Reviews
Function and mechanisms of memory destabilization and reconsolidation after retrieval
2020 年 96 巻 3 号 p. 95-106
詳細
2020 年 96 巻 3 号 p. 95-106
Memory retrieval is not a passive process. When a memory is retrieved, the retrieved memory is destabilized, similar to short-term memory just after learning, and requires memory reconsolidation to re-stabilize the memory. Recent studies characterizing destabilization and reconsolidation showed that a retrieved memory is not always destabilized and that there are boundary conditions that determine the induction of destabilization and reconsolidation according to certain parameters, such as the duration of retrieval and the memory strength and age. Moreover, the reconsolidation of contextual fear memory is not independent of memory extinction; rather, these memory processes interact with each other. There is an increasing number of findings suggesting that destabilization following retrieval facilitates the modification, weakening, or strengthening of the original memory, and the resultant updated memory is stabilized through reconsolidation. Reconsolidation could be targeted therapeutically to improve emotional disorders such as post-traumatic stress disorder and phobia. Thus, this review summarizes recent findings to understand the mechanisms and function of reconsolidation.
A memory of a recent experience, such as an episodic event, that should be memorized (short-term memory [STM]) is unstable. To store a memory as a stable long-term memory (LTM), it is stabilized through a consolidation process (Fig. 1).1)
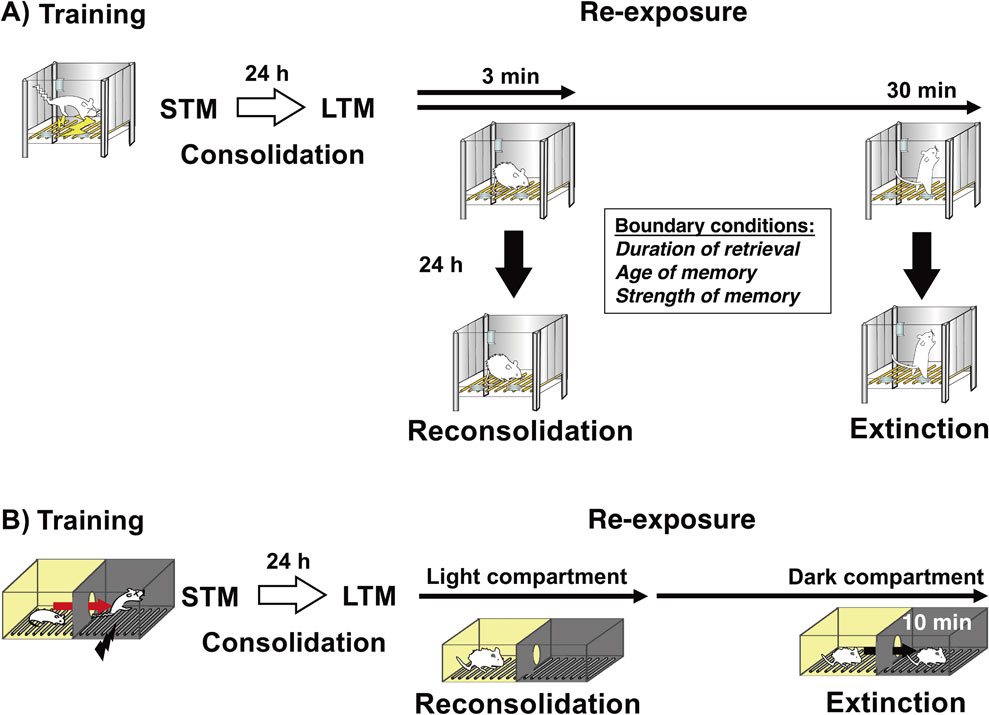
Comparison of consolidation, reconsolidation, and extinction. (A) Consolidation, reconsolidation, and extinction of contextual fear memory. Following training (contextual fear conditioning), an STM (short-term memory) is consolidated as an LTM (long-term memory) (consolidation). Following memory consolidation, short re-exposure to the context (e.g., 3 min) induces destabilization and reconsolidation, whereas extended re-exposure to the context (e.g., 30 min) induces memory extinction. There are boundary conditions that determine the induction of memory destabilization and reconsolidation according to certain parameters, such as the duration of retrieval and the strength and age of the memory. (B) Consolidation, reconsolidation, and extinction of inhibitory avoidance memory. Following training, an STM is consolidated as an LTM. Re-exposure to the light compartment induces destabilization and reconsolidation, whereas re-exposure to the dark compartment (e.g., 10 min) after entry from the light compartment induces memory extinction. This task allows us to discriminate between memory phases (reconsolidation or extinction) at the time point when the mice enter the dark compartment from the light compartment.
Importantly, memory retrieval is not a passive process. Nader et al. reported that a retrieved consolidated memory becomes labile, similar to an STM, via a destabilization process, and then that destabilized memory requires a reconsolidation process to re-stabilize it (re-storage of memory; Figs. 1, 2).2),3)
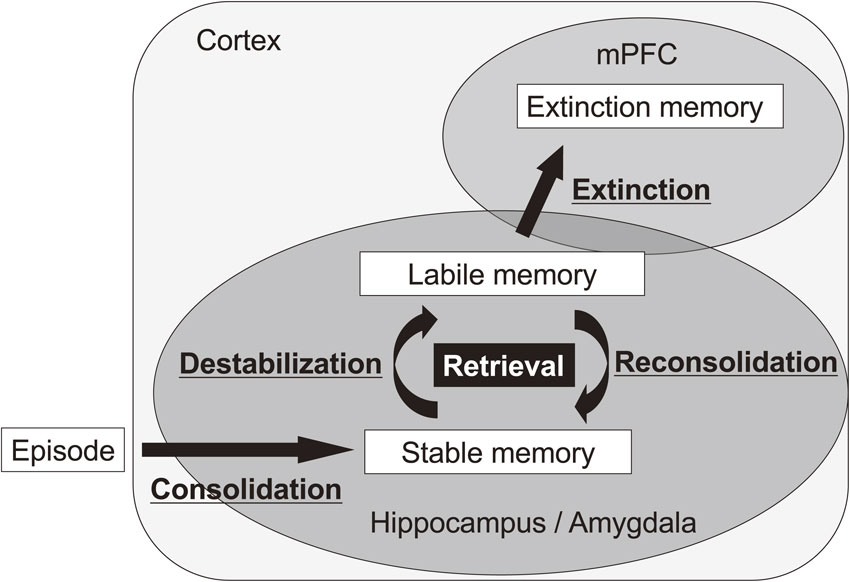
Memory processes after retrieval. To generate a stable memory, episodic memory including contextual fear memory is consolidated (consolidation) through the activation of gene expression. When a consolidated memory is retrieved, the retrieved memory is destabilized (destabilization) and then re-stabilized for re-storage (reconsolidation). Reconsolidation is also a gene expression-dependent process. A conditioned memory is extinguished when memory retrieval is extended by the long duration of re-exposure to the conditioned stimulus without an unconditioned stimulus. mPFC, medial prefrontal cortex.
From this finding, abundant questions have arisen in the field of learning and memory. For example, is memory reconsolidation a general and essential process after memory retrieval (is memory reconsolidation always required for the re-storage of retrieved memory)? Is memory reconsolidation observed for any memory type and in any species? What are the roles and function of memory reconsolidation (why is memory destabilized and reconsolidated after retrieval)? What are the differences in the mechanisms between consolidation and reconsolidation at the molecular, cellular, and circuit levels?
Notably, the retrieval of a fear memory initiates memory extinction, which is a process that weakens the memory (see below, Figs. 1, 2), whereas a retrieved fear memory is maintained or enhanced through memory reconsolidation. Therefore, memory retrieval induces two opposing processes (reconsolidation and extinction). The relationship between these processes has been investigated.
In this review, recent findings to characterize and understand memory reconsolidation are introduced and summarized to answer these fundamental questions about memory reconsolidation.
An STM lasts for a few hours after learning and is defined as a “labile” memory. To store an STM for a long period of time, a labile STM must be stabilized as a long-lasting LTM through a process known as “memory consolidation” (Fig. 1).1)
Memory consolidation consists of two sequential processes. The first is “cellular consolidation”, which allows a labile memory to become stable at the cellular level. The most important biochemical signature of this first process is the requirement for new gene expression. In rodents, amnestic drugs blocking gene expression, such as anisomycin, block memory consolidation, although this blockade does not affect STM.1),4) Of note, our previous study showed that blocking transcriptional activation by the transcription factor cAMP responsive element binding protein (CREB) in a genetically modified mouse model inhibits the formation of LTM (Fig. 3).4) This requirement for gene expression has been used as a marker to characterize or identify memory processes. Cellular consolidation induces changes in the plasticity of neurons/neural circuits to store a memory.1)
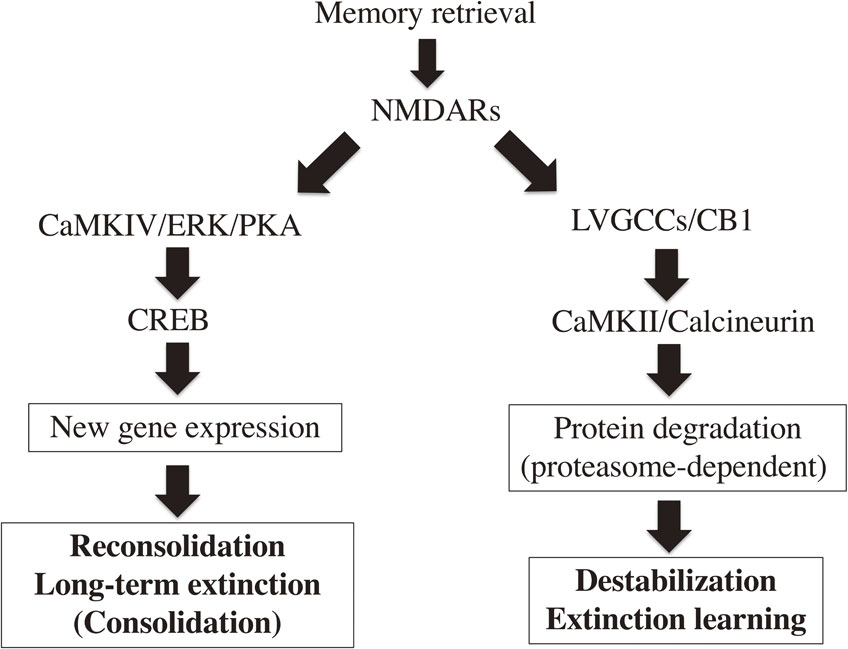
Signal transduction pathways regulating the destabilization, reconsolidation, and extinction of contextual fear memory. Activation of NMDA glutamate receptors (NMDARs) induces destabilization, reconsolidation, and extinction. Reconsolidation and long-term extinction require CREB-mediated gene expression through the phosphorylation of CREB by calcium/calmodulin-dependent protein kinase IV (CaMKIV), extracellular signal-regulated kinase (ERK), and protein kinase A (PKA). Destabilization and extinction learning require the activation of L-type voltage-gated calcium channels (LVGCCs), cannabinoid receptor B1 (CB1), calcineurin, and calcium/calmodulin-dependent protein kinase II (CaMKII) followed by proteasome-dependent protein degradation.
Memory consolidation involves a second process after cellular consolidation that is referred to as “systems consolidation”.5) Rodent studies showed that the hippocampus is required for the retrieval of an LTM that is formed within the previous few days. However, the hippocampus becomes no longer necessary for retrieval at 3–4 weeks after LTM formation.6) LTM retrieval requires the loss of hippocampal dependency and is dependent on cortical regions such as the anterior cingulate cortex with time after cellular consolidation.5)–7) Several studies have shown that systems consolidation requires the activity of alpha-calcium/calmodulin-dependent kinase II (αCaMKII),5),8) and it is also regulated by adult hippocampal neurogenesis.9) Thus, the current working hypothesis is that remote memories that had required the hippocampus for cellular consolidation are stored in various cortical regions following systems consolidation. In other words, the importance of the hippocampus is reduced in the neural network responsible for storing memory (memory circuit) as time passes.5) A study using optogenetic inhibition of hippocampal CA1 neurons showed that this inhibition blocks the retrieval of remote contextual fear memory,10) raising the possibility that the hippocampus contributes to the retrieval of remote memory when its function is not suppressed or inactivated during retrieval.
Since the 1960s, many reports have shown that consolidated memories are erased following retrieval under some conditions that block memory formation.11)–17) These phenomena suggest that consolidated memories become labile again following memory retrieval. Importantly, Nader et al. proposed that “reconsolidation” is required for re-storing a reactivated memory following memory retrieval (Fig. 2).2),3) Using rats, they showed that tone fear memory is disrupted when protein synthesis in the amygdala is inhibited immediately after retrieval by re-exposure to the tone as a conditioned stimulus, although this inhibition of gene expression does not affect the memory at 4 h following retrieval. This observation suggested that the activation of gene expression is required for re-storing a memory. Because this requirement for new gene expression is a feature that is shared with cellular consolidation, this process following memory retrieval is called “memory reconsolidation”. In conclusion, these findings suggested that when a fear memory is retrieved, it is destabilized and returns to a labile state, similar to an STM (i.e., destabilization), and then re-stored/re-stabilized through gene expression-dependent reconsolidation (Fig. 2). A large number of studies have shown that reconsolidation is necessary for re-storing various types of retrieved memories including contextual fear and spatial memories in various species from insects to humans.2)–4),18)–35) These findings suggested that memory reconsolidation is a common process among different types of memory and species.
Similar to the mechanism of consolidation, the reconsolidation of auditory and contextual fear memories requires gene expression in the amygdala or amygdala/hippocampus, respectively.2),36) Our study using CREB conditional mutant mice showed that the activation of CREB-mediated transcription is required for the consolidation and reconsolidation of contextual fear memory (Fig. 3). These observations and other experimental findings suggested that reconsolidation uses similar mechanisms as cellular consolidation at the molecular and cellular levels.37) However, differences between these mechanisms have been demonstrated at the molecular and anatomical levels. Brain-derived neurotrophic factor in the hippocampus is necessary for the consolidation, but not reconsolidation, of contextual fear memory, whereas the transcription factor Zif268 is required only for its reconsolidation.38) Notably, different brain regions are involved in the consolidation and re-consolidation of inhibitory avoidance memory. Gene expression in the hippocampus and amygdala is required for the consolidation of inhibitory avoidance memory, whereas gene expression only in the amygdala is necessary for its reconsolidation.21) These findings suggested that the mechanisms of memory consolidation and reconsolidation are similar but distinct.
These findings on the mechanism of reconsolidation suggested that a memory is destabilized when it is retrieved, thereby requiring gene expression to be re-stabilized (Fig. 2). One of the critical questions to understand the significance of memory reconsolidation is whether “memory destabilization” is an active or passive process. To answer this, molecules and/or signal transduction pathways to destabilize a retrieved memory have been identified by challenging the idea that their inactivation blocks the effects of amnestic drugs, such as anisomycin, which disrupt a retrieved memory. L-type voltage-gated calcium channels (LVGCCs), calcineurin, NMDA-type glutamate receptors (NMDARs), cannabinoid receptor B1 (CB1), proteasome-dependent protein degradation, dopamine signaling, CaMKII, muscarinic receptors, and dopamine D1/D5 receptors have been shown to be required for the destabilization of a retrieved memory (Fig. 3).39)–48) These findings indicated that memory destabilization is an active process requiring the activation of signal transduction in response to memory retrieval. A recent study showed that the levels of GluA2 subunit-containing Ca2+-impermeable and GluA2-lacking Ca2+-permeable AMPA-type glutamate receptors are decreased or increased, respectively, transiently at the post-synaptic surface in the lateral amygdala for a few hours following the retrieval of an auditory fear memory. This decrease in post-synaptic Ca2+-impermeable AMPA receptors is required for the destabilization of a retrieved memory, as blocking the endocytosis of Ca2+-impermeable AMPA receptors prevents the disruption of a retrieved memory with an amnestic drug. Conversely, Ca2+-permeable AMPA receptors are necessary for reconsolidation (restabilization), because blocking the function of Ca2+-permeable AMPA receptors disrupts a retrieved memory.49)
Memory destabilization and reconsolidation occur in the same brain regions, because destabilization of hippocampus-dependent memory is blocked by pharmacological inhibition of LVGCCs, calcineurin, or proteasome-dependent protein degradation in the hippocampus.40),43),47) Furthermore, the activation of gene expression and proteasome-dependent protein degradation occur in the same population of hippocampal neurons when a memory is reconsolidated.40)
Memory reconsolidation has been thought to be a process that updates an existing memory such as modifying memory strength, adding new information to an original memory, etc. Therefore, memory destabilization may be a process that opens a time window in which a memory can be updated. It is also possible that memory destabilization is crucial for behavioral flexibility. It is important to investigate the mechanisms of memory destabilization at the circuit and synaptic levels to understand further the roles and function of memory destabilization.
Do memory destabilization and reconsolidation always occur when a memory is retrieved? This was one of the general questions that arose when the reconsolidation process was identified in 2000.2),3) The following studies demonstrated that memory retrieval does not always destabilize a retrieved memory. These findings indicated that there are boundary conditions for the induction of memory destabilization and reconsolidation (Fig. 1).23)
In our contextual fear conditioning task, mice were placed in a contextual chamber (as a conditioned stimulus) for 3 min and given a 0.4-mA electric footshock (as an unconditioned stimulus) for 2 s in this chamber (training) (Fig. 1). To examine memory reconsolidation, the mice were returned to the contextual chamber for 3 min to retrieve the fear memory at 24 h after training (re-exposure). Inhibition of protein synthesis by systemic injection or micro-infusion of anisomycin after (or before) re-exposure disrupted the contextual fear memory when memory was tested at 24 h later. These observations indicated that re-exposure to the context (conditioned stimulus) for 3 min is sufficient to induce the destabilization and reconsolidation of a contextual fear memory.22) On the basis of these basic experimental procedures, we examined the parameters and conditions for the induction of destabilization and reconsolidation (Figs. 1, 2).
Duration of memory retrieval.When re-exposure was shortened to 1 min, the retrieved contextual fear memory was not disrupted, even when gene expression was inhibited. This re-exposure to the context for 1 min was sufficient for memory retrieval, because high levels of freezing responses were observed. This finding indicated that a retrieved memory is not destabilized when the duration of memory retrieval is short.23)
Memory strength.A stronger contextual fear memory generated by increasing the number of electric footshocks (3 footshocks) was not disrupted by inhibiting gene expression when mice were re-exposed to the context for 3 min. This observation suggests that a stronger memory is protected from destabilization following memory retrieval by this re-exposure duration.23) However, when the length of re-exposure was extended to 10 min, inhibition of gene expression disrupted the retrieved strong contextual fear memory. This observation suggested that longer memory retrieval is required to destabilize a strong memory.23) Of note, a similar phenomenon was observed for a strong auditory fear memory.50)
Memory age.A contextual fear memory becomes a hippocampus-independent “old memory” via systems consolidation at ∼4 weeks after memory formation.6) Similar to a strong fear memory, inhibition of gene expression failed to disrupt a retrieved old contextual fear memory (generated ∼4 weeks earlier) following re-exposure to the context for 3 min. However, this retrieved old memory was disrupted by inhibiting gene expression when re-exposure was extended to 10 min, suggesting that a longer re-exposure is necessary to destabilize an older memory, similar to a strong fear memory.23) Thus, the reconsolidation of a remote fear memory was observed. Previous studies have shown that the reconsolidation of a remote contextual fear memory depends on gene expression in the hippocampus, similarly with the reconsolidation of a recent memory.19),51) Additionally, the reconsolidation of recent and remote contextual fear memories depends on gene expression in the anterior cingulate cortex.52)
Taken together, these findings indicated that retrieved memories are protected more from destabilization as they get stronger or older, and that a longer retrieval duration is required for memory destabilization.23)
In 1927, Pavlov demonstrated the extinction of a conditioned memory. Re-exposure to a fear-conditioned context or tone (conditioned stimulus) induces fear responses such as freezing by memory retrieval.53),54) However, repeated or continuous representations of a conditioned stimulus without an unconditioned stimulus (e.g., electric footshock) extinguished fear responses induced by fear memory retrieval (memory extinction). Memory extinction has been thought to reflect new inhibitory learning rather than the erasure or forgetting of a conditioned memory; mice learn that they do not have to show a conditioned response by re-exposure to a conditioned stimulus.54) For example, contextual fear-conditioned mice showed intense freezing responses at the beginning of re-exposure to a conditioned stimulus, but showed a gradual decrease in freezing with time during re-exposure for 30 min (our experimental condition) (Fig. 1).23),54)
The spontaneous recovery of a conditioned response is observed even following extinction learning; animals with conditioned fear followed by extinction learning show fear relapse in response to re-exposure to the conditioned stimulus after a long period of time (e.g., 4 weeks later).54),55) Additionally, mice show reinstatement of fear; they show strong fear responses when they received even a weak electric footshock following memory extinction, which is insufficient to induce a strong fear response when received at the first fear conditioning. These behavioral findings supported the hypothesis that memory extinction does not consist of the forgetting, erasure, or disruption of a fear memory, unlike the disruption of a retrieved memory by blocking reconsolidation.54)
The consolidation of contextual fear extinction requires the activation of gene expression in the amygdala and medial prefrontal cortex (mPFC).36) Conversely, consolidation of the extinction of tone fear conditioning requires the activation of NMDARs and gene expression within the infralimbic cortex.56),57) These observations suggested that the consolidation of fear memory extinction (long-term extinction) shows similar molecular signatures as memory consolidation and reconsolidation (Fig. 3). Fear memory extinction requires the activation of CB1, LVGCCs, and proteasome-dependent protein degradation (data not shown).23),43),47),58) Of note, these molecules are required for the destabilization of a retrieved memory. Therefore, these findings indicated that memory destabilization and extinction share similar molecular mechanisms (Fig. 3).23),43),47),58),59)
As shown above, fear memory depends on the amygdala and hippocampus, whereas fear memory extinction requires the mPFC.36) Recent studies have suggested that the mPFC interacts with circuits in the amygdala and controls its function, thereby weakening fear expression.60),61) A recent study suggested that the dentate gyrus of the hippocampus encodes the extinction memory of contextual fear.62)
Comparisons of reconsolidation and extinction indicate that memory retrieval induces two opposite processes. Reconsolidation maintains or enhances a retrieved fear memory, while extinction weakens it; in contextual fear conditioning, brief memory retrieval (e.g., by 3 min re-exposure to the context) induces reconsolidation, whereas extended retrieval (30 min re-exposure) extinguished this memory (Figs. 1, 2).
Similar to memory consolidation, the activation of CREB-mediated transcription is required for the reconsolidation and extinction of a contextual fear memory. Phosphorylation of CREB at Ser133 was induced in the hippocampus and amygdala when a contextual fear memory is reconsolidated, whereas this increase occurred in the amygdala and mPFC when this memory is extinguished.36) Consistent with this, blocking gene expression in these brain regions or CREB-mediated transcription inhibited the reconsolidation and extinction, respectively, of a contextual fear memory.36)
On the basis of findings from a series of experiments on contextual fear memory, an important phenomenon has been identified.23) The inhibition of gene expression by the systemic injection of mice with the protein synthesis inhibitor anisomycin disrupted a retrieved contextual fear memory when reconsolidation is induced, whereas this inhibition blocked the consolidation of an extinction memory, “leaving the retrieved original contextual fear memory unaffected”, when a memory is extinguished. This is an important phenomenon because a retrieved fear memory is not disrupted when extinction learning is completed, even though gene expression is inhibited after memory retrieval. In other words, extinction learning prevents the destabilization of a retrieved contextual fear memory or cancels this destabilization, thereby blocking the disruption of this memory by inhibiting gene expression. These observations suggested that reconsolidation and extinction are not independent processes, but rather that both processes interact.
This conclusion was supported by other observations. First, similar experimental findings to those when using the systemic injection of anisomycin were observed when the amygdala receives a micro-infusion of anisomycin.36) The inhibition of protein synthesis in the amygdala disrupted a retrieved contextual fear memory following re-exposure to the context for 3 min; however, extended re-exposure for 30 min for extinction learning blocked this disruption, although the amygdaloid inhibition of gene expression blocked its long-term extinction. Second, a similar phenomenon was also observed at the molecular/cellular level. Phosphorylation of CREB at Ser133 was increased when a retrieved contextual fear memory was reconsolidated or extinguished. However, reconsolidation and extinction differ in the time course of the increase in Ser133-CREB phosphorylation in the amygdala, suggesting that the mechanisms of CREB activation in the amygdala are different between reconsolidation and extinction.36) Third, hippocampal CREB activation, including the induction of its phosphorylation and target gene expression such as c-Fos and Arc, was observed when a memory was reconsolidated. However, this CREB activation was no longer observed once extinction learning was completed.36) In other words, the extended retrieval of a fear memory for its extinction inhibited the transcriptional activation induced by CREB in the hippocampus. These experimental observations suggested that the interaction between reconsolidation and extinction occurred in the amygdala and hippocampus at the molecular level. It is important to identify the key molecules mediating this interaction and understand the mechanism for cancelling reconsolidation-induced signal transduction by extinction learning in the hippocampus.36)
Memory reconsolidation is required for the maintenance of an original memory. However, experimental evidence showing the boundary conditions of memory reconsolidation suggested functional roles for memory reconsolidation other than memory maintenance. Several reports suggested that memory reconsolidation is required for updating a memory, which enables the addition of new information to an original memory.3),16),31),63) Moreover, memory reconsolidation had been thought to play a role in enhancing a memory through the activation of signal transduction pathways such as mTOR and protein kinase A as well as gene expression.40),63)–65)
In contextual fear conditioning, the reconsolidation and extinction of a retrieved memory were induced in the same conditioning chamber. Therefore, we cannot discriminate between active memory processes after retrieval because it is difficult to clarify when reconsolidation and extinction learning are completed or initiated, respectively. In the inhibitory avoidance paradigm, mice received an electric footshock when they entered a dark compartment from a light compartment, generating a fear memory for the dark compartment (Fig. 1). Therefore, the mice retrieved this fear memory when they were re-exposed to the light compartment, but were unable to initiate extinction learning until they entered the dark compartment. Thus, this task allowed us to discriminate between “reconsolidation” and “extinction” at the time point when mice moved to the dark compartment from the light compartment because the subject would not know if they were going to be shocked or not until then. Therefore, in contrast to contextual fear conditioning, the inhibitory avoidance task allows us to examine the function of memory reconsolidation in an experimental condition in which memory retrieval triggers only reconsolidation without inducing extinction.
Our previous study using this task investigated the function and significance of memory reconsolidation.40) The retrieval of a fear memory by re-exposure to only the light compartment enhanced the memory. It is important to note that this enhancement was not additional learning because no footshock was delivered during the re-exposure session. Furthermore, inhibition of protein synthesis with re-exposure to the light compartment disrupted the retrieved fear memory, indicating that this re-exposure to the light compartment induced memory reconsolidation. Therefore, these observations suggested that memory reconsolidation functions as a memory enhancer in a condition that does not initiate extinction learning. Further studies showed that fear memory retrieval is enhanced through destabilization and reconsolidation via the activation of calcineurin-induced proteasome-dependent protein degradation and CREB-mediated gene expression, respectively, in the amygdala, hippocampus, and mPFC. Furthermore, the amygdala is required for the reconsolidation and enhancement of a fear memory, whereas the hippocampus and mPFC are required for its enhancement, but not reconsolidation, suggesting that the amygdala plays central and distinct roles from the hippocampus and mPFC in the enhancement/reconsolidation of this type of memory.40)
In summary, these experimental findings suggested that memory destabilization opens the window for the modification, weakening, or enhancement of an original memory, and the updated memory is then stabilized through reconsolidation.
Studies have suggested that humans and other animals such as rodents show similar mechanisms of fear memory regulation.66),67) Recent findings that fear memory retrieval opened the window to reconsolidate or extinguish an original memory and that blocking memory reconsolidation disrupted a retrieved memory suggest important clinical implications for retrieval-induced memory processes in the treatment of emotional disorders such as post-traumatic stress disorder (PTSD) and phobia.68) Therefore, findings from rodents studies have been applied to the development of PTSD treatment.69),70)
Prolonged exposure (PE) therapy is an effective cognitive treatment for PTSD.71)–73) PE therapy enables PTSD to be improved by the repeated and continuous retrieval of a memory of a traumatic experience. The biological basis for PE therapy is thought to reflect memory extinction.74)–78) Therefore, blocking reconsolidation and/or facilitating fear extinction are candidates for reducing fear in patients with emotional disorders to shorten the duration of PE therapy. For example, amygdaloid micro-infusion or systemic injection of the β-adrenergic blocker propranolol disrupts the retrieval of cued fear memory in rodents. PTSD pathologies such as fear responses to traumatic events are reduced by the administration of propranolol, perhaps by targeting reconsolidation.79) Of note, the administration of propranolol following the presentation of a tarantula improves the severity of phobia in patients with arachnophobia.80)
A recent report using rats showed that the systemic injection or amygdaloid micro-infusion of D-cycloserine, an agonist of the NMDA receptor glycine site, before re-exposure to a short (1 min) or long (1 min × 10 times with a 1-min interval) conditioned stimulus enhances or extinguishes, respectively, fear memory. This observation suggested that the activation of NMDARs enhanced reconsolidation following a short re-exposure, whereas this activation facilitates fear memory extinction following a long re-exposure.81) Therefore, it is important to estimate the phases in which memory is reconsolidated or extinguished when clinical trials are performed using drugs such as D-cycloserine. From this viewpoint, it is important to identify molecular markers that can be used to estimate memory status (reconsolidation or extinction). Nevertheless, clinical studies have suggested the efficacy of D-cycloserine for shortening the duration of PE therapy.73),78),82),83)
As described above, the activation of LVGCCs, CB1, and proteasome-dependent protein degradation weakens a fear memory by destabilizing or extinguishing this memory.23),40),43),49),58) These signal molecules might serve as therapeutic targets for the treatment of PTSD and phobia.
As described above, the spontaneous recovery of a fear response is observed even after fear memory extinction. A previous study using rodents showed that a fear response is not recovered when extinction learning is performed during a time window in which the fear memory is in the reconsolidation phase (within a few hours following fear memory retrieval).84) This “reconsolidation-update”, which prevents the spontaneous recovery of a fear memory, is also observed in humans.85) A recent finding showing that repeated extinction training prevents spontaneous recovery, suggesting that spontaneous recovery is the result of insufficient extinction learning.86) These experimental observations will help to develop treatment for PTSD.
Recently, we proposed that the enhancement of memory forgetting using a neurogenesis enhancer could be another therapeutic approach for PTSD, which may enable the development of PTSD treatment that does not require PE therapy.51),69),87) Further trials will be important to develop methods to facilitate PE therapy and/or PTSD treatment without PE therapy, by targeting memory processes including reconsolidation, extinction, and forgetting.
Memory destabilization and reconsolidation are processes that occur after memory retrieval. Memory destabilization/reconsolidation are not always induced when a memory is retrieved, but rather show boundary consolidation to induce these processes with parameters of retrieval duration and the strength and age of the memory. Memory extinction of contextual fear is a comparable process with memory reconsolidation after retrieval, because these processes are not independent, rather showing interactions at the molecular and cellular levels. Furthermore, memory reconsolidation is required for the enhancement and updating of an original memory. These findings from the past two decades suggest that memory destabilization following memory retrieval opens the window to modify, weaken, or strengthen an original memory, and the updated memory is then stabilized through memory reconsolidation. However, future studies are required to understand the molecular mechanisms of memory destabilization and reconsolidation at the synaptic level.
The author thanks members of the Kida lab for critical discussions on this manuscript. S. Kida was supported by The Science Research Promotion Fund, The Promotion and Mutual Aid Corporation for Private Schools of Japan; Grant-in-Aids for Scientific Research (A) [15H02488, 18H03944, 19H01047]; Grant-in-Aids for Scientific Research (B) [23300120, 20380078]; Grant-in-Aids for Challenging Exploratory Research [24650172, 26640014, 17K19464]; Grant-in-Aids for Scientific Research on Priority Areas-Molecular Brain Science [18022038, 22022039]; Grant-in-Aids for Scientific Research on Innovative Areas (Research in a proposed research area) [24116008, 24116001, 23115716, 17H06084, 17H05961, 17H05581, 18H05428, 18H05434, 19H04917]; Core Research for Evolutional Science and Technology (CREST), Japan; The Sumitomo Foundation; The Naito Foundation; The Uehara Memorial Foundation; Takeda Science Foundation, Japan.
Edited by Shigetada NAKANISHI, M.J.A.
Correspondence should be addressed: S. Kida, Graduate School of Agriculture and Life Sciences, The University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan (e-mail: akida@g.ecc.u-tokyo.ac.jp).

Satoshi Kida was born in Kanazawa in 1965. He graduated from the Department of Agricultural Chemistry at the University of Tokyo in 1989. He studied molecular endocrinology at the University of Tokyo, receiving his M.S. in 1991 and Ph.D. in 1994. Subsequently, he worked at the Institute of Molecular and Cellular Biosciences in the University of Tokyo as a postdoctoral fellow and received a fellowship from the Japan Society for the Promotion of Science (JSPS). He then moved to Cold Spring Harbor Laboratory as a postdoctoral fellow working with Dr. Alcino J. Silva. This postdoctoral work focused on genetic approaches to study the role of CREB in learning and memory. In 1997, he joined the Tokyo University of Agriculture (TUA) as an Associate Professor and became a professor in the Department of Bioscience of TUA in 2008. In 2019, Dr. Kida became a professor at the Graduate School of Agriculture and Life Sciences at the University of Tokyo. He has focused on understanding the mechanisms of memory consolidation, reconsolidation, extinction, and retrieval and tried to generate mouse models of post-traumatic stress disorders (PTSDs) and develop methods to improve PTSD. In 2009, he was elected to the council in the Molecular and Cellular Cognition Society (MCCS), which is a world-famous society for learning and memory, and is now a treasurer and Asian branch-president. He organized a grant group of Grant-in-Aid for Scientific Research on Innovative Areas (Research in a proposed research area) “Unraveling the micro-endophenotypes of psychiatric disorders at the molecular, cellular, and circuit levels” as a project leader from 2012.