weeks, the ghrelin-KO mice showed significantly increased cardiac hypertrophy versus WT mice (Fig. 9). Furthermore, the ghrelin-KO mice exhibited enhanced suppression of the cholinergic anti-inflammatory pathway, as indicated by reduced parasympathetic nerve activity and higher plasma IL-1β and IL-6 levels. Administration of either nicotine or ghrelin activated the cholinergic anti-inflammatory pathway and attenuated cardiac hypertrophy in ghrelin-KO mice.37)
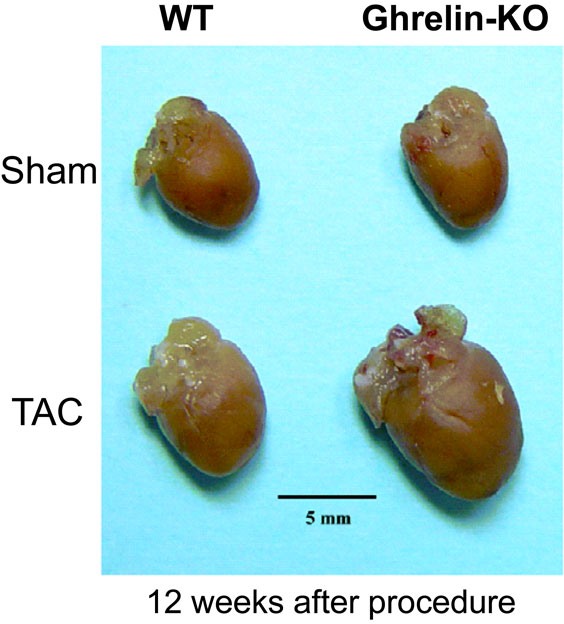
(Color online) Transverse aortic constriction (TAC) and cardiac hypertrophy. Typical gross heart samples from wild-type (WT) and ghrelin-knockout (KO) mice. Adapted from Mao et al.37)
Exogenous and endogenous ghrelin have potent beneficial effects on cardiovascular diseases, including MI, heart failure, pulmonary hypertension, and cardiac hypertrophy. These effects are exerted via modulation of the autonomic nervous system, and potentially by direct actions on cardiac cells (e.g., anti-apoptotic effects). Because ghrelin is an endogenous hormone, it has advantages over other medications, and, therefore, represents a promising new treatment for cardiovascular diseases.

Takeshi Tokudome was born in Kagoshima Prefecture, Japan, in 1971 and graduated from Kagawa Medical School in 1996. After graduation, he entered Kagawa Medical School Graduate School of Medicine under the directorship of Prof. Hirohide Matsuo, Second Department of Internal Medicine (Cardiology). He received his Ph.D. degree in 2000 and worked as a postdoctoral fellow at the National Cardiovascular Center Research Institute with Prof. Kenji Kanagawa. He is currently engaged in translational research on physiologically active peptides, in particular ghrelin and atrial natriuretic peptide. In 2010, he became Chief Investigator in the Department of Biochemistry, National Cerebral and Cardiovascular Center Research Institute.