Abstract
N-Glycanase 1 (NGLY1) deficiency is a congenital disorder caused by mutations in the NGLY1 gene. Because systemic Ngly1−/− mice with a C57BL/6 (B6) background are embryonically lethal, studies on the mechanism of NGLY1 deficiency using mice have been problematic. In this study, B6-Ngly1−/+ mice were crossed with Japanese wild mice-originated Japanese fancy mouse 1 (JF1) mice to produce viable F2 Ngly1−/− mice from (JF1×B6)F1 Ngly1−/+ mice. Systemic Ngly1−/− mice with a JF1 mouse background were also embryonically lethal. Hybrid F1 Ngly1−/− (JF1/B6F1) mice, however, showed developmental delay and motor dysfunction, similar to that in human patients. JF1/B6F1 Ngly1−/− mice showed increased levels of plasma and urinary aspartylglycosamine, a potential biomarker for NGLY1 deficiency. JF1/B6F1 Ngly1−/− mice are a useful isogenic animal model for the preclinical testing of therapeutic options and understanding the precise pathogenic mechanisms responsible for NGLY1 deficiency.
Introduction
N-Glycanase 1 (NGLY1), also known as peptide:N-glycanase, is an evolutionarily conserved enzyme among eukaryotes.1),2) NGLY1 is known to be involved in the quality control of newly synthesized N-glycoproteins. During the endoplasmic reticulum (ER)-associated degradation (ERAD) process, misfolded N-glycoproteins are retrotranslocated from the lumen of the ER to the cytosol. NGLY1 cleaves N-glycans that are attached to misfolded glycoproteins in the cytosol and the glycoproteins are then degraded by proteasomes.1)–5)
Mutations in the NGLY1 gene are associated with a rare congenital disorder that mainly affects young children.6)–12) Almost all NGLY1 missense and nonsense mutations studied thus far are characterized by reduced NGLY1 protein levels and enzymatic activity.13) In 2012, the first patient harboring mutations in NGLY1 was identified.12) Since then, more than 70 patients have been confirmed worldwide (Matt Wilsey, Grace Science Foundation, personal communication). The typical clinical features of NGLY1 deficiency include developmental delay, hypo/alacrimia, seizures, intellectual disability, motor function deficits, and movement disorders (including chorea, athetosis, dystonia, myoclonia, tremors, and dysmetria), as reported in several clinical reports.6)–11),14),15) Mutants of Ngly1 and its orthologs have been analyzed in various organisms to elucidate the molecular function of NGLY1.1),16)–28) However, the pathogenesis of NGLY1 deficiency is still poorly understood, and no effective therapy is currently available.
The establishment of systemic Ngly1-deficient rodent models that mimic the clinical features of NGLY1-deficient patients is essential for elucidating the mechanisms of NGLY1 deficiency and evaluating therapeutic options in preclinical studies. Rats and mice are typically the most commonly used rodent species for biomedical research. Rats are recognized as being more suitable for neurobehavioral studies than mice.29) Most behavioral phenotyping tasks have been developed in rats and are more popular. Indeed, Ngly1−/− rats develop neurological symptoms that are similar to subjects with NGLY1 deficiency.30) In contrast, the smaller size of mice also offers some advantages in drug development, because lower drug dosages and smaller breeding spaces are needed. Accordingly, they are much more cost-effective for use. Moreover, various genetic tools are available for use in studies with mice, such as a variety of tissue/cell-specific Cre-driver mouse lines.31) These driver mice enable researchers to investigate genes of interest in a tissue/cell (spatial control)-specific and/or time (temporal control)-specific manner and the detailed molecular pathological mechanisms equivalent to the human disease.31) The establishment of animal models for a disorder in multiple species can compare the phenotypic consequences among them and understand the disorder in a more precise manner.
A previous study reported that homozygous Ngly1 knockout (KO) mice derived from C57BL/6 (B6) mice showed embryonic lethality, whereas heterozygous mice were fertile and no recognizable phenotypes were observed.28) On the other hand, Ngly1-deficient mice, generated by mixing the genetic background between B6 and ICR mice, an outbred mice strain, were only partially embryonically lethal, indicating the importance of genetic background in rescuing the embryonic lethality of NGLY1 deficiency.28)
Both ICR and B6 mice and most experimental mouse strains have been established from the European wild mouse, Mus musculus domesticus. In contrast, strains from Japanese wild mouse (Mus musculus molossinus), including the Japanese fancy mouse 1 (JF1) strain, have high polymorphism levels against M. musculus domesticus and would be expected to better suppress various defects, including embryonic lethality, in the B6 background.
In this study, B6-Ngly1 hetero-KO mice were crossed with JF1. F1 Ngly1 hetero-KO mice and then crossed with each other. As anticipated, F2 viable Ngly1−/− mice were obtained, further demonstrating the importance of genetic background in the pathology of Ngly1−/− mice and the usefulness of M. musculus molossinus inbred strain established from Japanese wild mice for suppressing the embryonic lethality often found in the B6 background. A new Ngly1−/− type of mice with a JF1 background was then established using clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated 9 (Cas9) technology. Although the resulting mice were also embryonically lethal, F1 JF1/B6F1 Ngly1−/− mice obtained from the cross of Ngly1 hetero-KO B6 and JF1 mice partially avoided lethality through heterosis. JF1/B6F1 Ngly1−/− mice exhibited various phenotypic characteristics, including developmental delay and motor dysfunctions, which mimic NGLY1 deficiency symptoms. Histological abnormalities potentially associated with the neurologic phenotypes of JF1/B6F1 Ngly1−/− mice were also observed. B6 and JF1 mice are both inbred strains, and genetically homogeneous JF1/B6F1 Ngly1−/− mice provide yet another valuable rodent model for conducting preclinical studies related to NGLY1 deficiency as well as mechanistic insights into the pathophysiology of this devastating genetic disorder.
Materials and methods
Animals.
All animal care procedures and experiments conformed to the Association for Assessment and Accreditation of Laboratory Animal Care guidelines and were approved by either the Institutional Committee of RIKEN [approval no. H28-2-003(2)] or the Experimental Animal Care and Use Committee of Takeda Pharmaceutical Co., Ltd. (Kanagawa, Japan). All mice were housed in individual cages in a room with controlled temperature (23 °C), humidity (55%), and lighting (lights on from 7:00 am to 7:00 pm) and fed a normal chow diet (CE2 diet; CLEA Japan, Tokyo, Japan) with free access to water. For mice with a B6/JF1 mixed background, phenotypes were checked at monthly intervals. Once phenotypes were observed before death, the individuals were scored as positive; otherwise, the individuals were scored as negative. Mice were sacrificed by exsanguination. Spine curvature in each mouse was confirmed when they were sacrificed. Organs were eviscerated, and the weights of some were measured. The spinal cord was dissected. The thoracic and lumbar segments were identified using the ribs and vertebrae as a guide.
Generation of Ngly1−/− mice.
B6-Ngly1−/− mice were produced, as described previously.28) Similar to B6-Ngly1 KO mice,27),28) exons 11 and 12 and a 3′-flanking region of the JF1 mice Ngly1 genome were deleted using CRISPR/Cas9 genome editing, although this type of deletion was not reported in human patients.6)–12) JF1 mice were obtained from the National Institute of Genetics (Mishima, Japan). Two single-guide RNA (sgRNA) sequences targeting sites upstream (5′-sgRNA; 5′-TATGTCACTTCCACGGCCCT-3′) and downstream (3′-sgRNA; 5′-AGGGAACATTCACCTTGTCG-3′) from exons 11 and 12 and a 3′-flanking region of the JF1 mouse Ngly1 genome, respectively, were transcribed in vitro using the MEGAshortscript kit (MEGAshortscript T7 Transcription Kit; Life Technologies, Carlsbad, CA, U.S.A.). A mixture of Cas9 (100 ng/µL; New England Biolabs, Ipswich, MA, U.S.A.), 5′-sgRNA (50 ng/µL), and 3′-sgRNA (50 ng/µL) was microinjected into fertilized JF1 mouse eggs. The embryos were transferred to pseudo-pregnant females at the two-cell stage. The targeted genomic disruption of Ngly1 in mice was confirmed by polymerase chain reaction (PCR) using the primer set forward: aacccaggtagagatgtgagtttca and reverse: cctgagtctatggccttacacttca. To produce JF1/B6F1 Ngly1−/− mice, JF1-Ngly1−/+ mice were bred with B6-Ngly1−/+ mice. The procedures to produce Ngly1 KO mice were carried out by Axcelead Drug Discovery Partners, Inc. (Kanagawa, Japan). Ngly1−/− mice derived from JF1 mice will be deposited to and are available from the National BioResource Project - Mouse in Japan.
Motor function tests.
Hindlimb clasping test.
Mice were individually lifted by grasping their tails near the base, and their hindlimb positions were observed for 10 s. If both hindlimbs were partially or entirely retracted toward the abdomen for more than 5 s, mice were defined as having an abnormal hindlimb clasping reflex.
Rotarod test.
Motor performance and coordination behaviors were tested using an accelerating rotarod (MK610; Muromachi Kikai Co., Ltd., Tokyo, Japan) at different ages. Each mouse underwent 2 days of training session and 1 day of test session. Each session consisted of four separate 5-min trials with a 15-min interval between trials. The rotarod’s speed was accelerated from 4.5 to 45 rpm for 4 min and then kept at 45 rpm for another 1 min. The time required for mice to fall from the rod was measured. If mice remained on the rod until the end of the 5-min trial, a time of 300 s was recorded.
Grip-strength test.
During the grip-strength test, an animal was lifted by the tail, and its forelimbs or all limbs were allowed to grasp a metal mesh fixed to a force-electricity transducer (BS-TM-RM; Brain Science Idea Co., Ltd., Osaka, Japan). The animal was gently pulled upward while it grasped the mesh with its forelimbs or all limbs. The maximal force reached immediately before it released the mesh was taken as the grip-strength.
Gait analysis.
To assess the changes in the gait of mice, gait analyses were carried out. The forepaws and hind paws were coated with two nontoxic, water-soluble color inks (red for forepaws and blue for hind paws). Mice were then allowed to walk along 50-cm-long and 10-cm-wide runways. The floor of the runway was covered with sheets of white paper. The footprints were analyzed manually, and the stride length of each paw and the stance ratio (width ratio; hindlimbs to forelimbs) were quantified.
Histological analysis.
For light microscopy analysis, tissues from Ngly1−/− and wild-type (WT) mice at 5 and 42 weeks of age were dissected and fixed with 4% paraformaldehyde. Brains were trimmed coronally at four levels (levels 2, 3, 5, and 6) based on the STP position paper,32) paraffin-embedded, sectioned at 4 to 6 µm thickness, and mounted on slides. Sections were stained with hematoxylin and eosin (H&E). Sciatic nerves from Ngly1−/− and WT mice at 5 and 43 weeks of age were dissected and placed in 2% glutaraldehyde/2% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS; pH 7.4) overnight at 4 °C. After washing with PBS overnight at 4 °C, the samples were dehydrated and embedded in epoxy. For light microscopy, semi-thin plastic-embedded sections were prepared and stained with toluidine blue.
Immunoblotting analysis.
Brains were isolated and immediately frozen on dry ice. Whole brain lysates were prepared by homogenizing the tissue in T-PER buffer (Thermo Fisher Scientific, Waltham, MA, U.S.A.) with a protease inhibitor cocktail. The amount of protein in the lysates was quantified using the BCA Protein Assay Kit (Thermo Fisher Scientific). Tissue lysates (20 µg) were collected and resolved in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Bio-Rad, Hercules, CA, U.S.A.) and transferred onto polyvinylidene fluoride membranes (Bio-Rad) in accordance with the manufacturer’s instructions. Membranes were incubated with a primary antibody followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibody. These samples were then visualized with the ChemiDoc Imaging System (Bio-Rad) using a Western chemiluminescent HRP substrate (Merck Millipore, Burlington, MA, U.S.A.).
Immunostaining analysis.
For immunohistochemical analysis, all tissue sections were subjected to antigen retrieval using the microwave method [in Tris-EDTA (pH 9) for 15–20 min]. After blocking, the sections were incubated with primary antibodies overnight at 4 °C followed by 1 h incubation with fluorescently labeled secondary antibodies.
Antibodies.
The following antibodies were used at the indicated dilutions: rabbit anti-NGLY1 (1:200; HPA036825; Atlas Antibodies, Bromma, Sweden), guinea pig anti-p62 (1:1000; GP62-C; Progen, Heidelberg, Germany), mouse anti-glyceraldehyde 3-phosphate dehydrogenase (6C5; 1:5000; MAB374; Millipore, Burlington, MA, U.S.A.), rabbit anti-choline acetyltransferase (ChAT; 1:2000; ab178850; Abcam, Cambridge, United Kingdom), rabbit anti-glial fibrillary acidic protein (GFAP; 1:2000; ab7260; Abcam), goat anti-ionized calcium-binding adaptor molecule 1 (IbaI; 1:1000; ab5076; Abcam), rabbit anti-MAP2 (1:4000; ab183830; Abcam), rabbit anti-cleaved caspase-3 (1:200; #9661; CST, Danvers, MA, U.S.A.), rabbit anti-NeuN (1:3000; ab177487; Abcam), mouse anti-NeuN (1:1000; SIG-39860; BioLegend, San Diego, CA, U.S.A.), and mouse anti-ubiquitin (1B3; 1:200; MK-11-3; MBL, Nagoya, Japan) antibodies.
Measurement of urinary and plasma aspartylglycosamine (AsnGlcNAc) levels.
Urinary and plasma AsnGlcNAc levels were determined by liquid chromatography/tandem mass spectrometry. The sample was added to a ninefold volume of methanol and 1/10 of the internal standard (IS) solution (13C5-glutamic acid at 10 mmol/L in water). After vortex-mixing, the sample was centrifuged at 21,500 × g for 5 min. A 400 µL portion of the supernatant was evaporated to dryness. The dry residue was reconstituted with 50 µL of 0.1 mol/L aqueous Na2CO3 and 50 µL dansyl chloride solution (4 mg/mL in acetonitrile) and then incubated for 15 min at 60 °C. The derivatization was terminated by adding 5 µL of 10% (w/w) aqueous formic acid solution followed by centrifugation at 21,500 × g for 5 min. The supernatant was injected into a UHPLC Nexera liquid chromatography system (Shimadzu Co., Kyoto, Japan) equipped with a Gemini C18 column (100 × 2 mm, 3 µm, 110 Å; Phenomenex, Inc., Torrance, CA, U.S.A.) and maintained at 40 °C. AsnGlcNAc and IS were separated by gradient elution with mobile phase A (10 mmol/L HCOONH4 and 0.1% formic acid in water) and mobile phase B (acetonitrile). The gradient involved 5% B for 1 min, with a linear increase from 5% to 59% B in 4 min. The eluate was directly introduced under electrospray ionization into a 5500QTRAP mass spectrometer (AB Sciex Pte. Ltd., Toronto, Ontario, Canada). All target molecules were scanned in selected reaction monitoring (SRM) mode; the conditions are listed in Supplementary Table 1. For the calibration curve, working solutions were prepared by diluting a stock solution of AsnGlcNAc to concentrations of 0.01, 0.03, 0.1, 0.3, 1, 3, and 10 nmol/L with 0.2% bovine serum albumin (BSA) in saline. Five microliters of a working solution were mixed with 5 µL of the IS solution and 50 µL of 0.2% (w/v) BSA in saline and then processed as described above. SRM data were processed by Analyst 1.6.3 (AB Sciex). The linear calibration curve was obtained with a weight of 1/x and confirmed by assessing the accuracy within the range of ±20%.
Statistical analysis.
Data are presented as the mean ± standard error of the mean (SEM). Statistical significance was determined using Student’s t-test to compare two groups and Dunnett’s multiple comparison test to compare three groups at P < 0.05 and 0.01.
Results
B6 and JF1 mixed background genome partially rescues the embryonic lethality caused by the loss of the Ngly1 gene.
Ngly1-deficient mice with a B6 background genome showed embryonic lethality.28) B6-Ngly1−/+ mice were crossed with JF1 mice to produce JF1/B6F1 Ngly1−/+ mice. JF1/B6F1 Ngly1−/+ mice were further crossed to produce F2 mice (Fig. 1A). Ngly1−/− mice were obtained live from the F2 generation, although they were born in a proportion that deviated from the Mendelian ratio (WT/heterozygous/homozygous, 0.6:1.0:0.2; Supplementary Table 2). Therefore, this result suggested that the JF1 or B6 and JF1 mixed background genome may partially rescue the embryonic lethality caused by loss of the Ngly1 gene. More than 50% of Ngly1−/− F2 mice with a JF1/B6 mixed background survived after 30 weeks (Fig. 1B). This was in sharp contrast to Ngly1−/− F2 mice from the B6 and ICR cross, where >70% of mice died within 3 weeks, and the longest-living mice died at 29 weeks.28) Some mice that survived exhibited phenotypes with variable penetrance (Supplementary Table 3; see Supplementary Fig. 1A–1E for pictures of mice). Among them, hindlimb clasping was the most frequent phenotype for the JF1/B6 mixed background Ngly1−/− mice. All Ngly1−/− mice tested showed this phenotype (Supplementary Table 3). These results were consistent with previous observations for Engase Ngly1 double-KO mice28) or Ngly1−/− rat.30)

The results for the JF1/B6 mixed background led to the hypothesis that JF1 may have some modifier genes that were resistant to Ngly1 deletion in mice. Therefore, Ngly1−/− mice were generated in a JF1 background. To this end, the CRISPR/Cas9 genome editing technology was employed. Similar to B6-Ngly1−/− mice, JF1-Ngly1−/+ mice have approximately 2.4 kb of deletions in exons 11 and 12 and the 3′-flanking region of the Ngly1 gene (Fig. 2A). Surprisingly, however, no Ngly1−/− mice were produced when their littermate Ngly1−/+ and WT mice were weaned. This result clearly suggested that Ngly1 is also critical for mice with a JF1 background to be viable. The results of the mixed background mice indicated that both B6 and JF1 distinctly cause embryonic lethality, although heterosis cancels this lethality in both strains.

To produce JF1/B6F1 Ngly1−/− mice, JF1-Ngly1−/+ mice were bred with B6-Ngly1−/+ mice (Fig. 2B). The ratio of JF1/B6F1 Ngly1−/− mice that were weaned deviated from the Mendelian ratio (WT/heterozygous/homozygous, 0.5:1.0:0.3; Supplementary Table 4). However, once mice escaped embryonic lethality or early developmental death before weaning, no difference in mortality was found among the genotypes. Most JF1/B6F1 Ngly1−/− mice survived for 1.5 years, similar to WT and Ngly1−/+ mice. The targeted genomic disruption of Ngly1 in JF1/B6 Ngly1−/− mice was confirmed by PCR. There was no detectable Ngly1 protein in lysates of whole brains of JF1/B6F1 Ngly1−/− mice (Fig. 2C).
The mean body weight of male JF1/B6F1 Ngly1−/− mice was significantly lower than the values of their littermate WT and Ngly1−/+ mice (Fig. 2D). However, there was no significant difference in the mean body weight between female Ngly1−/− mice and their littermates (Supplementary Fig. 2A). As a distant possibility, the body weight difference may come from the difference in diet uptake between three groups, and further studies will be needed to clarify this issue. The brain weights of JF1/B6F1 Ngly1−/− mice were significantly lower than those of WT mice at 5 and 42 weeks of age [male (Fig. 2E) and female (Supplementary Fig. 2B)], implying microcephaly in JF1/B6F1 Ngly1−/− mice, as observed in NGLY1-deficient human patients.6)–11),15) Scoliosis, a characteristic of NGLY1-deficient subjects,6)–11),14) was also observed in 2 out of 9 older Ngly1−/− mice at 42 weeks of age (Fig. 2F) but not at 5 weeks of age.
JF1/B6F1 Ngly1−/− mice develop motor deficits.
The motor behaviors of JF1/B6F1 Ngly1−/− mice and their littermate WT mice were compared.
Hindlimb clasping test.
All JF1/B6F1 Ngly1−/− mice developed an abnormal hindlimb clasping reflex after 4 weeks of age when they were suspended by their tails, whereas WT mice showed the characteristic response, attempting to escape by splaying their hindlimbs away from the trunk of their body (Fig. 3A).
Rotarod test.
Motor coordination was assessed at 4, 8, 12, 21, and 36 weeks of age by rotarod tests. Male JF1/B6F1 Ngly1−/− mice showed a significantly reduced latency at 4 weeks of age (Fig. 3B). Female JF1/B6F1 Ngly1−/− mice also showed a significantly reduced latency at 4 weeks of age and showed a progressive decline in rotarod performance (Supplementary Fig. 3A).
Grip-strength test.
The grip-strength of forelimbs or forelimbs and hindlimbs were significantly reduced in both male and female JF1/B6F1 Ngly1−/− mice compared to their littermate WT mice (Fig. 3C and 3D; Supplementary Fig. 3B and 3C).
Gait analysis.
JF1/B6F1 Ngly1−/− mice showed obvious gait abnormalities. To further characterize the motor phenotype of JF1/B6F1 Ngly1−/− mice, their gait behavior was analyzed at several ages. Male JF1/B6F1 Ngly1−/− mice showed gait abnormalities, including a wide-based ataxic gait (Fig. 3E). Male Ngly1−/− mice had a significantly increased stance ratio (Fig. 3F) and shorter stride lengths (Fig. 3G) than their littermate WT mice at 4, 8, 12, 21, and 36 weeks of age.
Accumulation of ubiquitinated proteins in the central nervous system (CNS) of JF1/B6F1 Ngly1−/− mice.
NGLY1 is thought to play an important role in the efficient degradation of misfolded glycoproteins during ERAD.1)–5) The accumulation of ubiquitinated proteins is characteristic of impaired ERAD systems. Protein aggregation in the CNS was examined using an antibody against ubiquitin. Ubiquitin-positive proteins frequently accumulated in the cytoplasm of cells in the thalamus, hippocampus, cerebral cortex, and spinal cord of JF1/B6F1 Ngly1−/− mice, but this was not observed in WT mice (Fig. 4A–4C). There were few ubiquitin-positive cells in the cerebellum (e.g., Purkinje cells) of JF1/B6F1 Ngly1−/− mice. Notably, ubiquitin-positive proteins were observed exclusively in cells that were positive for the neural cell marker MAP2 (Supplementary Fig. 4). Immunohistochemical analyses indicated that ubiquitin-positive cells in the CNS were observed only in 5-week-old JF1/B6F1 Ngly1−/− mice but not obvious in 42-week-old mice (Supplementary Fig. 5). p62 colocalization with ubiquitin was also observed in the brain of Ngly1−/− mice at 5 weeks of age (Fig. 4D).

To elucidate the pathological mechanisms of NGLY1 deficiency, a histological examination of the brain, spinal cord, and sciatic nerves was carried out in JF1/B6F1 Ngly1−/− mice and their littermate WT mice at 5 and 42 weeks of age. Pathological abnormalities that could be associated with neurodegenerative phenotypes in JF1/B6F1 Ngly1−/− mice were identified. Histological examinations of H&E-stained sections revealed no clear Ngly1−/− mouse-specific neurodegenerative features in the CNS (Supplementary Fig. 6). Peripheral nerve abnormalities in JF1/B6F1 Ngly1−/− mice were also examined. Axonal degeneration in the sciatic nerve was observed in Ngly1−/− rats30); therefore, histological analysis of the sciatic nerves at different ages was carried out. Toluidine blue-stained semi-thin sections indicated the prominent atrophy of the sciatic nerve in JF1/B6F1 Ngly1−/− mice at 5 and 42 weeks of age (Supplementary Fig. 7). However, the number of axons in each area and the extent of myelination appeared normal in Ngly1−/− mice (Supplementary Fig. 7).
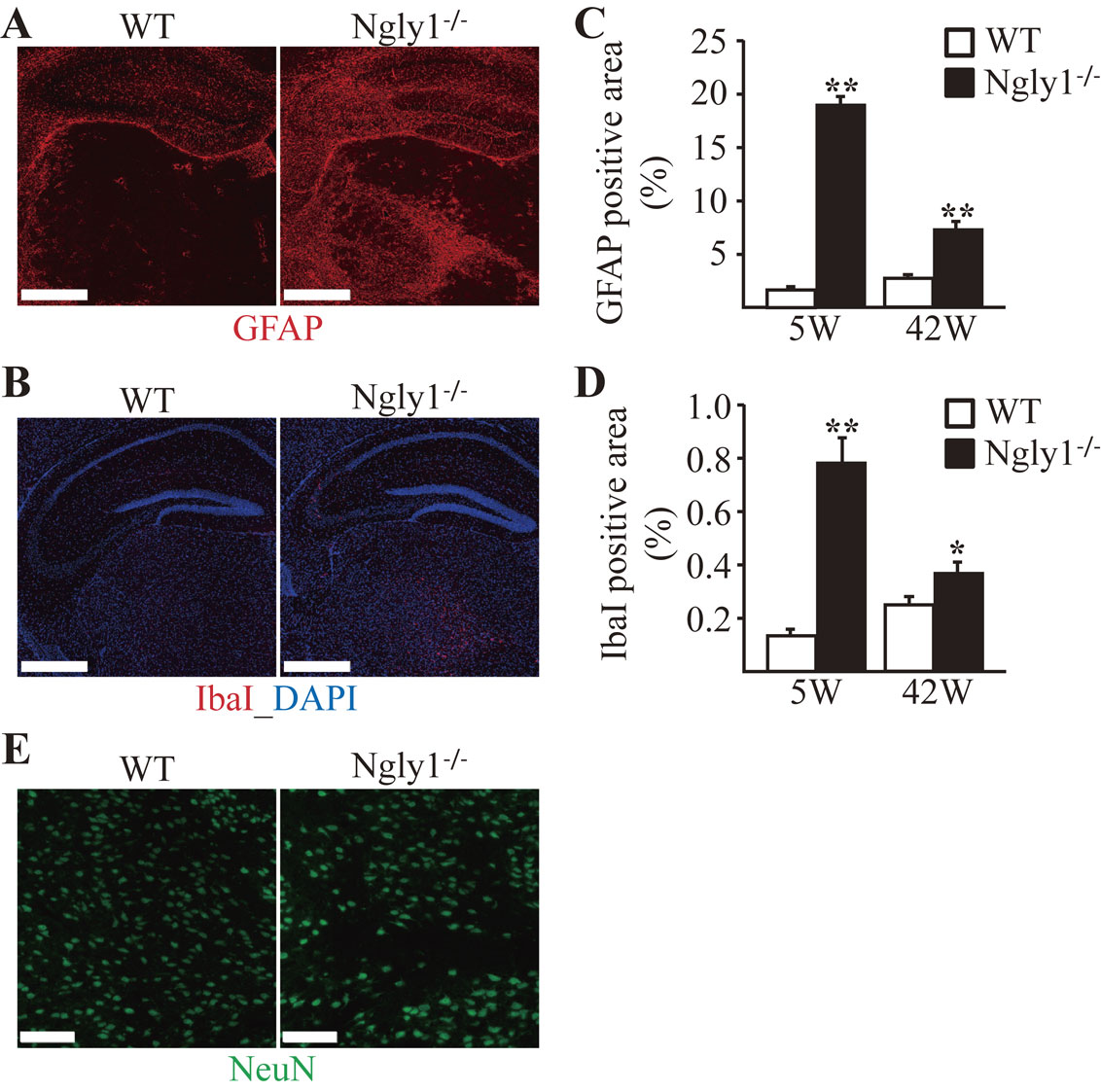
Astrogliosis and microgliosis in the CNS of JF1/B6F1 Ngly1−/− mice.
Reactive astrocytes and microglial activation are often regarded as indicators of neural toxicity or neuronal cell death.33) Because astrogliosis/microgliosis of the thalamus in Ngly1−/− rats was clearly observed,30) the GFAP expression level using immunofluorescence staining was quantified. Compared to their littermate WT mice, JF1/B6F1 Ngly1−/− mice developed enhanced GFAP immunoreactivity in the thalamus and hippocampus (Fig. 5A). Consistent with astrogliosis, microglial activation was also observed, as indicated by enhanced IbaI immunoreactivity in the same regions of the thalamus and hippocampus of JF1/B6F1 Ngly1−/− mice (Fig. 5B). Astrogliosis and microgliosis in the thalamus of Ngly1−/− mice were more prominent at 5 weeks of age than at 42 weeks of age (Fig. 5C and 5D). Although the Ngly1 gene is expressed ubiquitously in the entire CNS, the effects of Ngly1-KO on the CNS appear to be selective. Gliosis was most prominent in the lateral and medial parts of the ventroposterior (VPL/VPM), ventrolateral (VL), and ventromedial (VM) nuclei of the thalamus and the CA2 and CA3 regions of the hippocampus in JF1/B6F1 Ngly1−/− mice. Immunohistochemical staining with an antibody against mature neurons showed no significant difference in the number of neurons in the thalamus (Fig. 5E).
AsnGlcNAc was recently identified as a potential small-molecule biomarker for NGLY1 deficiency in human patients.34) Consistent with NGLY1-deficient subjects, JF1/B6F1 Ngly1−/− mice also showed increased plasma and urine AsnGlcNAc levels compared with their littermate WT mice (Fig. 6A and 6B). There was no significant difference in plasma AsnGlcNAc levels between JF1/B6F1 Ngly1−/+ and WT mice (Fig. 6A). No significant increases in AsnGlcNAc levels in plasma and urine with advancing age were found as far as this study has examined (Fig. 6A and 6B).
Discussion
The mechanisms that underlie neurological symptoms in NGLY1 deficiency are poorly understood, and no effective therapy is currently available. Animal models that recapitulate the clinical signatures of NGLY1-deficient patients are critical in elucidating relationships between the NGLY1 gene and the disease mechanism. The present work revealed that JF1/B6F1 Ngly1−/− mice, a novel systemic Ngly1-deficient model, developed symptoms that are characteristic of human NGLY1-deficient patients, including developmental delay, motor dysfunction, scoliosis, and lower brain weight. These phenotypes in JF1/B6F1 Ngly1−/− mice were also observed in Ngly1-deficient rats (Supplementary Table 5), suggesting the validity of JF1/B6F1 Ngly1−/− mice as another model animal for studying NGLY1 deficiency.
There were no obvious morphological changes in the CNS and PNS of JF1/B6F1 Ngly1−/− mice. In contrast, they showed pronounced microglial activation, a sign of neuroinflammation, within the VPM/VPL, VL, and VM nuclei of the thalamus. The motor thalamus is composed of VL and VM thalamic nuclei that function as a hub to transmit signals from the basal ganglia and cerebellum to the motor cortex in rodents. Thus, microgliosis in these nuclei could lead to motor dysfunction in JF1/B6F1 Ngly1−/− mice. Microglial activation in the thalamus was also observed to be a striking pathological feature in Ngly1-deficient rats.30) However, JF1/B6F1 Ngly1−/− mice did not show the neurodegenerative features observed in Ngly1−/− rats, including necrotic lesions, mineralization, and intracellular and extracellular eosinophilic bodies. These results in mouse and rat models of NGLY1 deficiency suggest that neuroinflammation in the thalamic nuclei could be associated with the motor deficit caused by the loss of the Ngly1 gene. The authors currently do not have any access to data of patients that specifically implicate a thalamus defect. Therefore, it is unclear if the enhanced gliosis in the VL and VM regions directly contributes to the phenotypic consequences of JF1/B6F1 Ngly1−/− mice. To confirm whether neuroinflammation may be a key contributor to the onset and progression of NGLY1 deficiency, pharmacological intervention by neuroprotective agents targeting neuroinflammatory pathways will be required in Ngly1 KO rodents (rats and mice). In recent years, increased interest has developed in finding appropriate tools to effectively target neuroinflammation in neurodegenerative diseases, amyotrophic lateral sclerosis (ALS), multiple sclerosis, and Alzheimer’s disease.35) For example, multiple compounds with anti-neuroinflammatory properties have been reported to enhance motor neuron survival in ALS animal models. However, it is not yet shown whether these compounds will be effective for human subjects.36)
NGLY1 is thought to play a pivotal role in ERAD processes by deglycosylating misfolded glycoproteins while they are degraded by proteasomes.1)–5) Consistent with this notion, JF1/B6F1 Ngly1−/− mice showed an accumulation of ubiquitinated proteins in the CNS. Impaired ubiquitin-proteasome systems (UPS) have been implicated in several neurodegenerative diseases based on the presence of ubiquitin-positive deposits in affected neurons.37) This suggested that ubiquitin-positive defective proteins could affect the symptoms of JF1/B6F1 Ngly1−/− mice with neuroinflammation in the brain. p62 mediates the formation of protein aggregates destined for autophagic turnover, which suggests that p62 facilitates the selective degradation of protein cargo via autophagy.38) It is now understood that p62 functions as an adaptor protein, binding ubiquitylated protein aggregates and delivering them to autophagosomes. There are two major pathways for accomplishing regulated protein catabolism, the UPS and the autophagy-lysosomal systems. The UPS serves as the primary route for the degradation of thousands of short-lived defective proteins and provides the exquisite specificity and temporal control needed for fine-tuning the steady-state levels of many regulatory proteins.39) Autophagy, in contrast, is primarily responsible for degrading long-lived proteins, although its contribution to the degradation of defective proteins may be comparable to that of the UPS.39) Recent studies have revealed the existence of a functional cross-talk between the UPS and autophagy, suggesting a coordinated and complementary relationship between these degradation systems that becomes critical during cellular stress. In this study, part of the ubiquitin-positive cells also stained positive for p62. Accumulation of ubiquitinated proteins was less observed in 42-week-old than 5-week-old JF1/B6F1 Ngly1−/− mice (Supplementary Fig. 5). Consistent with the number of ubiquitin-positive cells in the brain, gliosis in JF1/B6F1 Ngly1−/− mice was less pronounced at 42 weeks of age compared with that at 5 weeks of age (Fig. 5). These results may imply that there is a compensatory process, such as autophagy, to remove ubiquitinated proteins or ubiquitin-positive cells themselves in JF/B6F1 Ngly1−/− and, therefore, suppress neuro-inflammation over time, but the detailed mechanism will be a matter of further investigation.
AsnGlcNAc was recently identified as a potential small-molecule biomarker for NGLY1 deficiency, making the biochemical diagnosis for NGLY1 deficiency potentially feasible.34) As with human patients, JF1/B6F1 Ngly1−/− mice showed increased plasma and urinary AsnGlcNAc levels compared with their littermate WT mice. Considering the role of Ngly1, which catalyzes the cleavage of the amide bond between proximal N-acetylglucosamine residues in N-glycans and the asparagine residue of a protein, it is reasonable to assume that the loss of the Ngly1 gene may result in AsnGlcNAc accumulation. How and exactly where the AsnGlcNAc is formed and excreted into the plasma and urine remain unclear. To establish AsnGlcNAc as a promising marker for drug evaluation and patient screening, it is important to confirm whether AsnGlcNAc levels are correlated with the severity of disease and elucidate the precise molecular mechanism for the excretion of AsnGlcNAc.
In this study, the embryonic lethality caused by loss of the Ngly1 gene was partially rescued by mixing the genetic background between JF1 (M. musculus molossinus) and B6 (M. musculus domesticus) mice, demonstrating the usefulness of Japanese wild mouse-derived inbred strains for creating disease model animals. The results clearly showed that both JF1 and B6 mice have critical genetic factors for embryonic lethality, and it was suppressed by each other’s genetic factors through heterosis. Because some of the phenotypes observed in F2 mice (e.g., hair loss and swollen digits) were not obvious in JF1/B6F1 mice, the identification of modifier gene(s) by crossing JF1/B6F1 mice and scoring those phenotypes on resultant F2 mice may be possible, and efforts are currently under way along this line. Mouse genetics using inbred strains with very different genetic background provides a strong possibility to identify genes involved in the production of focused phenotypes, in this case modifying genes to mutated Ngly1. The number of available genetic markers due to the difference of genomic sequences between M. m. domesticus and M. m. molossinus is quite large and provides the means to trace the genes responsible, and this cannot be realized using popular inbred strains that are derived from domesticus. This beneficial approach was only realized by the efforts of mouse genetics researchers for the establishment of inbred mouse strains with molossinus origin and mentioning this issue would be appropriate to recognize their contributions.
In summary, JF1/B6F1 Ngly1−/− mice are a valuable, genetically homogeneous systemic Ngly1 KO rodent model that recapitulates the biological and behavioral features of human NGLY1 deficiency. JF1/B6F1 Ngly1−/− mice represent an experimentally reproducible in vivo model of NGLY1 deficiency that can be utilized to evaluate therapeutic options and study the cellular and molecular disease mechanisms responsible for this disease.
Acknowledgments
We wish to thank the members of the Suzuki Project in T-CiRA and the Grace Science Foundation for fruitful discussions. We also wish to thank Drs. Toshihiko Shiroishi and Toyoyuki Takada (RIKEN BRC) for valuable discussions. T.S. thanks Dr. Shinichi Nakagawa (Hokkaido University) for his suggestion on the use of M. musculus molossinus. This study was partly supported by the Glyco-lipidologue Initiative (RIKEN) and the Grace Science Foundation.
Notes
Edited by Kunihiko SUZUKI, M.J.A.
Correspondence should be addressed: T. Suzuki, Glycometabolic Biochemistry Laboratory, RIKEN Cluster for Pioneering Research, RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan (e-mail: tsuzuki_gm@riken.jp).
References
- 1) Suzuki, T., Park, H., Hollingsworth, N.M., Sternglanz, R. and Lennarz, W.J. (2000) PNG1, a yeast gene encoding a highly conserved peptide:N-glycanase. J. Cell Biol. 149, 1039–1052.
- 2) Suzuki, T., Huang, C. and Fujihira, H. (2016) The cytoplasmic peptide:N-glycanase (NGLY1) — Structure, expression and cellular functions. Gene 577, 1–7.
- 3) Hirsch, C., Blom, D. and Ploegh, H.L. (2003) A role for N-glycanase in the cytosolic turnover of glycoproteins. EMBO J. 22, 1036–1046.
- 4) Huang, C., Harada, Y., Hosomi, A., Masahara-Negishi, Y., Seino, J., Fujihira, H. (2015) Endo-β-N-acetylglucosaminidase forms N-GlcNAc protein aggregates during ER-associated degradation in Ngly1-defective cells. Proc. Natl. Acad. Sci. U.S.A. 112, 1398–1403.
- 5) Suzuki, T. (2015) The cytoplasmic peptide:N-glycanase (Ngly1) — Basic science encounters a human genetic disorder. J. Biochem. 157, 23–24.
- 6) Enns, G.M., Shashi, V., Bainbridge, M., Gambello, M.J., Zahir, F.R., Bast, T. (2014) Mutations in NGLY1 cause an inherited disorder of the endoplasmic reticulum-associated degradation pathway. Genet. Med. 16, 751–758.
- 7) Heeley, J. and Shinawi, M. (2015) Multi-systemic involvement in NGLY1-related disorder caused by two novel mutations. Am. J. Med. Genet. A. 167A, 816–820.
- 8) Caglayan, A.O., Comu, S., Baranoski, J.F., Parman, Y., Kaymakcalan, H., Akgumus, G.T. (2015) NGLY1 mutation causes neuromotor impairment, intellectual disability, and neuropathy. Eur. J. Med. Genet. 58, 39–43.
- 9) Lam, C., Ferreira, C., Krasnewich, D., Toro, C., Latham, L., Zein, W.M. (2016) Prospective phenotyping of NGLY1-CDDG, the first congenital disorder of deglycosylation. Genet. Med. 19, 160–168.
- 10) Abuduxikuer, K., Zou, L., Wang, L., Chen, L. and Wang, J.S. (2020) Novel NGLY1 gene variants in Chinese children with global developmental delay, microcephaly, hypotonia, hypertransaminasemia, alacrimia, and feeding difficulty. J. Hum. Genet. 65, 387–396.
- 11) Ge, H., Wu, Q., Lu, H., Huang, Y., Zhou, T., Tan, D. (2020) Two novel compound heterozygous mutations in NGLY as a cause of congenital disorder of deglycosylation: A case presentation. BMC Med. Genet. 21, 135.
- 12) Need, A.C., Shashi, V., Hitomi, Y., Schoch, K., Shianna, K.V., McDonald, M.T. (2012) Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 49, 353–361.
- 13) He, P., Grotzke, J.E., Ng, B.G., Gunel, M., Jafar-Nejad, H., Cresswell, P. (2015) A congenital disorder of deglycosylation: Biochemical characterization of N-glycanase 1 deficiency in patient fibroblasts. Glycobiology 25, 836–844.
- 14) Cahan, E.M. and Frick, S.L. (2019) Orthopaedic phenotyping of NGLY1 deficiency using an international, family-led disease registry. Orphanet J. Rare Dis. 14, 148.
- 15) Bosch, D.G., Boonstra, F.N., de Leeuw, N., Pfundt, R., Nillesen, W.M., de Ligt, J. (2016) Novel genetic causes for cerebral visual impairment. Eur. J. Hum. Genet. 24, 660–665.
- 16) Funakoshi, Y., Negishi, Y., Gergen, J.P., Seino, J., Ishii, K., Lennarz, W.J. (2010) Evidence for an essential deglycosylation-independent activity of PNGase in Drosophila melanogaster. PLoS One 5, e10545.
- 17) Seiler, S. and Plamann, M. (2003) The genetic basis of cellular morphogenesis in the filamentous fungus Neurospora crassa. Mol. Biol. Cell 14, 4352–4364.
- 18) Habibi-Babadi, N., Su, A., de Carvalho, C.E. and Colavita, A. (2010) The N-glycanase png-1 acts to limit axon branching during organ formation in Caenorhabditis elegans. J. Neurosci. 30, 1766–1776.
- 19) Maerz, S., Funakoshi, Y., Negishi, Y., Suzuki, T. and Seiler, S. (2010) The Neurospora peptide:N-glycanase ortholog PNG1 is essential for cell polarity despite its lack of enzymatic activity. J. Biol. Chem. 285, 2326–2332.
- 20) Gosain, A., Lohia, R., Shrivastava, A. and Saran, S. (2012) Identification and characterization of peptide:N-glycanase from Dictyostelium discoideum. BMC Biochem. 13, 9.
- 21) Lehrbach, N.J. and Ruvkun, G. (2016) Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. eLife 5, e17721.
- 22) Galeone, A., Han, S.Y., Huang, C., Hosomi, A., Suzuki, T. and Jafar-Nejad, H. (2017) Tissue-specific regulation of BMP signaling by Drosophila N-glycanase 1. eLife 6, e27612.
- 23) Owings, K.G., Lowry, J.B., Bi, Y., Might, M. and Chow, C.Y. (2018) Transcriptome and functional analysis in a Drosophila model of NGLY1 deficiency provides insight into therapeutic approaches. Hum. Mol. Genet. 27, 1055–1066.
- 24) Rodriguez, T.P., Mast, J.D., Hartl, T., Lee, T., Sand, P. and Perlstein, E.O. (2018) Defects in the neuroendocrine axis contribute to global development delay in a Drosophila model of NGLY1 deficiency. G3 (Bethesda) 8, 2193–2204.
- 25) Yang, K., Huang, R., Fujihira, H., Suzuki, T. and Yan, N. (2018) N-glycanase NGLY1 regulates mitochondrial homeostasis and inflammation through NRF1. J. Exp. Med. 215, 2600–2616.
- 26) Lehrbach, N.J., Breen, P.C. and Ruvkun, G. (2019) Protein sequence editing of SKN-1A/Nrf1 by peptide:N-glycanase controls proteasome gene expression. Cell 177, 737–750.
- 27) Fujihira, H., Masahara-Negishi, Y., Akimoto, Y., Hirayama, H., Lee, H.C., Story, B.A. (2020) Liver-specific deletion of Ngly1 causes abnormal nuclear morphology and lipid metabolism under food stress. Biochim. Biophys. Acta 1866, 165588.
- 28) Fujihira, H., Masahara-Negishi, Y., Tamura, M., Huang, C., Harada, Y., Wakana, S. (2017) Lethality of mice bearing a knockout of the Ngly1-gene is partially rescued by the additional deletion of the Engase gene. PLoS Genet. 13, e1006696.
- 29) Lindsey, J.R. (2013) Historical foundations. In The Laboratory Rat: Biology and Diseases (1st ed.) (eds. Baker, H.J., Lindsey, J.R. and Weisbroth, S.H.). Elsevier, Amsterdam, pp. 1–36.
- 30) Asahina, M., Fujinawa, R., Nakamura, S., Yokoyama, K., Tozawa, R. and Suzuki, T. (2020) Ngly1−/− rats develop neurodegenerative phenotypes and pathological abnormalities in their peripheral and central nervous systems. Hum. Mol. Genet. 29, 1635–1647.
- 31) Kim, H., Kim, M., Im, S.K. and Fang, S. (2018) Mouse Cre-LoxP system: General principles to determine tissue-specific roles of target genes. Lab. Anim. Res. 34, 147–159.
- 32) Bolon, B., Garman, R.H., Pardo, I.D., Jensen, K., Sills, R.C., Roulois, A. (2013) STP position paper: Recommended practices for sampling and processing the nervous system (brain, spinal cord, nerve, and eye) during nonclinical general toxicity studies. Toxicol. Pathol. 41, 1028–1048.
- 33) Pekny, M. and Pekna, M. (2016) Reactive gliosis in the pathogenesis of CNS diseases. Biochim. Biophys. Acta 1862, 483–491.
- 34) Haijes, H.A., de Sain-van der Velden, M.G.M., Prinsen, H.C.M.T., Willems, A.P., van der Ham, M., Gerrits, J. (2019) Aspartylglycosamine is a biomarker for NGLY1-CDDG, a congenital disorder of deglycosylation. Mol. Genet. Metab. 127, 368–372.
- 35) Ransohoff, R.M. (2016) How neuroinflammation contributes to neurodegeneration. Science 353, 777–783.
- 36) Liu, J. and Wang, F. (2017) Role of neuroinflammation in amyotrophic lateral sclerosis: Cellular mechanisms and therapeutic implications. Front. Immunol. 8, 1005.
- 37) Ross, C.A. and Poirier, M.A. (2004) Protein aggregation and neurodegenerative disease. Nat. Med. 10, S10–S17.
- 38) Komatsu, M., Waguri, S., Koike, M., Sou, Y.S., Ueno, T., Hara, T. (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163.
- 39) Nedelsky, N.B., Todd, P.K. and Taylor, J.P. (2008) Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta 1782, 691–699.

