Abstract
In pancreatic islet cell culture models and animal models, we studied the molecular mechanisms involved in the development of insulin-dependent diabetes. The diabetogenic agents, alloxan and streptozotocin, caused DNA strand breaks, which in turn activated poly(ADP-ribose) polymerase/synthetase (PARP) to deplete NAD+, thereby inhibiting islet β-cell functions such as proinsulin synthesis and ultimately leading to β-cell necrosis. Radical scavengers protected against the formation of DNA strand breaks and inhibition of proinsulin synthesis. Inhibitors of PARP prevented the NAD+ depletion, inhibition of proinsulin synthesis and β-cell death. These findings led to the proposed unifying concept for β-cell damage and its prevention (the Okamoto model). The model met one proof with PARP knockout animals and was further extended by the discovery of cyclic ADP-ribose as the second messenger for Ca2+ mobilization in glucose-induced insulin secretion and by the identification of Reg (Regenerating gene) for β-cell regeneration. Physiological and pathological events found in pancreatic β-cells have been observed in other cells and tissues.
1. Introduction
The islets of Langerhans are located in the pancreas and are the only organs that produce insulin for the target tissues. Loss or dysfunction of the islets results in the development of diseases exemplified by diabetes mellitus. Recent advances in molecular biology1) along with improved procedures for isolating the islets of Langerhans from pancreases2) have made it possible to study the insulin synthesis and secretion at the molecular level.
The biosynthesis of insulin in the β-cells of pancreatic islets of Langerhans involves the transcription of proinsulin mRNA from the insulin gene, translation of the proinsulin mRNA by ribosomes associated with rough endoplasmic reticulum, conversion of proinsulin to insulin in the Golgi apparatus, and the formation of β-granules.3) Both proinsulin synthesis and insulin secretion in the islets of Langerhans are known to be markedly induced by glucose.3)
Insulin-dependent (Type 1) diabetes is caused by a destructive process affecting the insulin-producing β-cells of the islets of Langerhans. Destruction of β-cells may be induced by inflammatory tissue damage and β-cytotoxins such as alloxan and streptozotocin.
Since the early 1980s, we have proposed a hypothesis concerning β-cell damage and its prevention, according to which poly(ADP-ribose) polymerase/synthetase (PARP) activation induced by DNA strand breaks is critically involved in the consumption of NAD+, leading to energy depletion, decreased cell functions including proinsulin synthesis and cell death by necrosis.2),4)–7) This hypothesis was reconfirmed by results using PARP knockout (KO) mice8)–11) and has been recognized as providing the basis for necrotic cell death of various cells and tissues. Since then, the hypothesis has become known as the Okamoto model for β-cell damage and its prevention.12)–14) Based on this model, we have proposed two novel signal systems in β-cells: one is the CD38-cyclic ADP-ribose (cADPR) signal system for insulin secretion, and the other is the Regenerating gene protein (Reg)-Reg receptor system for β-cell regeneration. The physiological and pathological significance of the two signal systems in a variety of cells and tissues as well as in pancreatic β-cells has recently been recognized.
Here, we describe the Okamoto model and its expansions, the CD38-cADPR signal system and the Reg-Reg receptor system, focusing on recent advances and how their significance came to light. PARP activation reduces the cellular NAD+, leading to decrease the formation of cADPR (which is synthesized from NAD+ and is a second messenger for insulin secretion) as well as causing necrotic β-cell death, and PARP is involved in Reg gene transcription inducing β-cell regeneration. Therefore, it is reasonable to assume that PARP and its inhibitors have key roles in the prevention of β-cell death, the maintenance of insulin synthesis and secretion, and the induction of β-cell regeneration.
This article shows the achievements gained by a long exploration with the islets of Langerhans, during which we further clarified the physiology of insulin production and the pathogenesis of diabetes and its complications, as described below. In particular, interest in the physiological or pathological event observed in pancreatic β-cells of the islets of Langerhans has been hightened by its possible extension to other cells and tissues, as shown in Figs. 5, 6, 11, 14, 17, 18, 19, 20, 21, and 22 in this article.
2. Isolation of the islets of Langerhans from pancreas
The procedure for isolating the islets of Langerhans from rodent pancreas was established by Lacy and Kostianovsky.15) This group introduced collagenase digestion to dissect endocrine tissues in the pancreas from the surrounding exocrine tissues. However, the possibility that some functions of the islets are impaired during the isolation may not be totally ignored. Nevertheless, little attention has been fixed on this possibility. We at first examined the possible impairment of the islet functions by determining the insulin secreting ability, since if any damage occurred to islets during the isolation process, it would be reflected first by the insulin secreting ability rather than by the insulin synthesizing function. Okamoto et al.2),16) improved the method of isolating rat islets by adding bovine serum albumin to the medium for collagenase-treatment of rat pancreas and employing a Buchler’s vigorously rotatory incubator-shaker. As shown in Fig. 1A, compared to the islets obtained after 25 min treatment, those obtained after 10 min treatment exhibited a steeper response of insulin secretion to glucose stimulation. The islets obtained after a relatively brief treatment with collagenase also displayed a distinct susceptibility to somatostatin, which was no longer observed in islets obtained after relatively long treatment. Somatostatin (10 ng/mL) inhibited insulin secretion from the islets obtained after 10 min collagenase treatment by approximately 50%, whereas the insulin secretion from islets obtained after 25 min treatment was not affected at all by the somatostatin (Fig. 1B). The modified method consistently yielded hundreds of intact islets per one pancreas. The amount of insulin secreted reached approximately 600 µU/islet/90 min as the treatment period decreased. We were thus led to conclude that the value obtained by extrapolation of the treatment period to 0 min gives the amount of insulin secreted from an ‘intact’ islet. Therefore, that almost ‘intact’ pancreatic islets were obtained was confirmed by examining the amount of insulin secreted from an isolated islet, the response curve of insulin secreted corresponding to change in glucose concentration, and the sensitivity to somatostatin.

It should be noted here that somatostatin inhibited the glucose-induced insulin secretion from the islets by at most 50% and the inhibitory effect of somatostatin was not significant when the Ca2+ concentration in the incubation medium was raised from 5 to 10 mEq/L.2),16) These results also suggested the importance of Ca2+ mobilization from the intracellular Ca2+ pool in the insulin secretion as well as from the extracellular Ca2+ influx, as described in Sections 5 and 7.
3. Translational control of proinsulin synthesis by glucose in the islets of Langerhans
Proinsulin synthesis in the islets of Langerhans of rats as well as in many other animals is known to be induced by glucose,3) in marked contrast to non-insulin proteins, the synthesis of which is only slightly affected by glucose.2) Thus, there must be a highly selective mechanism in the islets for proinsulin synthesis, either at the level of insulin gene transcription or at the level of proinsulin mRNA translation. If proinsulin induction by glucose is regulated at the transcriptional level, the amount of proinsulin mRNA may be increased by the administration of glucose. Alternatively, if proinsulin induction is regulated at the translational level, the rate of proinsulin synthesis may not be correlated with the amount of proinsulin mRNA. Itoh and Okamoto17) assayed the de novo synthesis of proinsulin with concomitant quantification of proinsulin mRNA in the islets of Langerhans, using nucleic acid hybridization with cDNA synthesized from purified rat proinsulin mRNA.2),17),18) The approach revealed that, while the islet synthesis of proinsulin is significantly stimulated by exposure to high glucose, the amount of proinsulin mRNA in the glucose-stimulated or unstimulated islets remained unchanged during the period of proinsulin induction up to 60 min (Fig. 2). In a wheat-germ cell-free system, the amount of proinsulin mRNA in islets as determined by the in vitro translation assay remained unchanged during the period of proinsulin induction by glucose.19),20)

We examined the transcriptional activity of the insulin gene in rat islets using recombinant DNA containing a rat proinsulin cDNA sequence as a probe. Although proinsulin synthesis was stimulated 8.4-fold by glucose, proinsulin mRNA synthesis was stimulated only 1.8-fold.21),22) Cycloheximide, an inhibitor of eukaryotic translation elongation, completely inhibited the glucose-induced rise in proinsulin synthesis. α-Amanitin, an inhibitor of RNA polymerase II (weakly RNA polymerase III), had essentially no effect on the proinsulin mRNA level in pancreatic islets of rats.21),22) These results clearly indicate that proinsulin synthesis is regulated at the translational level (translational control).2),17)
As insulin regulates the glucose level in the blood, the rates of both insulin synthesis and insulin secretion need to change rapidly according to the change in the blood glucose concentrations. This regulation of proinsulin synthesis should be the means by which the rapid regulation of insulin synthesis by glucose is ensured.23) In fact, Itoh23) estimated the number of proinsulin mRNA molecules per rat pancreatic islet β-cell to be approximately 2.7 × 105. The number of proinsulin mRNA molecules per rat pancreatic β-cell was comparable to that of globin mRNA per rabbit reticulocyte24) (1.4 × 105 per cell) and ovalbumin mRNA per chicken oviduct cell25) (1.5 × 105 per cell), which had been thought to represent the most abundant mRNA species in eukaryotic cells. The control of proinsulin synthesis was assumed to be achieved by the selective enhancement of the translation of proinsulin mRNA at the elongation step.17),22) The 5′-untranslated region of proinsulin mRNA was reported to be necessary for the glucose-induced translational control in mouse and human β-cells.26),27)
There are some well-documented examples of the translational control of specific gene products. In such examples as ornithine aminotransferase,28) ornithine decarboxylase29) and ferritin,30) translational control is regulated at the translation initiation step. The translational control of proinsulin synthesis, which seems to be regulated mainly at the elongation step, is a unique regulation of gene expression in eukaryotic cells.
4. The Okamoto model for β-cell damage and its prevention
Diabetes can be usefully studied at an experimental level by methods that make use of either surgical techniques or chemical agents.
Many of the acute metabolic derangements of severe human insulinopenic diabetes can be reproduced by removing the insulin-producing pancreatic β-cells of the islets of Langerhans. This artificial form of diabetes was first produced by von Mering and Minkowski31) when they removed the pancreases of dogs. The same model of diabetes was adopted by Banting and Best32) for their historic observation on the hypoglycemic properties of a crude pancreatic extract of insulin.
For chemical agents, alloxan33) (2,4,5,6-tetraoxohexahydropyrimidine) and streptozotocin34) (2-deoxy-2-(3-methyl-3-nitrosoureido)-D-glucopyranose) are particularly instructive; they exert selective cytotoxic effects on pancreatic β-cells in animals and are extremely potent diabetogenic substances. Since the original discoveries were made by Dunn et al.33) and Rakieten et al.,34) alloxan and streptozotocin have been widely used to induce diabetes in experimental animals because of their advantages in terms of specificity, convenience and reproducibility. How the β-cytotoxins cause functional deteriorations and degenerative changes in the β-cells, however, had been the subject of much debate, until we proposed a unifying concept for the diabetogenicity of alloxan and streptozotocin.2),4)–7),35)–37)
4.1. Alloxan and streptozotocin induce DNA strand breaks and poly(ADP-ribose) polymerase/synthetase (PARP) that depletes NAD+ in pancreatic islets.
In 1981, Yamamoto et al.4) demonstrated that streptozotocin and alloxan cause DNA strand breaks in isolated rat islets. As shown in Fig. 3, DNA from intact islets incubated in the absence of the diabetogenic agents was recovered as a single peak near the bottom of the alkaline sucrose gradient, the position at which undamaged DNA is deposited as a sediment. However, after only 5 min incubation with streptozotocin or alloxan, a considerable amount of DNA is deposited as a broad peak in the middle of the gradient with a concomitant decrease in undamaged DNA; after 10–20 min incubation, the islet DNA was almost completely fragmented. The results indicated that streptozotocin and alloxan produce strand breaks in islet DNA.

Next, Yamamoto et al.4) prepared a nuclear fraction from islets incubated in conditions that cause the DNA breaks, and assayed the activity of PARP, a nuclear enzyme that catalyzes the polymerization of the ADP-ribosyl moiety of NAD+ to form poly(ADP-ribose); the enzyme was described by three independent groups in France and Japan, namely, Chambon et al.,38) Sugimura et al.39) and Nishizuka et al.,40) and was implicated in DNA repair.41) Both streptozotocin and alloxan were found to induce islet PARP activity with a peak at 10 min (Fig. 4 upper panel). The increase in nuclear PARP activity was associated with a concomitant decrease in cellular NAD+, the substrate of PARP; the NAD+ content of the islets showed a sharp drop within 20 min of incubation with either streptozotocin or alloxan (Fig. 4 lower panel).

The diabetogenic doses of alloxan and streptozotocin did induce in vivo DNA strand breaks and NAD+ depletion in the islets of Langerhans.5) From these results, it is reasonable to assume that the β-cytotoxins cause DNA breaks to induce PARP, thereby depleting islet NAD+.
Human genome sequence projects revealed the occurrence of similar DNA sequences to PARP. At least seventeen PARP-related genes (PARP-1, -2, -3, -4, -5a, -5b, -5c, -6, -7, -8, -9, -10, -11, -12, -14, -15, and -16) were revealed to constitute a multigene family, the PARP gene family.42)–44) PARP-1 activation by extensive DNA damage is the major pathway in necrotic cell death, and therefore “PARP” will be used from here on to indicate “PARP-1”.
4.2. Protection by radical scavengers and PARP inhibitors against alloxan- and streptozotocin-induced islet DNA strand breaks, the NAD+ depletion and the inhibition of proinsulin synthesis.
Yamamoto et al.4) incubated isolated rat pancreatic islets in the presence of alloxan or streptozotocin with or without the addition of PARP inhibitors such as nicotinamide and picolinamide. The inhibitors almost completely abolished the alloxan- and streptozotocin-induced decreases in the islet proinsulin synthesis as well as in the islet NAD+ level.4) The various PARP inhibitors (benzamide, 3-aminobenzamide, 3-nitrobenzamide, 3-methoxybenzamide, picolinamide, nicotinamide, theophylline, and IBMX) were found to reverse the alloxan- or streptozotocin-induced inhibition of proinsulin synthesis in a dose-dependent manner, and the stronger inhibitors exerted protective effects at lower concentrations.1),6),36) In vivo administrations of nicotinamide or 3-aminobenzamide to rats effectively protected against the alloxan- or streptozotocin-induced decrease in the islet ability to synthesize proinsulin.7)
It has been suggested that alloxan may work through the formation of the hydroxyl radical (OH•),45) which is produced by the interaction between superoxide (O2−•) and peroxide (H2O2)46):
| \begin{equation*}
\text{O$_{2}{}^{-\bullet}$} + \text{H$_{2}$O$_{2}$} \rightarrow \text{OH$^{\bullet}$} + \text{OH$^{-}$} + \text{O$_{2}$}
\end{equation*}
|
Superoxide dismutase and catalase catalyze the removal of O
2−• and H
2O
2, respectively,
47) and hence may inhibit the formation of OH
•. Uchigata
et al.6) showed that combined administration of superoxide dismutase and catalase more effectively protects against alloxan-induced islet DNA strand breaks, as well as against proinsulin synthesis inhibition, than administration of either of these scavenging enzymes alone. The results indicate that it is OH
• that is ultimately generated from alloxan to attack DNA. On the other hand, streptozotocin-induced islet DNA strand breaks were not affected by the radical scavengers.
7) The breakage of DNA by streptozotocin is probably associated with its alkylating activity, as suggested with nitrosoureas.
48)
4.3. The proposal of the Okamoto model for β-cell damage and its prevention.
From the experimental results described above, Okamoto et al.2),6),14),36),37) proposed a basic model for the action of alloxan and streptozotocin in the induction of experimental diabetes. As shown in Fig. 5, the first step is the generation of free radicals by alloxan and streptozotocin, which attack DNA to produce strand breaks. PARP then acts to repair the DNA breaks,2),36),37) consuming β-cell NAD+. Since NAD+ is the most abundant of cellular coenzymes and participates in many biological reactions in the cell, a severe reduction in intracellular NAD+ to non-physiological levels can adversely affect β-cell functions, including ATP production, proinsulin synthesis and insulin secretion, and thus the β-cell ultimately dies. Therefore, in diabetes induction, the β-cells seem to be making “a suicide response” in their attempt to repair the damaged DNA.2),36),37)

Here the problem arises as to why only pancreatic β-cells are specifically damaged by alloxan and streptozotocin. It has been conjectured that alloxan and streptozotocin have an affinity to the cell membranes of β-cells because of their chemical structures.49) 14C-Labeled alloxan or streptozotocin injected into mice was recovered in pancreatic islets.50),51) The NAD+ content per DNA of normal pancreatic islets was approximately one half of that of the liver5) and, therefore, pancreatic β-cells may be more susceptible to a reduction in NAD+ levels. Malaisse et al.52) reported that the ability to provide protection against potent reactive radicals may be weak in islet cells given the low glutathione peroxidase activity in islets.
This model for the mechanism of action of alloxan and streptozotocin has received much attention because of its possible relevance to the effects of viruses and inflammation, especially those related to inflammatory damage of β-cells,2),14),36),37) because biological events like inflammation and virus infection, physical insults like radiation, and chemical insults may independently or interactively produce β-cell DNA strand breaks. Repeated administration of subdiabetogenic doses of streptozotocin to rats induced insulitis and a diabetic state similar to Type 1 diabetes.53) Tsubouchi et al.54) have reported that a large dose of X-ray irradiation provoked the necrosis of pancreatic islets in hamsters, and that the hamsters exhibited a diabetic state. Cumulative β-cell damage induced by subdiabetogenic doses of streptozotocin and by encephalomyocarditis or Coxsackie virus infection was shown to result in the development of diabetes.55)
Therefore, although insulin-dependent Type 1 diabetes can be caused by a variety of internal and external environmental factors, including immunologic abnormalities, inflammatory tissue damage, viruses, irradiation, and chemical insults, the final pathway may be one and the same (Fig. 5), involving DNA damage, PARP activation, and NAD+ depletion. Accordingly, it may be possible to prevent insulin-dependent Type 1 diabetes by blocking immune reactions, scavenging free radicals, and inhibiting PARP. Actually, diabetes in non-obese diabetic (NOD) mice,56) a murine model that spontaneously develops clinical and pathological manifestations similar to those seen in human Type 1 diabetes, is reported to be preventable by treatment with immunosuppressive agents,57) an immunomodulator OK-432,58),59) radical scavengers,12) and PARP inhibitors.12),60) Concerning nitric oxide (NO) (see Fig. 5), Takamura et al.61) found that in transgenic mice expressing type 2 nitric oxide synthase (NOS2) constitutively in pancreatic β-cells, the β-cell mass was markedly reduced resulting in the development of severe diabetes. NOS2 is usually induced by proinflammatory/inflammatory cytokines such as interleukin-1β, interferon-γ, and tumor necrosis factor-α.
4.4. Proof and extension of the Okamoto model.
In 1999, three independent groups in Germany, Japan and the U.S. provided irrefutable support using PARP deficient mice for the model shown in Fig. 5. The mice were extremely resistant to streptozotocin and the β-cell death was prevented.8)–11) PARP deficient mice were resistant against not only single-high-dose streptozotocin-induced diabetes, as described above, but also multiple-low-dose streptozotocin-induced diabetes.62)
PARP is one of the best known proteins with DNA-damage scanning activity, and poly(ADP-ribosyl)ation by PARP has been proposed to function in DNA repair by modifying architectural proteins proximal to DNA breaks, thus facilitating the opening of the condensed chromatin structure required for the recruitment of the repairing complex.41) Paradoxically, in spite of this beneficial effect, PARP can induce necrotic cell death (“necrosis”) through NAD+ depletion as described above. In spite of the apparent Mr. Hyde-like side of this enzyme, it may represent an evolutionary strategy adopted by multicellular organisms to prevent the survival of cells that would otherwise transmit potentially dangerous genetic material. Many other tissues and cells such as those involved in cerebral ischemia,63) myocardial ischemia,64) abdominal aortic aneurysm,65) laryngeal injury,66) intestinal mucosal injury,67) organ injury by hemorrhagic shock,68) ischemic renal injury,69) acute pancreatitis,70) acetoaminophen-induced liver cell death,71) benzo[a]pyrene-induced liver cell death,72) diabetic kidney disease,73) and diabetic myocardial and endothelial injury74) have been reported to die by the same mechanism as that in pancreatic β-cell death (see Fig. 5). Accordingly, the inhibition of PARP activity may be a possible therapeutic approach in a wide number of disorders other than diabetes.
5. The CD38-cyclic ADP-ribose (cADPR) signal system for glucose-induced insulin secretion in pancreatic β-cells of the islets of Langerhans
Glucose increases the intracellular Ca2+ concentration in pancreatic β-cells to cause the secretion of insulin. Ashcroft et al.75) proposed in 1984 that this increase in the Ca2+ concentration is provided extracellularly. That is, in the process of glucose metabolism, the millimolar concentrations of ATP produced inhibit the potassium (KATP) channel, causing membrane depolarization and the opening of the voltage-dependent Ca2+ channels. In 1995, KATP channels were reconstituted in vitro, and the molecular structure of the channels was determined.76) KATP channels are hetero-octameric proteins composed of pore-forming Kir6.x subunits and regulatory sulfonylurea receptor (SUR) subunits.76),77)
In a series of studies beginning in 199278)–103) (see also Ref. 104), we proposed another model of insulin secretion by glucose, as shown in Fig. 6 in red; ATP, produced in the process of glucose metabolism, inhibits the cADPR hydrolase of CD38, causing the accumulation of cADPR, which acts as a second messenger for Ca2+ mobilization from an intracellular Ca2+ pool, the endoplasmic reticulum, for insulin secretion.
5.1. Cyclic ADP-ribose, a second messenger for intracellular Ca2+ mobilization in glucose-induced insulin secretion.
Cyclic ADP-ribose (cADPR) is a cyclic compound synthesized from NAD+. This compound was first found in 1987 by Lee and associates105) when studying Ca2+ release in sea urchin eggs. However, the physiological significance of cADPR in mammalian systems was not then understood. According to the Okamoto model, the decrease in the NAD+ level should cause a decrease in cADPR in β-cells. Thus, in 1993, we79),80) proposed that insulin secretion by glucose occurred via cADPR-mediated Ca2+ mobilization from an intracellular Ca2+ pool, the endoplasmic reticulum (Fig. 6). Using rat islet microsomes as a cell-free system to study Ca2+ release, cADPR was found to induce Ca2+ release from islet microsomes as indicated by the observed prompt increase in fluo 3 (1-[2-Amino-5-(2,7-dichloro-6-hydroxy-3-oxo-9-xanthenyl)phenoxy]-2-(2-amino-5-methylphenoxy)ethane-N,N,N′,N′-tetraacetic acid) fluorescence (Fig. 7). Repeated additions of cADPR resulted in attenuation of the Ca2+-release response to cADPR. IP3 was not found to release Ca2+ from islet microsomes, and at this point islet microsomes were still responsive to cADPR. In cerebellum microsomes, IP3 induced Ca2+ release from the microsomes, and repeated additions of IP3 resulted in attenuation of the Ca2+-release response. cADPR also induced Ca2+ release from cerebellum microsomes, and repeated additions of cADPR resulted in attenuation of the Ca2+-release response to cADPR. At this point, cerebellum microsomes were still responsive to the addition of IP3. Moreover, heparin, an inhibitor of IP3 binding to its receptor, blocked IP3-induced Ca2+ release. The cADPR-induced Ca2+ release from islet microsomes showed a steep dose-response relationship with a clear effect at concentrations as low as 0.1 µM and a near-maximum release at 0.5 µM.79) However, IP3 was not found to cause Ca2+ release from islet microsomes at these concentrations. These results indicated that islet microsomes did respond to cADPR but not to IP3. In contrast with islet microsomes, cerebellum microsomes responded to both cADPR and IP3, but cADPR induced Ca2+ release from cerebellum microsomes via a different mechanism than that utilized by IP3. These results suggested that responses to cADPR and IP3 varied according to the tissue or cell type (see also Section 5.5 and Fig. 8).


In Section 4, we explain that diabetogenic agents such as streptozotocin deplete intracellular NAD+ in islets. PARP inhibitors such as nicotinamide and 3-aminobenzamide prevent the NAD+ depletion through PARP.2),4),6),36),37) We therefore incubated islets with glucose and streptozotocin, prepared the islet extract and measured its Ca2+ mobilizing activity.79) A prominent Ca2+ release was found with the extract from islets treated with 20 mM glucose, but not with extracts from islets treated with 2.8 mM glucose. Streptozotocin caused a great reduction in the Ca2+ mobilizing activity of the extract from islet treated with 20 mM glucose, and PARP inhibitors such as nicotinamide and 3-aminobenzamide reversed the reduction. The results also suggested that the active component for Ca2+ release in the glucose-stimulated islet extract was cADPR. In fact, we measured the cADPR content in normal rat and mouse islets by radioimmunoassay with an anti-cADPR antibody and found an increase in the cADPR content by glucose in the islets.89) The increase in the cADPR concentration in response to high glucose was confirmed using BALB/c mouse islets.106)
Moreover, cADPR stimulated insulin secretion from digitonin-permeabilized islets.79) Near maximal secretion of insulin by cADPR was observed at 0.5 µM and half-maximal secretion at 0.1 µM. The dose-response curve was well fitted to that of Ca2+ release from islet microsomes. In contrast with cADPR, IP3 did not induce insulin secretion.
5.2. CD38 as a major enzyme for the cADPR synthesis.
The next issue concerns the mechanism by which the glucose stimulus induces the formation of cADPR. We80),81) found that CD38, a 300-amino acid protein in human first recognized as a leukocyte antigen, was expressed in a variety of tissues including pancreatic β-cells. In 1993, CD38 was found to have both ADP-ribosyl cyclase, synthesizing cADPR from NAD+, and cADPR hydrolase to decompose cADPR to yield ADP-ribose.80),107)–111) We purified human CD38 protein and found that millimolar concentrations of ATP inhibited the cADPR hydrolase of CD38, competing with the substrate, cADPR.80) Based on the competitive inhibition of the cADPR hydrolysis by ATP, cADPR and ATP appear to bind to the same site of CD38. By labeling the recombinant CD38 with an ATP analogue, 5′-p-fluorosulfonylbenzoyladenosine (FSBA), Tohgo et al.86) identified the binding site for ATP and/or cADPR as the lysine-129 of CD38. Neither the Ala- nor Arg-129 mutant was labeled by FSBA nor catalyzed the hydrolysis of cADPR to ADP-ribose. Furthermore, the mutants did not bind cADPR, whereas they still used NAD+ as a substrate to form cADPR.86) These results indicate that Lys-129 of CD38 participates in cADPR binding and ATP, produced in the process of glucose metabolism, competes with cADPR for the binding site, resulting in the inhibition of the cADPR hydrolase activity of CD38 and then in the accumulation of cADPR in β-cells (Fig. 9). Tohgo et al. also showed that cysteine-119 and -201 of CD38 were essential for the hydrolysis of cADPR to ADP-ribose.82)

cADPR is believed to activate a ryanodine receptor (RyR) to release Ca2+ from the intracellular stores, the endoplasmic reticulum.104) We confirmed that type 2 RyR was expressed in rat pancreatic islets.98),101) Experiments with rat islets revealed that cADPR did not bind directly to the RyR but acted on the receptor through a mediator such as FK506 (Tacrolimus)-binding protein 12.6 (FKBP12.6) to release Ca2+. The cellular target for FK506, one of the most widely used immunosuppressive agents, is thought to be FKBP12 and FKBP12.6. Rat FKBP12 is composed of 108 amino acids and is highly conserved among human, mouse, bovine, and rabbit FKBP12. Rat FKBP12.6 is also a 108-amino acid protein as are human and bovine FKBP12.6. Noguchi et al.87) isolated microsomes from rat islets, carried out immunoblot analyses, and found that rat islet microsomes contained FKBP12.6, but did not contain FKBP12. Interestingly, cADPR was found to bind to FKBP12.6 at a Kd value of 35 nM. The cADPR-binding was inhibited by FK506, and neither structurally nor functionally related analogues of cADPR inhibited the cADPR binding to FKBP12.6. These results not only indicate that FKBP12.6 acts as a cADPR-binding protein, but also strongly suggest that cADPR is the physiological ligand of FKBP12.6 since FK506 does not normally exist in mammalian cells. As mentioned above, FKBP12.6 occurs in rat islet microsomes. When rat islet microsomes were treated with cADPR, FKBP12.6 was dissociated from the microsomes and moved to the supernatant, releasing Ca2+ from the intracellular stores. The microsomes that had been treated with FK506 or cADPR and were then devoid of FKBP12.6, did not show Ca2+ release by cADPR. These results and those from other experiments suggest that, when cADPR binds to FKBP12.6 in the RyR and causes the dissociation of FKBP12.6 from the RyR to form the FKBP12.6-cADPR complex, the RyR channel activity is thereby increased to release Ca2+ from the endoplasmic reticulum. When FK506 is present, cADPR cannot act on the RyR to release Ca2+, and the glucose-induced insulin secreting machinery ceases to function. Using FKBP12.6-deficient mice, Noguchi et al.87),97) confirmed that FKBP12.6 plays a role in glucose-induced insulin secretion downstream of ATP production, independently of ATP-sensitive K+ channels, in pancreatic β-cells. In human, when FK506 was used as an immuno-suppressant in kidney transplantation, hyperglycemia was observed in 20–35% of the recipients.112) The diabetogenic side effect of FK506 may be explained by the mechanism shown in Fig. 6. It should be also noted that, in the presence of calmodulin (CaM), islet microsomes become sensitized to cADPR at much lower concentrations for Ca2+ release, and the Ca2+ release is greatly increased.83) Inhibitors for CaM-dependent protein kinase II (CaM kinase II) completely abolished the glucose-induced insulin secretion as well as the cADPR-mediated and CaM-activated Ca2+ mobilization. Western blot analysis revealed that rat microsomes contained CaM kinase IIα but did not contain CaM. When the active 30 kDa chymotryptic fragment of CaM kinase II was added to the microsomes, fully activated cADPR-mediated Ca2+ release was observed in the absence of CaM.83) These results suggest that CaM kinase II is required to phosphorylate and activate the RyR for the cADPR-mediated Ca2+ release. As also shown in Fig. 6, cyclic AMP-dependent protein kinase (A-kinase) activated the cADPR-mediated Ca2+ release from islet microsomes.94) In the absence of A-kinase, only a small amount of Ca2+ was released from the microsomes by low concentrations of cADPR. On the other hand, when the catalytic subunit of A-kinase was added to the islet microsomes, the Ca2+ release was sensitized at lower concentrations of cADPR. As incretin peptide hormones such as glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP, also known as glucose-dependent insulinotropic polypeptide) increase the intracellular concentrations of cyclic AMP to activate A-kinase and exchange protein activated by cAMP2 (EPAC2)/cAMP-guanine nucleotide exchange factor II,113) it is quite possible the cADPR-mediated Ca2+ mobilization for insulin secretion is activated by CaM kinase II and A-kinase/EPAC2. Possibly, the activated kinases phosphorylate the RyR to sensitize the Ca2+ channel for the cADPR signal. Kim et al. also showed the involvement of cADPR in the enhancement of insulin secretion by GLP-1.114)
5.4. Deficiency of CD38 resulted in decreased intracellular cADPR, Ca2+ and insulin secretion.
To verify the role of the CD38-cADPR signal system in the regulation of insulin secretion, Kato et al.84),91) created CD38 knockout (KO) and transgenic mice.
The glucose-induced increase in the cADPR content was greatly reduced, the Ca2+ concentration was impaired more than 50% in the KO mouse islets (Fig. 10), and the glucose-induced insulin secretion was decreased more than 50%.91) CD38 KO mice showed impaired glucose tolerance, and the serum insulin level was lower than in the control. Further, whether the observed phenotype could be rescued by the pancreatic β-cell specific expression of CD38 cDNA was tested by crossbreeding transgenic mice carrying a human CD38 cDNA under the rat insulin promoter and the CD38 KO mice. By intercrossing their offspring, CD38 KO mice carrying the human CD38 transgene were generated. In the results, the human CD38 transgene ameliorated the glucose intolerance and the decreased insulin secretion. CD38 KO mice did not show insulin resistance, suggesting that the observed phenotype is indeed caused by the absence of CD38 in pancreatic β-cells.91) The KO islets, however, responded normally to the extracellular Ca2+ influx stimulants tolbutamide and KCl to secrete insulin.91) Thus, as shown in Fig. 6, the CD38-cADPR signal system as well as the ATP-sensitive K+ channel system appeared to function almost equally in insulin secretion by glucose stimulation.

This paradigm of insulin secretion based on the CD38-cADPR signal system has been supported by a wide body of evidence obtained in rats and mice. Recent results indicate that the CD38-cADPR signal system also functions in insulin secretion in human. As a result of a missense mutation (Arg140Trp) found in the CD38 gene in Japanese diabetic patients (4 in 31 Type 2 diabetic patients but none in 90 control subjects), the CD38 protein showed altered catalytic activities and a decreased production of cADPR.115) The decreased function of the CD38 mutant may have contributed to the impairment of glucose-stimulated insulin secretion in Type 2 diabetes patients. It is also significant that circulating anti-CD38 autoantibodies were detected in 10–14% of Japanese116) as well as Caucasian diabetic patients.117)–120) The autoantibody altered the in vitro enzymic activity of islet CD38, glucose-induced increase of cADPR levels, and insulin secretion.116),117) These findings provide further support for the concept that the CD38-cADPR signal system functions in insulin secretion in human.
5.5. cADPR versus IP3 in intracellular Ca2+ release for insulin secretion (Fig. 11).
Since 1984, Berridge and Irvine121) had proposed that IP3 induces Ca2+ release from the intracellular pool, the endoplasmic reticulum. They studied the Ca2+ release from the intracellular Ca2+ store in pancreatic acinar cells by IP3.122)
Concerning the Ca2+ release from the intracellular pool in pancreatic β-cells of the islets of Langerhans, the CD38-cADPR signal system proposed by the Okamoto group was the focus of intense debate.88),104),107),123)–126) Discrepant results were reported in diabetic β-cells such as ob/ob mouse islets and RINm5F cells, which have been often used for studying Ca2+ release. However, the Ca2+ release responses of these diabetic β-cell microsomes differed greatly from those of normal islet microsomes.89) Microsomes from normal C57BL mouse islets released Ca2+ in response to cADPR, but scarcely in response to IP3. This response to cADPR was completely attenuated by the prior addition of 8-amino-cADPR. In ob/ob mouse islet microsomes, however, only a small amount of Ca2+ was released by cADPR, but much Ca2+ was released by IP3 (Fig. 8). All the RyR2 mRNAs in C57/BL islets were the islet-type98),101) (see also Section 5.6). RINm5F cell microsomes also responded well to IP3 to release Ca2+ but did not respond to cADPR (Fig. 8). However, RINm5F cells are rat insulinoma-derived immortal cells that show almost no ability for glucose-induced insulin secretion. Concerning intracellular Ca2+ release channels, the mRNA expression of type 2 RyR, which is postulated to be a Ca2+ release channel for cADPR, was clearly detected in normal islets but not in ob/ob islets (Fig. 8A). In contrast, IP3 receptor (IP3R-1, IP3R-2, IP3R-4 and IP3R-5) mRNAs were not detected in normal islets but were clearly detected in ob/ob islets (Fig. 8B) and, although IP3R-3 mRNA was slightly detected in normal islets, the mRNA was significantly increased in ob/ob islets, consistent with the observation that IP3-induced Ca2+ mobilization preferentially worked in ob/ob islet microsomes. More importantly, the CD38 mRNA level was significantly decreased in ob/ob islets,80) and CD38 mRNA was not expressed in RINm5F cells.89) The decrease of CD38 mRNA in ob/ob islets may explain the low response in the cADPR content of β-cells by glucose stimulation. Decreased CD38 mRNA was also reported in islets of Goto-Kakizaki (GK) diabetic rats,127) which showed impaired glucose-induced insulin secretion. These results show that the CD38-cADPR signal system for insulin secretion acts under normal physiological conditions but the IP3 system becomes dominant in diabetic β-cells such as ob/ob mouse islets and RINm5F cells (Fig. 11). In fact, it was confirmed by Rutter and co-workers and Ohta et al. using aequorin chimera that MIN6 cells, which retained the ability of glucose-induced insulin section and showed dramatic Ca2+ mobilization in response to cADPR128)–130) via the RyR despite the fact that no response to IP3 was observed. Kim and co-workers demonstrated that BALB/c mouse islets exhibited distinct increases in intracellular cADPR, Ca2+, and insulin secretion by glucose stimulation.106)

The CD38-cADPR signal system can be divided into two parts, the cADPR synthesis and metabolism and the Ca2+ release by cADPR from RyR (Fig. 6). There are three types of RyR in cells, RyR1 (type 1, skeletal type), RyR2 (type 2, cardiac type), and RyR3 (type 3, brain type). The three isoforms of RyR (RyR1, RyR2, and RyR3) are encoded by different genes and located in different chromosomal regions. RyR1 and RyR2 are predominantly expressed in skeletal muscles and cardiac muscles, respectively. On the other hand, RyR3 is ubiquitously expressed including in brain. Despite extensive physiological research, the type of RyR that is the target for cADPR has remained elusive. In pancreatic islets, RyR2 appeared to be expressed.89),101),104) Takasawa, Kuroki et al.98) isolated a novel RyR (islet-type RyR) from rat islet cDNA library, which was generated from type 2 RyR gene by alternative splicing of exons 4 and 75. The deduced protein of the cDNA was 4,947 amino acids with a calculated molecular weight of 562,291 daltons. These two regions, corresponding to the 7 amino acids in exon 4, and the 12 amino acids in exon 75 of cardiac RyR2, were not found in the rat islet RyR mRNA. Interestingly, neither exon 4 nor exon 75 was found in human and mouse islet RyR2 mRNA. Further analysis of the alternative splicing patterns of RyR2 mRNA in several rat tissues revealed that the islet-type RyR was expressed not only in islets but also in cerebrum, cerebellum, pituitary gland, and adrenal gland (Fig. 12). The cardiac-type RyR was expressed in atrium, ventriculum, and kidney. In many other tissues, both types are expressed. In the human RYR2 gene, the tissue-specific alternative splicing pattern was essentially similar to that of the rat gene, and the islet-type RYR mRNA was also expressed in the brain as well as in islets.98),101) Interestingly, the splice donor consensus “gt” was substituted by “gg” in the 0.8 kbp intron, whereas all the other splice donor and acceptor sequences conformed to the consensus “gt/ag rule” for splicing.98),101)

By RT-PCR analyses of human and mouse RNAs, the expression of (alternatively spliced) islet-type RyR2 mRNA was also found in human and mouse islet RNAs. Furthermore, human and mouse islet-type RyR mRNA expressed in islets were also generated from the RyR2 gene by alternative splicing. The splice donor consensus “gt” was also substituted by “gg”. As shown in Fig. 13, intron 75 of RyR2 gene was spliced using “gg” as a donor for splicing instead of canonical “gt” in generating rat, mouse, and human RyR2 mRNAs. RyR2 gene for the CD38-cADPR Ca2+ signal system produced two different messenger RNAs by alternative splicing; one is for pancreatic islet β-cells and neuro-endocrine cells using “gt/ag” splicing, and the other is for heart and blood vessels using not only the canonical “gt/ag” intron splicing site but also using the novel “gg/ag” site. Therefore, the heart/blood vessel type RyR2 can contain exon 4 and exon 75 (Fig. 14).

The islet-type RyR caused a greater increase in the Ca2+ release by caffeine when expressed in HEK293 cells pretreated with cADPR, suggesting that the novel islet-type RyR was an intracellular target for the CD38-cADPR signal system in mammalian cells, playing many important physiological roles in the functioning of the cADPR-sensitive Ca2+ release.101) Most recently, the islets-type RyR2 was reported to be essential for insulin biosynthesis in human 1.1B4 insulin-producing cells via Ca2+ homeostasis.131) The use of gg as a splice donor has not been reported in disease-free conditions.132),133) Utilization of the gg splice donor was only reported in cholesteryl ester transfer protein gene in a patient with cholesteryl ester protein deficiency.134)
5.7. The CD38-cADPR signal system in other tissues and cells.
Although IP3 has been thought to be a second messenger for Ca2+ mobilization from intracellular stores, cADPR induces Ca2+ release from pancreatic islet microsomes but IP3 does not, as described above. In microsomes of the cerebellum, Ca2+ release is induced by both cADPR and IP3. It is, therefore, apparent that cells can utilize two second messengers, IP3 and cADPR, for Ca2+ mobilization, depending on the types of cells as well as differences in cellular conditions, physiological or pathological, performing a variety of cellular functions (Fig. 11). Recently, various physiological phenomena in animal, plant, and bacterial cells have been found to utilize this novel signal system.93),94),99),101),135)–150) In pancreatic acinar cells of CD38 KO mice, the acetylcholine-induced Ca2+ oscillation was greatly reduced or completely disappeared under a physiological concentration of acetylcholine.144) Furthermore, acetylcholine induced cADPR formation in normal acinar cells but not in CD38 KO acinar cells. The IP3 formation was very small in the presence of a physiological concentration of acetylcholine and showed no difference between normal and CD38 KO cells. It appears likely that acetylcholine induces the cADPR formation via the G-protein coupled CD38 system.145) In pancreatic β-cells, glucose is metabolized and induces the CD38-cADPR signal system to secrete insulin. In other cells, hormones and neurotransmitters may regulate the CD38-cADPR signal system in a receptor-coupled manner, such as in a G-protein-coupled manner, for various other types of physiological responses.94),96)
The possible involvement of the CD38-cADPR signal system in cardiovascular abnormalities has been reported. Myocardial hypertrophy was observed in male mice with null mutations of CD38, and FKBP12.6, a cADPR binding protein146),147) and the altered stoichiometry of FKBP12.6 versus type 2 RyR as a cause of abnormal Ca2+ leak through RyR in heart failure in man.148) As diabetic complications in the cardiovascular system are frequently observed, screening of the abnormalities in the CD38-cADPR signal system may provide a clue for the underlying molecular mechanism.
Jin et al. revealed that adult CD38 KO female and male mice showed marked defects in maternal nurturing and social behavior, respectively, with high locomotor activity.149) Consistently, the plasma level of oxytocin was strongly decreased in CD38 KO mice. Oxytocin injection or lentiviral-vector-mediated delivery of CD38 in the hypothalamus rescued social memory and maternal care in CD38 KO mice but the introduction of CD38 Arg140Trp (rs1800561), which had been found in Japanese diabetic patients,115) failed to recover the social memory and maternal care. Depolarization-induced oxytocin secretion and Ca2+ elevation in oxytocinergic neurohypophysial axon terminals were disrupted in the CD38 KO mice. These results indicate that CD38 has a key role in neuropeptide release, thereby critically regulating maternal and social behaviors, and may be an element in neurodevelopmental disorders. The mutation that caused tryptophan to replace arginine at amino acid residue 140 (CD38R140W; rs1800561[4693C>T])115) was found in 0.6–4.6% of the Japanese population and was associated with autism spectrum disorder in a smaller case-control study.151) In addition, maternal CD38R140W (rs1800561[4693C>T]) polymorphism was reported to be associated with an increased risk of admission to the neonatal intensive care unit due to preterm birth from Nara Medical University.152) These reports strongly suggest that the CD38R140W (rs1800561[4693C>T]) polymorphism is a disease prone genotype, although the polymorphism is restricted in Mongoloid.153)
6. The Reg (Regenerating gene protein)-Reg receptor system for pancreatic β-cell regeneration of the islets of Langerhans
Insulin is synthesized in pancreatic β-cells in the islets of Langerhans and is the only hypoglycemic factor in human and animals. When food is eaten, large amounts of insulin are immediately secreted from β-cells, and then additional insulin is synthesized and stored in β-cells.2),17),23) Therefore, the maintenance of the β-cell mass, as well as the insulin-synthesizing and -releasing ability of β-cells, as described in Sections 3, 4 and 5, is important in the process of insulin production.
According to the Okamoto model, streptozotocin- and alloxan-diabetes can be prevented by PARP inhibitors. In 1984, Yonemura et al.154) successfully made 90% depancreatized rats and then we extended the model by proposing an integral function of poly ADP-ribosylation in cell regeneration.36),96),154)–167)
6.1. Pancreatic β-cell regeneration in 90% depancreatized rats by the administration of PARP inhibitors.
In 1984, we demonstrated that PARP inhibitors induced the regeneration of pancreatic β-cells in 90% depancreatized rats, thereby ameliorating the surgical diabetes.154),156) Male Wistar rats were 90% depancreatized and, beginning 7 days before the partial pancreatectomy and continuing post-operatively, nicotinamide (0.5 g/kg body weight) or 3-aminobenzamide (0.05 g/kg body weight) was injected intraperitoneally every day. The 90% depancreatized rats exhibited glucosuria 1 to 3 months after the operation, but in the rats receiving nicotinamide or 3-aminobenzamide, the urinary glucose excretion level decreased markedly. Plasma glucose levels in rats receiving PARP inhibitors were also significantly lower than those in control 90% depancreatized rats.36),154),160),163) The islets in the remaining pancreases of rats that had received the PARP inhibitors for 3 months were extremely large.36),154),164) When the remaining pancreases were cytochemically stained, almost all the area of the enlarged islets was densely stained for insulin (Fig. 15), and an increase in the number of β-cells was found to be responsible for the increase in islet size (Table 1).36),154),164) A small number of islet cells, however, were stained for insulin in the remaining pancreas of the control 90% depancreatized rats. On the other hand, cells stained for glucagon (α-cells) were localized only on the periphery of enlarged islets and their number was almost the same as that in islets in the parabiliary segment of normal rat pancreatic tissues. The number of cells stained for somatostatin (δ-cells) was also unchanged. These cytochemical findings indicate that it is the β-cell population that increased in the islets of the remaining pancreas of nicotinamide- and 3-aminobenzamide-treated rats, and thus the increased β-cell mass ameliorated the surgical diabetes.

Table 1. Cell numbers in the parabiliary segment of rat pancreatic tissues
| Treatment of rats |
β-cells
(number/cm2) |
α-cells
(number/cm2) |
δ-cells
(number/cm2) |
| Normal untreated |
2086 ± 507 |
285 ± 85 |
191 ± 42 |
| 90% Depancreatized |
442 ± 482 |
359 ± 120 |
185 ± 72 |
90% Depancreatized+
nicotinamide
(0.5 g/kg per day) |
3204 ± 1412** |
310 ± 207 |
133 ± 119 |
90% Depancreatized+
3-aminobenzamide
(0.05 g/kg per day) |
2286 ± 1173* |
310 ± 161 |
92 ± 42 |
*P < 0.05 vs. 90% depancreatized rats (control); **P < 0.01 vs. 90% depancreatized rats (control).
The number of cells was determined in the parabiliary segment of at least five rats and expressed as the mean ± SD. Adapted from Refs. 156 and 164.
We isolated the rat regenerating islets and constructed a cDNA library. By differential screening of the regenerating islet-derived cDNA library, we found a novel gene expressed in regenerating islets. The cDNA had one large open reading frame encoding a 165-amino acid protein and the deduced protein has a signal sequence. We named the novel gene Reg, that is, regenerating gene, with the implication that the gene may be involved in islet regeneration.157) We then isolated human REG gene.157),168) Watanabe et al.169) administered a recombinant rat Reg protein to 90% depancreatized rats and observed increased [3H]thymidine incorporation and frequent mitosis in islets of the remaining pancreas. On the 30th and 60th postoperative day, the fasting plasma glucose level of the rats that had received daily intraperitoneal injection of Reg protein (1 mg/kg/day) was significantly lower than that of the 90% depancreatized control rats. After 2 months, almost all the islets in the 90% depancreatized control rats were destroyed, whereas the islets of the remaining pancreas in the Reg protein-treated rats were enlarged and almost entirely stained for insulin.169) Human REG protein administration also ameliorated diabetes in NOD mice with an increase in the β-cell mass.170) These results indicate that Reg protein stimulates the regeneration and/or growth of pancreatic β-cells, thereby ameliorating animal diabetes.
Transgenic mice expressing Reg in β-cells showed increased [3H]thymidine incorporation in the islets.171) The development of diabetes was significantly retarded in the Reg transgene-carrying NOD mice. On the other hand, Reg KO mice created by homologous recombination showed the decreased [3H]thymidine incorporation in the islets.171) Further, when hyperplastic islets were induced by the injection of goldthioglucose, the islet sizes of Reg KO mice were significantly smaller than those from control Reg+/+ mice.171)
6.3. Reg protein signal.
As Reg protein was thought to act on pancreatic β-cells as an autocrine and/or paracrine growth factor, we tried to isolate a cDNA for the Reg protein receptor from a rat islet expression cDNA library using 125I-labeled rat Reg protein as a probe.165) The isolated cDNA encoded a cell surface 919-amino acid protein, and a homology search revealed that the cDNA was a homologue to multiple exostoses-like gene, the function of which had hitherto been unknown. The COS-7 cells into which the cDNA had been introduced bound Reg protein with high affinity. When the cDNA was introduced into RINm5F β-cells, the transformants exhibited significant increases in the incorporation of 5′-bromo-2′-deoxyuridine as well as in the cell numbers in response to Reg protein, suggesting that the receptor mediated a growth signal of Reg protein for β-cell regeneration.165)
To determine what intracellular signal transduction events are induced in pancreatic β-cells by Reg protein, Takasawa et al. tested several signal transduction pathways in pancreatic β-cells by Reg protein stimulation using a PathDetect trans-reporting systems and found that activating transcription factor 2 (ATF-2) was activated by Reg protein stimulation.167) The ATF-2 activation was also observed by the cotransfection of the Reg receptor expression plasmid instead of by Reg protein addition to the medium. Phosphorylation of ATF-2 at Thr-71 was increased by the stimulation of Reg protein. These results indicate that the Reg-Reg receptor system activates ATF-2, suggesting that genes under the control of ATF-2 play an important role in the cell cycle progression in pancreatic β-cells.
Cyclin D1 promoter activation by Reg protein was induced in a dose-dependent manner. In the electrophoretic mobility assay using cyclin D1 promoter sequence, Reg protein-stimulated β-cell nuclear proteins formed a specific complex with the ATF-2 binding site. Addition of the antibody against ATF-2 resulted in the formation of a supershift, suggesting the presence of ATF-2 in the complex.167) The addition of phosphoinositide 3-kinase (PI(3)K) inhibitors LY294002 and wortmannin attenuated the Reg protein-induced ATF-2 phosphorylation/activation. Moreover, the immunoprecipitated PI(3)K phosphorylated ATF-2 in a time-dependent and dose-dependent fashion, suggesting that PI(3)K directly phosphorylated ATF-2 to activate the cyclin D1 promoter.167)
The Reg and Reg-related genes were isolated and revealed to constitute a multigene family, the Reg gene family96),164) as later mentioned (Section 6.5). We made a knockout (KO) mice disrupting the gene of the Reg family, Reg I. The BrdU incorporation in Reg I KO islets without the addition of Reg I protein in the culture medium was reduced. The levels of phospho-ATF-2, cyclin D1 protein, and phospho-Rb in Reg I-deficient mouse islets were compared with those in normal littermates. Wild-type islets secreted Reg I in the culture medium and the phospho-Rb level in the islets was much higher than that in Reg I KO islets. This well fitted the decreased BrdU incorporation in Reg I KO islets. As expected, both the levels of phospho-ATF-2 and cyclin D1 were decreased in Reg I KO islets, whereas the levels of other proteins such as PARP96),166) and CD3880),91),96),101) as controls were unchanged.
These results indicate that the activation of the cyclin D1 gene promoter in response to Reg I protein stimulation is mediated by the PI(3)K/ATF-2 signal transduction pathway in pancreatic β-cells, as shown in Fig. 16. Reg protein activated the cyclin D1 promoter for the cell cycle progression, and ATF-2 is an essential transcription factor in the process of Reg protein-induced cyclin D1 promoter activation. The PI(3)K activation is involved in the ATF-2 activation for cyclin D1 expression. It has been reported that disruption of the CDK4 gene resulted in the development of insulin-deficient diabetes due to a reduction in the number of pancreatic β cells, and that the expression of a mutant CDK4, which escaped from the inhibitory regulation of CDK4, caused islets to become hyperplastic.172) The results of the CDK4 disruption well explained the result that the islets from the Reg I KO mice showed reduced BrdU incorporation,167) and that the islet hyperplasia induced by the goldthioglucose treatment was attenuated by Reg I gene disruption171) because Reg protein induced a regulatory subunit of CDK4, cyclin D1, for the cell cycle progression (Fig. 16).

Reg was induced in insulin producing pancreatic β-cells by inflammatory stimulation such as by interleukin-6 (IL-6)/glucocorticoids and IL-22166),173)–175) and acted as an autocrine/paracrine growth factor for β-cell regeneration via a cell surface Reg receptor (see Figs. 16 and 17)165),167) to ameliorate experimental diabetes.154),156),169) Further the Reg gene is only expressed during islet regeneration,157),161) suggesting that the regeneration and proliferation of pancreatic β-cells for the increase of the cell mass were primarily regulated by the Reg gene expression. In this context, Akiyama et al.166) found that Reg gene was activated by IL-6, dexamethasone, and PARP inhibitors. The combined addition of IL-6 and dexamethasone increased the Reg mRNA level, and further addition of nicotinamide or 3-aminobenzamide increased the mRNA even more. Progressive deletion of the 5′-flanking region of rat Reg gene revealed that the region between nucleotides -81 and -70 “TGCCCCTCCCAT” is essential for the Reg gene promoter activity. In gel mobility shift assays with the Reg promoter, the intensity of the band, which was detected in the nuclear extracts of RINm5F cells treated with IL-6, dexamethasone and/or nicotinamide, was correlated with the luciferase activity, and the protein binding to the sequence was revealed to be PARP.166) The inhibition of PARP activity was shown to facilitate Reg transcription by preventing the excessive self-poly(ADP-ribosyl)ation of PARP and the consequent dissociation of PARP from the active transcriptional DNA/protein complex. The transcriptional regulation by PARP activity and the transcription-regulating PARP-binding sequences were subsequently described in other genes such as chemokine (C-X-C motif) ligand 1,176) nuclear factor κ-light-chain-enhancer of activated B cells-regulated genes,177) α-synuclein,178) V-type proton ATPase 116 kDa subunit a isoform 3,179) B-cell lymphoma 6 protein,180) tatrate-resistant acid phosphatase,181) vimentin,182) cyclooxygenase-2,183) hemochromatosis protein,184) cellular communication network factor 2,185) micro opioid receptor,186) microsomal epoxide hydrolase,187) FoxO1188) and PARP.189) So far, the novel view that PARP acts as a transcriptional regulator has been established.
Thus, as shown in Fig. 17, inflammatory mediators, IL-6 and glucocorticoids, induce the formation of an active transcriptional complex involving PARP for Reg gene, and the Reg gene transcription therefore proceeds. However, during inflammation, superoxide (O2•) and nitric oxide (NO•) are produced, causing DNA damage. In this situation, PARP is activated by DNA nicks to repair the DNA. Then, PARP poly(ADP-ribosyl)ates PARP itself, the poly(ADP-ribose) chains on the PARP protein inhibit the formation of the active transcriptional complex, and the Reg gene transcription is halted. In the presence of PARP inhibitors such as nicotinamide, the PARP is not poly(ADP-ribosyl)ated, the transcriptional complex is stabilized, and the Reg gene transcription proceeds. Therefore, PARP inhibitors maintain the function of PARP as a transcription factor for β-cell regeneration. This can account for the islet regeneration found in 90% depancreatized rats treated with PARP inhibitors154) and also supports the notion that the restriction of β-cell replication is relieved by PARP inhibitors.36) When DNA is massively damaged, PARP is rapidly activated to repair the DNA, as described in Section 4, and the complex for Reg gene transcription is not formed at all.

The Reg and Reg-related genes were isolated and revealed to constitute a multigene family, the Reg gene family.96),164),190),191) Based on the primary structures of the Reg proteins, the members of the family are grouped into four subclasses, types I, II, III, and IV (Fig. 18). In human, four REG family genes, i.e., REG Iα,157),168) REG Iβ,192) REG-related sequence (RS),168) HIP193)/PAP,194) and REG III195) are tandemly ordered in the 95 kbp region of chromosome 2p12,196) whereas REG IV is located on chromosome 1.197) In the mouse genome, all the Reg family genes except for Reg IV, i.e., Reg I, Reg II, Reg IIIα, Reg IIIβ, Reg IIIγ, and Reg IIIδ were mapped to a contiguous 75 kbp region of chromosome 6C,198) whereas Reg IV was mapped on chromosome 3. Type I and mouse Type II Reg proteins are expressed in regenerating islets157),190) and are involved in β-cell regeneration.169),199)–208) Reg family proteins have been suggested to be involved in cellular proliferation in exocrine pancreatic cells,209),210) gastrointestinal cells,211)–222) Helicobacter pylori-infected gastric mucosa,223),224) hepatic cells,225)–229) cardiovascular cells,230)–232) salivary ductal cells,233)–235) bone and muscle cells,236),237) and neuronal cells.238) Importantly, mouse Reg III was shown to be a Schwann cell mitogen that accompanied the regeneration of motor neurons,239) and Reg protein functions as a neurotrophic factor for motor neurons.240),241) Reg was also shown to mediate gastric mucosal proliferation in rats.242),243)

In 2000, a group of St. James’s University Hospital reported that death from early colorectal cancer was predicted by the presence of transcripts of the REG gene family.244) In 2003, Yonemura et al.245) showed that the expression of the REG Iα gene is closely related to the infiltrating property of gastric carcinomas, and may be a prognostic indicator of differentiated adenocarcinoma of the stomach (Fig. 19). Since then, a correlation between REG family gene expression and cancer prognosis has been reported.246)–256) These observations suggest that the Reg gene family is involved in a variety of cell types including cancer cells for regeneration/proliferation.
7. Concluding remarks
Based on the results of Sections 2, 3, and 4 we described that PARP activation, which was induced by damaged DNA, caused the NAD+ depletion to form poly(ADP-ribose) and the decreased β-cell function such as proinsulin synthesis, resulting in necrotic cell death (Fig. 5). Since induction of insulin biosynthesis in islet β-cells is achieved at the level of translation (as discussed in Section 3), an immediate decrease in insulin biosynthetic activity cannot be attributed to DNA damage itself. Rather, the β-cell, it may reasonably be assumed, commits suicide in its attempt to repair the DNA strand breaks.1),2),35)–37)
In Section 5, we described that cADPR formation from NAD+ was essential for cell functions, such as insulin secretion in pancreatic β-cells. An increase in the intracellular Ca2+ concentration, which then triggers insulin secretion, has conventionally been explained by Ca2+ influx from extracellular sources. On the other hand, in 1993, we showed a novel mechanism of insulin secretion in which the Ca2+ release from the endoplasmic reticulum, an intracellular Ca2+ pool, induced insulin secretion (Fig. 6).79),80) Rojas et al. examined the Ca2+ influx from extracellular sources and the intracellular pool in human β-cells, and showed that 42–75% of the increase in the intracellular Ca2+ concentration by glucose stimulation was due to the release of Ca2+ from the intracellular stores.257) Thus, the importance of Ca2+ release from the endoplasmic reticulum in insulin secretion by glucose has been recognized.
In Section 6, we described that PARP inhibitors induced the regeneration of pancreatic β-cells in 90% depancreatized rats, thereby ameliorating the surgical diabetes.154),156) Then, we isolated Reg (Regenerating gene) from the regenerating islet-derived cDNA library157),160) and established the Reg-Reg receptor system for the cell regeneration/proliferation (Figs. 16 and 17). Further, PARP was found to act as a transcription factor for Reg gene, and the active transcription complex for Reg gene was not formed when PARP was activated and auto-poly(ADP-ribosyl)ated (Fig. 17). Therefore, the inhibition of the PARP activity by PARP inhibitors results in at least three important events in the cell (Fig. 20): PARP inhibitors prevent cell death (Fig. 5), maintain the formation of a second messenger, cADPR, to achieve the cell function, and keep PARP active as a transcription factor for cell regeneration or proliferation (Fig. 17).

In Section 4, PARP inhibitors were shown to prevent necrotic β-cell death. In 1987, Vague et al.258) reported that nicotinamide, a PARP inhibitor, could extend the remission phase of insulin-dependent Type 1 diabetes. Then, clinical trials with nicotinamide have been performed by several groups in the world. The results suggested that the nicotinamide treatment may be promising one for the prevention and the cure of Type 1 diabetes,259)–264) whereas in another study nicotinamide was reported to be somewhat ineffective to prevent Type 1 diabetes.265) Concerning this, Uchigata et al.7) have already reported that it is essentially important when the nicotinamide administration against Type 1 diabetes should be started: before, just after, or while β-cells are being damaged. It is also important that nicotinamide266),267) can protect the necrotic cell death but cannot protect the apoptotic cell death as shown in Figs. 21 and 22. Actually, pretreatment of islet grafts with nicotinamide prevented their deterioration in a mouse islet transplantation model.268) Furthermore, in human islet isolation for islet transplantation, nicotinamide significantly improved islet yields in both the University of Wisconsin and the two-layer method preservation protocols.269) PARP inhibitors were also shown in Section 6 to induce Reg gene expression by stabilizing the transcriptional complex for Reg gene, resulting in the β-cell regeneration and neogenesis from the progenitor cells such as pancreatic duct cells.270) In generating insulin-producing endocrine β-cells from mouse embryonic stem cells and human induced pluripotent stem cells, nicotinamide was found to be an indispensable factor for differentiation/proliferation into insulin-producing endocrine β-cells.271),272) It is, therefore, possible that the amelioration of diabetes observed in the clinical nicotinamide trials could be caused not only by the prevention of necrotic β-cells but also by the induction of β-cell regeneration/neogenesis. Furthermore, as described in Section 5, because cADPR is produced from NAD+ by ADP-ribosyl cyclase of CD38, and because PARP is the major enzyme for NAD+ degradation/consumption, the inhibition of PARP by inhibitors such as nicotinamide may maintain the cADPR formation from NAD+ for β-cells to achieve their functions, such as glucose-stimulated insulin secretion.

Therefore, PARP inhibitors and their derivatives appear to be one of the promising therapeutic approaches for diabetes treatment, although some toxic effects such as reversible liver toxicity,273) teratogenicity,274) growth-retardation,275) and oncogenicity35),36),276) were reported in animal models at very high doses of the inhibitor. However, it has been revealed that there is no evidence in man of most of the toxic effects of nicotinamide that have been reported in animal models.264)
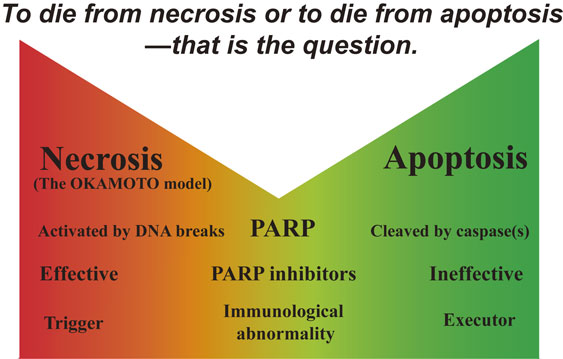
Some experiments using cytokines as β-cytotoxic agents showed that nicotinamide did not effectively inhibit human β-cell death.277),278) However, there are two kinds of cell death, necrosis and apoptosis. PARP activation results in necrotic cell death36),37),100),155),279) whereas, in apoptotic cell death, PARP is cleaved by caspases and inactivated.280)–282) Therefore, PARP inhibitors can prevent necrosis but not apoptosis (Fig. 21). The type of cell death, whether necrosis or apoptosis, would depend on the severity and duration of the insult, differences in signaling pathways, and the type of cell. It has been suggested that dendritic cells/macrophages can distinguish between the two types of cell death, and that necrosis may provide the critical control for the initiation of immunity283),284) upon recognition of necrotic cells, and the proinflammatory responses of activated macrophages are increased. In contrast, apoptotic cells have a strong inhibitory effect on phlogistic macrophage responses. Thus, immunological abnormalities, which are frequently observed in Type 1 diabetes, may be triggered by preceding necrotic cell death, followed by apoptotic death of β-cells (Fig. 22).
More recently, roles of the NAD+ preservation in the islet β-cell survival and of the CD38-cADPR signal system in insulin secretion (described in Sections 4 and 5) with Imeglimin ((6R)-N2,N2,6-Trimethyl-3,6-dihydro-1,3,5-triazine-2,4-diamine monohydrochloride) have been recognized.285) Imeglimin is the first in a new class of oral anti-Type 2 diabetes agent286) and is scheduled to be launched in 2021 in Japan for the first time in the world.
Never advance anything which cannot be proved in a simple and decisive fashion.
—Louis Pasteur (1888)—
Acknowledgements
The authors are grateful to Mr. Brent Bell for critical reading of the manuscript.
We thank all of the past and present colleagues for contributions to the studies described herein especially Drs. Susumu Miyamoto, Yutaka Noto, Ryoyu Takeda, Junji Koizumi, Hiroshi Mabuchi, Gunji Yamazaki, Kiyoshi Nose, Nobuyuki Itoh, Terunobu Sei, Yasumi Ohshima, Hiroshi Yamamoto, Yasuko Uchigata, Makoto Watanabe, Yutaka Yonemura, Yumiko Hayakawa, Norio Hayashi, Kenzo Ohsawa, Koji Nata, Hideto Yonekura, Kimio Terazono, Kiyoto Shiga, Chiyoko Inoue, Motoo Kitagawa, Michiaki Unno, Hiroshi Munakata, Akira Tohgo, Akira Sugawara, Takuo Watanabe, Shigeki Moriizumi, Ichiro Kato, Tetsuhiko Koguma, Kan-ichi Nakagawara, Hikari Miyashita, Takao Anzai, Yu Suzuki, Atsuya Akabane, Toshinari Takamura, Tadahiro Karasawa, Shinichi Nakamura, Naoya Noguchi, Yoichi Narushima, Fumiko Ikehata, Jo Satoh, Takayoshi Toyota, Yoshio Goto, Takako Akiyama, Kei Nakagawa, Seiichi Kobayashi, Michiaki Abe, Nausheen Jamal Shervani, Yasuhiko Yamamoto, Shoko Kawaguchi, Michio Kuroki, Takayuki Ikeda, Akiyo Yamauchi, Takeo Yoshikawa, Yoshikazu Kinoshita, Yoichi Yasunami, Osamu Hori, and Haruhiro Higashida.
It is also a pleasure to express our sincere appreciation to late Prof. Osamu Hayaishi, his encouragement and generous support were driving force in initiating and continuing the work. The study was supported in part by Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (especially 60065004 and 08102003), Japan Society for the Promotion of Science, and Japan Science and Technology Agency for grants with which those studies were able to be conducted.
Notes
Edited by Shigetada NAKANISHI, M.J.A.
Correspondence should be addressed: H. Okamoto, Department of Biochemistry and Molecular Vascular Biology, Kanazawa University Graduate School of Medical Sciences, 13-1 Takara-machi, Kanazawa, Ishikawa 920-8640, Japan (e-mail: okamotoh@med.tohoku.ac.jp).
References
- 1) Okamoto, H. (ed.) (1990 & 2008) Molecular Biology of the Islets of Langerhans. Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney.
- 2) Okamoto, H. (1981) Regulation of proinsulin synthesis in pancreatic islets and a new aspect to insulin-dependent diabetes. Mol. Cell. Biochem. 37, 43–61.
- 3) Steiner, D.F., Kemmler, W., Clark, J.L., Oyer, P.E. and Rubenstein, A.H. (1972) The biosynthesis of insulin. In Handbook of Physiology, section 7, vol. 1 (eds. Steiner, D.F. and Freinkel, N.). American Physiology Society, Washington, D.C., pp. 175–198.
- 4) Yamamoto, H., Uchigata, Y. and Okamoto, H. (1981) Streptozotocin and alloxan induce DNA strand breaks and poly(ADP-ribose) synthetase in pancreatic islets. Nature 294, 284–286.
- 5) Yamamoto, H., Uchigata, Y. and Okamoto, H. (1981) DNA strand breaks in pancreatic islets by in vivo administration of alloxan or streptozotocin. Biochem. Biophys. Res. Commun. 103, 1014–1020.
- 6) Uchigata, Y., Yamamoto, H., Kawamura, A. and Okamoto, H. (1982) Protection by speroxide dismutase, catalase, and poly(ADP-ribose) synthetase inhibitors against alloxan- and streptozotocin-induced islet DNA strand breaks and against the inhibition of proinsulin synthesis. J. Biol. Chem. 257, 6084–6088.
- 7) Uchigata, Y., Yamamoto, H., Nagai, H. and Okamoto, H. (1983) Effect of poly(ADP-ribose) synthetase inhibitor administration to rats before and after injection of alloxan and streptozotocin on islet proinsulin synthesis. Diabetes 32, 316–318.
- 8) Charron, M.J. and Bonner-Weir, S. (1999) Implicating PARP and NAD+ depletion in type I diabetes. Nat. Med. 5, 269–270.
- 9) Burkart, V., Wang, Z.Q., Radons, J., Heller, B., Herceg, Z., Stingl, L. et al. (1999) Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic beta-cell destruction and diabetes development induced by streptozocin. Nat. Med. 5, 314–319.
- 10) Masutani, M., Suzuki, H., Kamada, N., Watanabe, M., Ueda, O., Nozaki, T. et al. (1999) Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. U.S.A. 96, 2301–2304.
- 11) Pieper, A.A., Brat, D.J., Krug, D.K., Watkins, C.C., Gupta, A., Blackshaw, S. et al. (1999) Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. U.S.A. 96, 3059–3064.
- 12) Nomikos, I.N., Prowse, S.J., Carotenuto, P. and Lafferty, K.J. (1986) Combined treatment with nicotinamide and desferrioxamine prevents islet allograft destruction in NOD mice. Diabetes 35, 1302–1304.
- 13) Colman, P.G., Wang, Y. and Lafferty, K.J. (1989) Molecular biology and autoimmunity of Type I diabetes mellitus. In Molecular Cellular Biology of Diabetes Mellitus, Vol. 1, Insulin Secretion (eds. Draznin, B., Melmed, S. and LeRoith, D.). Alan Liss, New York, pp. 125–137.
- 14) Renold, A.E. (1988) Animal models: an aid to the understanding of the etiology and pathogenesis of diabetes mellitus. In The Diabetes Annual/4 (eds. Alberti, K.G.M.M. and Krall, L.P.). Elsevier, Amsterdam-New York-Oxford, pp. 592–608.
- 15) Lacy, P.E. and Kostianovsky, M. (1967) Methods for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes 16, 35–39.
- 16) Okamoto, H., Noto, Y., Miyamoto, S., Mabuchi, H. and Takeda, R. (1975) Inhibition by somatostatin of insulin release from isolated pancreatic islets. FEBS Lett. 54, 103–105.
- 17) Itoh, N. and Okamoto, H. (1980) Translational control of proinsulin synthesis by glucose. Nature 283, 100–102.
- 18) Itoh, N., Nose, K. and Okamoto, H. (1979) Purification and characterization of proinsulin mRNA from rat B-cell tumor. Eur. J. Biochem. 97, 1–9.
- 19) Itoh, N., Sei, T., Nose, K. and Okamoto, H. (1978) Glucose stimulation of the proinsulin synthesis in isolated pancreatic islets without increasing amount of proinsulin mRNA. FEBS Lett. 93, 343–347.
- 20) Okamoto, H., Nose, K., Itoh, N., Sei, T. and Yamamoto, H. (1979) Translational control of proinsulin synthesis in pancreatic islets. In Proinsulin, Insulin, C-peptide. Congress Series No. 468 (eds. Baba, S., Kaneko, T. and Yanaihara, N.). Excepta Medica, Amsterdam-Oxford, pp. 27–35.
- 21) Itoh, N., Ohshima, Y., Nose, K. and Okamoto, H. (1982) Glucose stimulates proinsulin synthesis in pancreatic islets without a concomitant increase in proinsulin mRNA synthesis. Biochem. Int. 4, 315–321.
- 22) Watanabe, M., Itoh, N. and Okamoto, H. (1982) Translational control of proinsulin synthesis by glucose. In Endocrinology, International Congress Series No. 598 (eds. Shizume, K., Imura, H. and Shimizu, N.). Elsevier, Amsterdam, pp. 266–270.
- 23) Itoh, N. (1990 & 2008) The translational control of proinsulin synthesis by glucose. In Molecular Biology of the Islets of Langerhans (ed. Okamoto, H.). Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 49–65.
- 24) Humphries, S., Windass, J. and Williamson, R. (1976) Mouse globin gene expression in erythroid and non-erythroid tissues. Cell 7, 267–277.
- 25) Harris, S.E., Rosen, J.M., Means, A.R. and O’Malley, B.W. (1975) Use of a specific probe for ovalbumin messenger RNA to quantitate estrogen-induced gene transcript. Biochemistry 14, 2072–2081.
- 26) Wicksteed, B., Uchizono, Y., Alarcon, C., McCuaig, J.F., Shalev, A. and Rhodes, C.J. (2007) A cis-element in the 5′ untranslated region of the preproinsulin mRNA (ppIGE) is required for glucose regulation of proinsulin translation. Cell Metab. 5, 221–227.
- 27) Takasawa, S., Itaya-Hironaka, A., Yamauchi, A., Sakuramoto-Tsuchida, S., Ota, H., Fujimura, T. et al. (2015) Nucleotide sequence responsible for translational control of proinsulin synthesis in beta cells by glucose. In World Diabetes Congress 2015 (Vancouver, Canada). Abstract: VA-0256.
- 28) Mueckler, M.M., Merrill, M.J. and Pitot, H.C. (1983) Translational and pretranslational control of ornithine aminotransferase synthesis in rat liver. J. Biol. Chem. 258, 6109–6114.
- 29) White, M.W., Kameji, T., Pegg, A.E. and Morris, D.R. (1987) Increased efficiency of translation of ornithine decarboxylase mRNA in mitogen-activated lymphocytes. Eur. J. Biochem. 170, 87–92.
- 30) Rogers, J. and Munro, H. (1987) Translation of ferritin light and heavy subunit mRNAs is regulated by intracellular chelatable iron levels and rat hepatoma cells. Proc. Natl. Acad. Sci. U.S.A. 84, 2277–2281.
- 31) von Mering, J. and Minkowski, O. (1898) Diabetes mellitus nach Pankreasextirpation. Centralbl. Klin. Med. 10, 393–394.
- 32) Banting, F.G., Best, C.H., Collip, J.B., Campbell, W.R. and Fletcher, A.A. (1922) Pancreatic extracts in the treatment of diabetes mellitus: preliminary report. Can. Med. Assoc. J. 12, 141–146.
- 33) Dunn, J.S., Sheehan, H.L. and McLetchie, N.G.B. (1943) Necrosis of islets of Langerhans produced experimentally. Lancet 241, 484–487.
- 34) Rakieten, N., Rakieten, M.L. and Nadkarni, M.V. (1963) Studies on the diabetogenic action of streptozotocin. Cancer Chemother. Rep. 29, 695–696.
- 35) Okamoto, H. and Yamamoto, H. (1983) DNA strand breaks and poly(ADP-ribose) synthetase activation in pancreatic islets — a new aspect to development of insulin-dependent diabetes and pancreatic B-cell tumors. In ADP-ribosylation, DNA Repair and Cancer (eds. Miwa, M., Hayaishi, O., Shall, S., Smulson, M. and Sugimura, T.). Japan Scientific Societies Press, Tokyo, pp. 297–308.
- 36) Okamoto, H. (1985) Molecular basis of experimental diabetes: degeneration, oncogenesis, and regeneration of pancreatic B-cells of islets of Langerhans. BioEssays 2, 15–21.
- 37) Okamoto, H. (1990 & 2008) The molecular basis of experimental diabetes. In Molecular Biology of the Islets of Langerhans (ed. Okamoto, H.). Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 209–231.
- 38) Chambon, P., Weil, J.D., Doly, J., Strosser, M.T. and Mandel, P. (1966) On the formation of a novel adenylic compound by enzymatic extracts of liver nuclei. Biochem. Biophys. Res. Commun. 25, 638–643.
- 39) Sugimura, T., Fujimura, S., Hasegawa, S. and Kawamura, Y. (1967) Polymerization of the adenosine 5′-diphosphate ribose moiety of NAD by rat liver nuclear enzyme. Biochim. Biophys. Acta 138, 438–441.
- 40) Nishizuka, Y., Ueda, K., Nakazawa, K. and Hayaishi, O. (1967) Studies on the polymer of adenosine diphosphate ribose. J. Biol. Chem. 242, 3164–3171.
- 41) Ueda, K. and Hayaishi, O. (1985) ADP-ribosylation. Annu. Rev. Biochem. 54, 73–100.
- 42) Shall, S. (2002) Poly(ADP-ribosylation) — a common control process? BioEssays 24, 197–201.
- 43) Amé, J.-C., Spenlehauer, C. and de Murcia, G. (2004) The PARP superfamily. BioEssays 26, 882–893.
- 44) Cohen, M.S. (2020) Interplay between compartmentalized NAD+ synthesis and consumption: a focus on the PARP family. Genes Dev. 34, 254–262.
- 45) Heikkila, R.E., Winston, B., Cohen, G. and Barden, H. (1976) Alloxan-induced diabetes — evidence for hydroxyl radical as a cytotoxic intermediate. Biochem. Pharmacol. 25, 1085–1092.
- 46) Haber, F. and Weiss, J. (1934) The catalytic decomposition of hydrogen peroxide by iron salts. Proc. R. Soc. Lond. A Math. Phys. Sci. 147, 332–351.
- 47) McCord, J.M. and Fridovich, I. (1969) Superoxide dismutase. An enzymic function for erythrocuprein (Hemocuprein). J. Biol. Chem. 244, 6049–6055.
- 48) Erickson, L.C., Bradley, M.O. and Kohn, K.W. (1977) Strand breaks in DNA from normal and transformed human cells treated with 1,3-bis(2-chloroethyl)-1-nitrosourea. Cancer Res. 37, 3744–3750.
- 49) Cooperstein, S.J. and Watkins, D. (1981) Actions of toxic drugs on islet cells. In The Islets of Langerhans (eds. Cooperstein, S.J. and Watkins, D.). Academic Press, New York, pp. 387–425.
- 50) Hammerström, L. and Ullberg, S. (1966) Specific uptake of labeled alloxan in the pancreatic islets. Nature 212, 708–709.
- 51) Tjälve, H., Wilander, E. and Johansson, E. (1976) Distribution of labeled streptozotocin in mice: uptake and retention in pancreatic islets. J. Endocrinol. 69, 455–456.
- 52) Malaisse, W.J., Malaisse-Lagae, F., Sener, A. and Pipeleers, D.G. (1982) Determinants of the selective toxicity of alloxan to the pancreatic B cell. Proc. Natl. Acad. Sci. U.S.A. 79, 927–930.
- 53) Leiter, E.H. (1982) Multiple low-dose streptozotocin-induced hyperglycemia and insulitis in C57BL mice: influence of inbred background, sex, and thymus. Proc. Natl. Acad. Sci. U.S.A. 79, 630–634.
- 54) Tsubouchi, S., Suzuki, H., Ariyoshi, H. and Matsuzawa, T. (1981) Radiation-induced acute necrosis of the pancreatic islet and the diabetic syndrome in the golden hamster. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 40, 95–106.
- 55) Toniolo, A., Onodera, T., Yoon, J.-W. and Notkins, A.L. (1980) Induction of diabetes by cumulative environmental insulitis from viruses and chemicals. Nature 288, 383–385.
- 56) Makino, S., Kunimoto, K., Muraoka, Y., Mizushima, Y., Katagiri, K. and Tochino, Y. (1980) Breeding of a non-obese diabetic strain of mice. Jikken Dobutsu 29, 1–13.
- 57) Mori, Y., Suko, M., Okudaira, H., Matsuba, I., Tsuruoka, A., Yokoyama, H. et al. (1986) Preventive effects of cyclosporine on diabetes in NOD mice. Diabetologia 29, 244–247.
- 58) Okamoto, H., Sr. (1976) Antitumor activity of streptolysin S-forming streptococci. In Mechanisms in Bacterial Toxicology (ed. Bernheimer, A.W.). John Wiley & Sons, New York, pp. 237–257.
- 59) Toyota, T., Satoh, J., Oya, K., Shintani, S. and Okano, T. (1986) Streptococcal preparation (OK-432) inhibits development of type I diabetes in NOD mice. Diabetes 35, 496–499.
- 60) Yamada, K., Nonaka, K., Hanafusa, T., Miyazaki, A., Toyoshima, H. and Tarui, S. (1982) Preventive and therapeutic effects of large-dose nicotinamide injections on diabetes associated with insulitis. An observation in nonobese diabetic (NOD) mice. Diabetes 31, 749–753.
- 61) Takamura, T., Kato, I., Kimura, N., Nakazawa, T., Yonekura, H., Takasawa, S. et al. (1998) Transgenic mice overexpressing type 2 nitric oxide synthase in pancreatic β cells develop insulin-dependent diabetes without insulitis. J. Biol. Chem. 273, 2493–2496.
- 62) Mabley, J.G., Suarez-Pinzon, W.L., Haskó, G., Salzman, A.L., Rabinovitch, A., Kun, E. et al. (2001) Inhibition of poly (ADP-ribose) synthetase by gene disruption or inhibition with 5-iodo-6-amino-1,2-benzopyrone protects mice from multiple-low-dose-streptozotocin-induced diabetes. Br. J. Pharmacol. 133, 909–919.
- 63) Eliasson, M.J., Sampei, K., Mandir, A.S., Hurn, P.D., Traystman, R.J., Bao, J. et al. (1997) Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 3, 1089–1095.
- 64) Pieper, A.A., Walles, T., Wei, G., Clements, E.E., Verma, A., Snyder, S.H. et al. (2000) Myocardial postischemic injury is reduced by polyADPripose polymerase-1 gene disruption. Mol. Med. 6, 271–282.
- 65) Liang, E.S., Bai, W.W., Wang, H., Zhang, J.N., Zhang, F., Ma, Y. et al. (2018) PARP-1 (poly[ADP-ribose] polymerase 1) inhibition protects from Ang II (angiotensin II)-induced abdominal aortic aneurysm in mice. Hypertension 72, 1189–1199.
- 66) Stern, Y., Salzman, A., Cotton, R.T. and Zingarelli, B. (1999) Protective effect of 3-aminobenzamide, an inhibitor of poly (ADP-ribose) synthetase, against laryngeal injury in rats. Am. J. Respir. Crit. Care Med. 160, 1743–1749.
- 67) Jijon, H.B., Churchill, T., Malfair, D., Wessler, A., Jewell, L.D., Parsons, H.G. et al. (2000) Inhibition of poly(ADP-ribose) polymerase attenuates inflammation in a model of chronic colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 279, G641–G651.
- 68) Liaudet, L., Soriano, F.G., Szabó, É., Virág, L., Mabley, J.G., Salzman, A.L. et al. (2000) Protection against hemorrhagic shock in mice genetically deficient in poly(ADP-ribose)polymerase. Proc. Natl. Acad. Sci. U.S.A. 97, 10203–10208.
- 69) Zheng, J., Devalaraja-Narashimha, K., Singaravelu, K. and Padanilam, B.J. (2005) Poly(ADP-ribose) polymerase-1 gene ablation protects mice from ischemic renal injury. Am. J. Physiol. Renal Physiol. 288, F387–F398.
- 70) Mota, R.A., Sánchez-Bueno, F., Saenz, L., Hernández-Espinosa, D., Jimeno, J., Tornel, P.L. et al. (2005) Inhibition of poly(ADP-ribose) polymerase attenuates the severity of acute pancreatitis and associated lung injury. Lab. Invest. 85, 1250–1262.
- 71) Cover, C., Fickert, P., Knight, T.R., Fuchsbichler, A., Farhood, A., Trauner, M. et al. (2005) Pathophysiological role of poly(ADP-ribose) polymerase (PARP) activation during acetaminophen-induced liver cell necrosis in mice. Toxicol. Sci. 84, 201–208.
- 72) Lin, T. and Yang, M.S. (2008) Benzo[a]pyrene-induced necrosis in the HepG2 cells via PARP-1 activation and NAD+ depletion. Toxicology 245, 147–153.
- 73) Shevalye, H., Maksimchyk, Y., Watcho, P. and Obrosova, I.G. (2010) Poly(ADP-ribose) polymerase-1 (PARP-1) gene deficiency alleviates diabetic kidney disease. Biochim. Biophys. Acta 1802, 1020–1027.
- 74) Pacher, P., Liaudet, L., Soriano, F.G., Mabley, J.G., Szabó, É. and Szabó, C. (2002) The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes 51, 514–521.
- 75) Ashcroft, F.M., Harrison, D.E. and Ashcroft, S.J.H. (1984) Glucose induced closure of single potassium channels in isolated rat pancreatic β-cells. Nature 312, 446–448.
- 76) Inagaki, N., Gonoi, T., Clement, J.P. 4th, Namba, N., Inazawa, J., Gonzalez, G. et al. (1995) Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270, 1166–1170.
- 77) Inagaki, N., Gonoi, T., Clement, J.P., Wang, J.Z., Aguilar-Bryan, L., Bryan, J. et al. (1996) A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron 16, 1011–1017.
- 78) Okamoto, H., Takasawa, S., Nata, K. and Yonekura, H. (1992) Cyclic ADP-ribose, a novel second messenger for intracellular Ca2+ mobilization in pancreatic islets. In The First Conference of the International Union of Biochemistry and Molecular Biology; Biochemistry of Diseases (June 1–6, 1992, Nagoya, Japan, Abstract). Bridge Ltd., Nagoya, p. 218.
- 79) Takasawa, S., Nata, K., Yonekura, H. and Okamoto, H. (1993) Cyclic ADP-ribose in insulin secretion from pancreatic β-cells. Science 259, 370–373.
- 80) Takasawa, S., Tohgo, A., Noguchi, N., Koguma, T., Nata, K., Sugimoto, T. et al. (1993) Synthesis and hydrolysis of cyclic ADP-ribose by human leukocyte antigen CD38 and inhibition of the hydrolysis by ATP. J. Biol. Chem. 268, 26052–26054.
- 81) Koguma, T., Takasawa, S., Tohgo, A., Karasawa, T., Furuya, Y., Yonekura, H. et al. (1994) Cloning and characterization of cDNA encoding rat ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase (homologue to human CD38) from islets of Langerhans. Biochim. Biophys. Acta 1223, 160–162.
- 82) Tohgo, A., Takasawa, S., Noguchi, N., Koguma, T., Nata, K., Sugimoto, T. et al. (1994) Essential cysteine residues for cyclic ADP-ribose synthesis and hydrolysis by CD38. J. Biol. Chem. 272, 3879–3882.
- 83) Takasawa, S., Ishida, A., Nata, K., Nakagawa, K., Noguchi, N., Tohgo, A. et al. (1995) Requirement of calmodulin-dependent protein kinase II in cyclic ADP-ribose-mediated intracellular Ca2+ mobilization. J. Biol. Chem. 270, 30257–30259.
- 84) Kato, I., Takasawa, S., Akabane, A., Tanaka, O., Abe, H., Takamura, T. et al. (1995) Regulatory role of CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) in insulin secretion by glucose in pancreatic β cells: Enhanced insulin secretion in CD38 transgenic mice. J. Biol. Chem. 270, 30045–30050.
- 85) Okamoto, H., Takasawa, S. and Tohgo, A. (1995) New aspects of the physiological significance of NAD, poly ADP-ribose and cyclic ADP-ribose. Biochimie 77, 356–363.
- 86) Tohgo, A., Munakata, H., Takasawa, S., Nata, K., Akiyama, T., Hayashi, N. et al. (1997) Lysine 129 of CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) participates in the binding of ATP to inhibit the cyclic ADP-ribose hydrolase. J. Biol. Chem. 272, 3879–3882.
- 87) Noguchi, N., Takasawa, S., Nata, K., Tohgo, A., Kato, I., Ikehata, F. et al. (1997) Cyclic ADP-ribose binds to FK506-binding protein 12.6 to release Ca2+ from islet microsomes. J. Biol. Chem. 272, 3133–3136.
- 88) Okamoto, H., Takasawa, S. and Nata, K. (1997) The CD38—cyclic ADP-ribose signaling system in insulin secretion: Molecular basis and clinical implications. Diabetologia 40, 1485–1491.
- 89) Takasawa, S., Akiyama, T., Nata, K., Kuroki, M., Tohgo, A., Noguchi, N. et al. (1998) Cyclic ADP-ribose and inositol 1,4,5-trisphosphate as alternate second messengers for intracellular Ca2+ mobilization in normal and diabetic β-cells. J. Biol. Chem. 273, 2497–2500.
- 90) Okamoto, H., Takasawa, S., Tohgo, A. Nata, K., Kato, I. and Noguchi, N. (1997) Synthesis and hydrolysis of cyclic ADP-ribose by human leukocyte antigen CD38: Inhibition of hydrolysis by ATP and physiological significance. In Methods in Enzymology Volume 280, Vitamins and Coenzymes Part J (eds. McCormick, D.B., Suttie, J.W. and Wagner, C.). Academic Press, San Diego, London, Boston, New York, Sydney, Tokyo, Toronto, pp. 306–318.
- 91) Kato, I., Yamamoto, Y., Fujimura, M., Noguchi, N., Takasawa, S. and Okamoto, H. (1999) CD38 disruption impairs glucose-induced increases in cyclic ADP-ribose, [Ca2+]i and insulin secretion. J. Biol. Chem. 274, 1869–1872.
- 92) Okamoto, H. (1999) Cyclic ADP-ribose-mediated insulin secretion and Reg, regenerating gene. J. Mol. Med. (Berl.) 77, 74–79.
- 93) Okamoto, H., Takasawa, S., Nata, K., Kato, I., Tohgo, A. and Noguchi, N. (2000) Physiological and pathological significance of the CD38-cyclic ADP-ribose signaling system. Chem. Immunol. 75, 121–145.
- 94) Takasawa, S. and Okamoto, H. (2002) The CD38-cyclic ADP-ribose signal system in pancreatic β-cells. The discovery and biological significance of a novel signal system in mammalian cells. In Cyclic ADP-Ribose and NAADP: Structure, Metabolism and Functions (ed. Lee, H.C.). Kluwer Academic Publishers, Dordrecht, Netherlands, pp. 269–299.
- 95) Okamoto, H. and Takasawa, S. (2002) CD38. In Wiley Encyclopedia of Molecular Medicine, Vol. 5 John Wiley & Sons, Inc., Hoboken, NJ, pp. 601–604.
- 96) Okamoto, H. and Takasawa, S. (2002) Recent advances in the Okamoto model. The CD38-cyclic ADP-ribose signal system and the regenerating gene protein (Reg)-Reg receptor system in β-cells. Diabetes 51, S462–S473.
- 97) Noguchi, N., Yoshikawa, T., Ikeda, T., Takahashi, I., Shervani, N.J., Uruno, A. et al. (2008) FKBP12.6 disruption impairs glucose-induced insulin secretion. Biochem. Biophys. Res. Commun. 371, 735–740.
- 98) Takasawa, S., Kuroki, M., Nata, K., Noguchi, N., Ikeda, T., Yamauchi, A. et al. (2010) A novel ryanodine receptor expressed in pancreatic islets by alternative splicing from type 2 ryanodine receptor gene. Biochem. Biophys. Res. Commun. 397, 140–145.
- 99) Ikeda, T., Takasawa, S., Noguchi, N., Nata, K., Yamauchi, A., Takahashi, I. et al. (2012) Identification of a major enzyme for the synthesis and hydrolysis of cyclic ADP-ribose in amphibian cells and evolutional conservation of the enzyme from human to invertebrate. Mol. Cell. Biochem. 366, 69–80.
- 100) Takasawa, S. and Okamoto, H. (2002) Pancreatic β-cell death, regeneration and insulin secretion: Roles of poly(ADP-ribose) polymerase and cyclic ADP-ribose. Int. J. Exp. Diabetes Res. 3, 79–96.
- 101) Okamoto, H., Takasawa, S. and Yamamoto, Y. (2017) From insulin synthesis to secretion: Alternative splicing of type 2 ryanodine receptor gene is essential for insulin secretion in pancreatic β cells. Int. J. Biochem. Cell Biol. 91, 176–183.
- 102) Okamoto, H. and Takasawa, S. (2003) Recent advances in physiological and pathological significance of NAD+ metabolites: roles of poly(ADP-ribose) and cyclic ADP-ribose in insulin secretion and diabetogenesis. Nutr. Res. Rev. 16, 253–266.
- 103) Okamoto, H., Takasawa, S. and Sugawara, A. (2014) The CD38-cyclic ADP-ribose system in mammals: historical background, pathophysiology and perspective. Messenger (Los Angel.) 3, 27–34.
- 104) Galione, A. (1993) Cyclic ADP-ribose: A new way to control calcium. Science 259, 325–326.
- 105) Clapper, D.L., Walseth, T.F., Dargie, P.J. and Lee, H.C. (1987) Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J. Biol. Chem. 262, 9561–9568.
- 106) An, N.H., Han, M.K., Um, C., Park, B.H., Park, B.J., Kim, H.K. et al. (2001) Significance of ecto-cyclase activity of CD38 in insulin secretion of mouse pancreatic islet cells. Biochem. Biophys. Res. Commun. 282, 781–786.
- 107) Takasawa, S., Nata, K., Yonekura, H. and Okamoto, H. (1993) Cyclic ADP-ribose in β cells. Science 262, 584–586.
- 108) Howard, M., Grimaldi, J.C., Bazan, J.F., Bazan, J.F., Lund, F.E., Santos-Argumedo, L. et al. (1993) Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 262, 1056–1059.
- 109) Summerhill, R.J., Jackson, D.G. and Galione, A. (1993) Human lymphocyte antigen CD38 catalyzes the production of cyclic ADP-ribose. FEBS Lett. 335, 231–233.
- 110) Zocchi, E., Franco, L., Bebattii, U., Bargellesi, A., Malavasi, F., Lee, H.C. et al. (1993) A single protein immunologically identified as CD38 displays NAD+ glycohydrolase, ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase activities at the outer surface of human erythrocytes. Biochem. Biophys. Res. Commun. 196, 1459–1465.
- 111) Nata, K., Takamura, T., Karasawa, T., Kumagai, T., Hashioka, W., Tohgo, A. et al. (1997) Human gene encoding CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase): organization, nucleotide sequence and alternative splicing. Gene 186, 285–292.
- 112) Pirsch, J.D., Miller, J., Deierhoi, M.H., Vincenti, F. and Filo, R.S. (1997) A comparison of tacrolimus (FK506) and cyclosporine for immunosuppression after cadaveric renal transplantation: FK506 Kidney Transplant Study Group. Transplantation 63, 977–983.
- 113) Seino, Y., Fukushima, M. and Yabe, D. (2010) GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 1, 8–23.
- 114) Kim, B.J., Park, K.H., Yim, C.Y., Takasawa, S., Okamoto, H., Im, M.J. et al. (2008) Generation of NAADP and cADPR by glucagon-like peptide-1 evokes Ca2+ signal that is essential for insulin secretion in mouse pancreatic islets. Diabetes 57, 868–878.
- 115) Yagui, K., Shimada, F., Miura, M., Hashimoto, N., Suzuki, Y., Tokuyama, Y. et al. (1998) A missense mutation in the CD38 gene, a novel factor for insulin secretion: Association with Type II diabetes mellitus in Japanese subjects and evidence of abnormal function when expressed in vitro. Diabetologia 41, 1024–1028.
- 116) Ikehata, F., Satoh, J., Nata, K., Tohgo, A., Nakazawa, T., Kato, I. et al. (1998) Autoantibodies against CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) which impair glucose-induced insulin secretion in non-insulin dependent diabetes patients. J. Clin. Invest. 102, 395–401.
- 117) Pupilli, C., Giannini, S., Marchetti, P., Lupi, R., Antonelli, A., Malavasi, F. et al. (1999) Autoanitibodies to CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) in Caucasian patients with diabetes: Effects on insulin release from human islets. Diabetes 48, 2309–2315.
- 118) Mallone, R., Ortolan, E., Baj, G., Funaro, A., Giunti, S., Lillaz, E. et al. (2001) Autoantibody response to CD38 in Caucasian patients with type 1 and type 2 diabetes: immunological and genetic characterization. Diabetes 50, 752–762.
- 119) Antonelli, A., Baj, G., Marchetti, P., Fallahi, P., Surico, N., Pupilli, C. et al. (2001) Human anti-CD38 autoantibodies raise intracellular calcium and stimulate insulin release in human pancreatic islets. Diabetes 50, 985–991.
- 120) Antonelli, A., Tuomi, T., Nannipieri, M., Fallahi, P., Nesti, C., Okamoto, H. et al. (2002) Autoimmunity to CD38 and GAD in Type I and Type II diabetes: CD38 and HLA genotypes and clinical phenotypes. Diabetologia 45, 1298–1306.
- 121) Berridge, M.J. and Irvine, R.F. (1984) Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature 312, 315–321.
- 122) Streb, H., Irvine, R.F., Berridge, M.J. and Schulz, I. (1983) Release of Ca2+ from a nonmitochodrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature 306, 67–69.
- 123) Islam, M.S., Larsson, O., Berggren, P.O., Takasawa, S., Nata, K., Yonekura, H. et al. (1993) Cyclic ADP-ribose in β cells. Science 262, 584–586.
- 124) Rutter, G.A., Theler, J.-M. and Wollheim, C.B. (1994) Ca2+ stores in insulin-secreting cells: lack of effect of cADP ribose. Cell Calcium 16, 71–80.
- 125) Webb, D.-L., Islam, M.S., Efanov, A.M., Brown, G., Kohler, M., Larsson, O. et al. (1996) Insulin exocytosis and glucose-mediated increase in cytoplasmic free Ca2+ concentration in the pancreatic β-cell are independent of cyclic ADP-ribose. J. Biol. Chem. 271, 19074–19079.
- 126) Islam, M.S. and Berggren, P.O. (1997) Cyclic ADP-ribose and the pancreatic beta cell: where do we stand? Diabetologia 40, 1480–1484.
- 127) Matsuoka, T., Kajimoto, Y., Watada, H., Umayahara, Y., Kubota, M., Kawamori, R. et al. (1995) Expression of CD38 gene, but not of mitochondrial glycerol-3-phosphate dehydrogenase gene, is impaired in pancreatic islets of GK rats. Biochem. Biophys. Res. Commun. 214, 239–246.
- 128) Mitchell, K.J., Pinton, P., Varadi, A., Tacchetti, C., Ainscow, E.K., Pozzan, T. et al. (2001) Dense core secretory vesicles revealed as a dynamic Ca2+ store in neuroendocrine cells with a vesicle-associated membrane protein aequorin chimaera. J. Cell Biol. 155, 41–51.
- 129) Varadi, A. and Rutter, G.A. (2002) Dynamic imaging of endoplasmic reticulum Ca2+ concentration in insulin-secreting MIN6 cells using recombinant targeted cameleons: Roles of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA)-2 and ryanodine receptors. Diabetes 51, S190–S201.
- 130) Ohta, Y., Kitanaka, A., Mihara, K., Imataki, O., Ohnishi, H., Tanaka, T. et al. (2011) Expression of CD38 with intracellular enzymic activity: a possible explanation for the insulin release induced by cADPR. Mol. Cell. Biochem. 352, 293–299.
- 131) Makino, M., Itaya-Hironaka, A., Yamauchi, A., Sakuramoto-Tsuchida, S. and Takasawa, S. (2020) Tissue-specific alternative splicing of type 2 ryanodine receptor gene affects insulin biosynthesis in pancreatic β cells. Diabetologia 63 (Suppl. 1), Abstract No. 385.
- 132) Mount, S.M. (1982) A catalogue of splice junction sequences. Nucleic Acids Res. 10, 459–472.
- 133) Nakai, K. and Sakamoto, H. (1994) Construction of a novel database containing aberrant splicing mutations of mammalian genes. Gene 141, 171–177.
- 134) Sakai, N., Santmaria-Fojo, S., Yamashita, S., Matsuzawa, Y. and Brewer, H.B. Jr. (1996) Exon 10 skipping caused by intron 10 splice donor site mutation in cholesteryl ester transfer protein gene results in abnormal downstream splice site selection. J. Lipid Res. 37, 2065–2073.
- 135) Hua, S.Y., Tokimasa, T., Takasawa, S., Furuya, Y., Nohmi, M., Okamoto, H. et al. (1994) Cyclic ADP-ribose modulates Ca2+ release channels for activation by physiological Ca2+ entry in bullfrog sympathetic neurons. Neuron 12, 1073–1079.
- 136) Ebihara, S., Sasaki, T., Hida, W., Kikuchi, Y., Oshiro, T., Shimura, S. et al. (1997) Role of cyclic ADP-ribose in ATP-activated potassium currents in alveolar macrophages. J. Biol. Chem. 272, 16023–16029.
- 137) Sasamori, K., Sasaki, T., Takasawa, S., Tamada, T., Nara, M., Irokawa, T. et al. (2004) Cyclic ADP-ribose, a putative Ca2+-mobilizing second messenger, operates in submucosal gland acinar cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 287, L69–L78.
- 138) Ota, H., Tamaki, S., Itaya-Hironaka, A., Yamauchi, A., Sakuramoto-Tsuchida, S., Morioka, T. et al. (2012) Attenuation of glucose-induced insulin secretion by intermittent hypoxia via down-regulation of CD38. Life Sci. 90, 206–211.
- 139) Ota, H., Fujita, Y., Yamauchi, M., Muro, S., Kimura, H. and Takasawa, S. (2019) Relationship between intermittent hypoxia and Type 2 diabetes in sleep apnea syndrome. Int. J. Mol. Sci. 20, 4756.
- 140) Wu, Y., Kuzma, J., Maréchal, E., Graeff, R., Lee, H.C., Foster, R. et al. (1997) Abscisic acid signalling through cyclic ADP-ribose in plats. Science 278, 2126–2130.
- 141) Leckie, C.P., McAinsh, M.R., Allen, G.J., Sanders, D. and Hetherington, A.M. (1998) Abscisic acid-induced stomatal closure mediated by cyclic ADP-ribose. Proc. Natl. Acad. Sci. U.S.A. 95, 15837–15842.
- 142) Dodd, A.N., Gardner, M.J., Hotta, C.T., Hubbard, K.E., Delchau, N., Love, J. et al. (2007) The Arabidopsis circadian clock incorporates a cADPR-based feedback loop. Science 318, 1789–1792.
- 143) Fellner, S.K. and Parker, L. (2005) Endothelin-1, superoxide and adeninediphosphate ribose cyclase in shark vascular smooth muscle. J. Exp. Biol. 208, 1045–1052.
- 144) Fukushi, Y., Kato, I., Takasawa, S., Sasaki, T., Ong, B.H., Ohsaga, A. et al. (2001) Identification of cyclic ADP-ribose-dependent mechanisms in pancreatic muscarinic Ca2+ signaling using CD38 knockout mice. J. Biol. Chem. 276, 649–655.
- 145) Higashida, H., Yokoyama, S., Hashii, M., Taketo, M., Higashida, M., Takayasu, T. et al. (1997) Muscarinic receptor-mediated dual regulation of ADP-ribosyl cyclase in NG108-15 neuronal cell membranes analyzed by thin layer chromatography. J. Biol. Chem. 272, 31272–31277.
- 146) Takahashi, J., Kagaya, Y., Kato, I., Ohta, J., Isoyama, S., Miura, M. et al. (2003) Deficit of CD38/cyclic ADP-ribose is differentially compensated in hearts by gender. Biochem. Biophys. Res. Commun. 312, 434–440.
- 147) Xin, H.-B., Senbonmatsu, T., Cheng, D.-S., Wang, Y.-X., Copello, J.A., Ji, G.-J. et al. (2002) Oestrogen protects FKBP12.6 null mice from cardiac hypertrophy. Nature 416, 334–337.
- 148) Yano, M., Ono, K., Ohkusa, T., Suetsugu, M., Kohno, M., Hisaoka, T. et al. (2000) Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca2+ leak through ryanodine receptor in heart failure. Circulation 102, 2131–2136.
- 149) Jin, D., Liu, H.-X., Hirai, H., Torashima, T., Nagai, T., Lopatina, O. et al. (2007) CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 446, 41–45.
- 150) Karasawa, T., Takasawa, S., Yamakawa, K., Yonekura, H., Okamoto, H. and Nakamura, S. (1995) NAD+-glycohydrolase from Streptococcus pyogenes shows cyclic ADP-ribose forming activity. FEMS Microbiol. Lett. 130, 201–204.
- 151) Munesue, T., Yokoyama, S., Nakamura, K., Anitha, A., Yamada, K., Hayashi, K. et al. (2010) Two genetic variants of CD38 in subjects with autism spectrum disorder and controls. Neurosci. Res. 67, 181–191.
- 152) Enami, N., Itaya-Hironaka, A., Yamauchi, A., Sakuramoto-Tsuchida, S., Takasawa, S. and Takahashi, Y. (2015) The CD38 genotype (rs1800561 (4693C>T): R140W) is associated with an increased risk of admission to the neonatal intensive care unit. Early Hum. Dev. 91, 467–470.
- 153) rs1800561 in dbSNP (The Single Nucleotide Polymorphism Database) (2021) Release Version: 20201027095038. NCBI, Bethesda, MD, USA. https://www.ncbi.nlm.nih.gov/snp/rs1800561#frequency_tab.
- 154) Yonemura, Y., Takashima, T., Miwa, K., Miyazaki, I., Yamamoto, H. and Okamoto, H. (1984) Amelioration of diabetes mellitus in partially depancreatized rats by poly(ADP-ribose) synthetase inhibitors: Evidence of islet B-cell regeneration. Diabetes 33, 401–404.
- 155) Okamoto, H., Yamamoto, H., Takasawa, S., Terazono, K., Shiga, K. and Kitagawa, M. (1988) Molecular mechanism of degeneration, oncogenesis and regeneration of pancreatic B-cells of islets of Langerhans. In Frontiers in Diabetes Research: Lessons from Animal Diabetes II (eds. Shafrir, E. and Renold, A.E.). John Libbery & Company, London, pp. 149–157.
- 156) Okamoto, H., Yamamoto, H. and Yonemura, Y. (1985) Poly(ADP-ribose) synthetase inhibitors induce islet B-cell regeneration in partially depancreatized rats. In ADP-ribosylation of Proteins (eds. Althaus, F.R., Hilz, H. and Shall, S.). Springer-Verlag, Berlin, Heidelberg, pp. 410–416.
- 157) Terazono, K., Yamamoto, H., Takasawa, S., Shiga, K., Yonemura, Y., Tochino, Y. et al. (1988) A novel gene activated in regenerating islets. J. Biol. Chem. 263, 2111–2114.
- 158) Okamoto, H. (1985) The role of poly(ADP-ribose) synthetase in the development of insulin-dependent diabetes and islet B-cell regeneration. Biomed. Biochim. Acta 44, 15–20.
- 159) Okamoto, H. (1991) Islet B-cell regeneration and reg genes. In Diabetes 1991 (Proceedings of the 14th International Diabetes Federation Congress, Washington, D.C., 23–28 June 1991) (eds. Rifkin, H., Colwell, J.A. and Taylor, S.I.). Excerpta Medica, Amsterdam, London, New York, Tokyo, pp. 268–270.
- 160) Terazono, K., Watanabe, T. and Yonemura, Y. (1990 & 2008) A novel gene, reg, expressed in regenerating islets. In Molecular Biology of the Islets of Langerhans (ed. Okamoto, H.). Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, pp. 301–313.
- 161) Terazono, K., Uchiyama, M., Ide, M., Watanabe, T., Yonekura, H., Yamamoto, H. et al. (1990) Expression of reg protein in rat regenerating islets and its co-localization with insulin in the Beta cell secretory granules. Diabetologia 33, 250–252.
- 162) Yonekura, H., Unno, M., Watanabe, T., Moriizumi, S., Suzuki, Y., Miyashita, H. et al. (1994) Structure and expression of reg gene family in pancreatic islets. In Insulin Secretion and Pancreatic B-cell Research (eds. Flatt, P.R. and Lenzen, S.). Smith-Gordon, London, pp. 581–588.
- 163) Okamoto, H. (1997) New aspects to the functioning and regeneration of pancreatic β-cells: Cyclic ADP-ribose and Reg gene. In Physiology and Pathophysiology of the Islets of Langerhans (ed. Soria, B.). Plenum Press, New York, pp. 301–311.
- 164) Okamoto, H. (1999) The Reg gene family and Reg proteins: With special attention to the regeneration of pancreatic β-cells. J. Hepatobiliary Pancreat. Surg. 6, 254–262.
- 165) Kobayashi, S., Akiyama, T., Nata, K., Abe, M., Tajima, M., Shervani, N.J. et al. (2000) Identification of a receptor for Reg (Regenerating Gene) protein, a pancreatic β-cell regeneration factor. J. Biol. Chem. 275, 10723–10726.
- 166) Akiyama, T., Takasawa, S., Nata, K., Kobayashi, S., Abe, M., Shervani, N.J. et al. (2001) Activation of Reg gene, a gene for insulin-producing β-cell regeneration: Poly(ADP-ribose) polymerase binds Reg promoter and regulates the transcription by autopoly(ADP-ribosyl)ation. Proc. Natl. Acad. Sci. U.S.A. 98, 48–53.
- 167) Takasawa, S., Ikeda, T., Akiyama, T., Nata, K., Nakagawa, K., Shervani, N.J. et al. (2006) Cyclin D1 activation through ATF-2 in Reg-induced pancreatic β-cell regeneration. FEBS Lett. 580, 585–591.
- 168) Watanabe, T., Yonekura, H., Terazono, K., Yamamoto, H. and Okamoto, H. (1990) Complete nucleotide sequence of human reg gene and its expression in normal and tumoral tissues: The reg protein, pancreatic stone protein, and pancreatic thread protein are one and the same product of the gene. J. Biol. Chem. 265, 7432–7439.
- 169) Watanabe, T., Yonemura, Y., Yonekura, H., Suzuki, Y., Miyashita, H., Sugiyama, K. et al. (1994) Pancreatic beta-cell replication and amelioration of surgical diabetes by Reg protein. Proc. Natl. Acad. Sci. U.S.A. 91, 3589–3592.
- 170) Gross, D.J., Weiss, L., Reibstein, I., van den Brand, J., Okamoto, H., Clark, A. et al. (1998) Amelioration of diabetes in nonobese diabetic mice with advanced disease by Linomide-induced immunoregulation combined with Reg protein treatment. Endocrinology 139, 2369–2374.
- 171) Unno, M., Noguchi, N., Narushima, Y., Nata, K., Akiyama, T., Ikeda, T. et al. (2002) Production and characterization of Reg knockout mice: Reduced proliferation of pancreatic β-cells in Reg knockout mice. Diabetes 51, S478–S483.
- 172) Rane, S.G., Dubus, P., Mettus, R.V., Galbreath, E.J., Boden, G., Reddy, E.P. et al. (1999) Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat. Genet. 22, 44–52.
- 173) Hill, T., Krougly, O., Nikoopour, E., Bellemore, S., Lee-Chan, E., Fouser, L.A. et al. (2013) The involvement of interleukin-22 in the expression of pancreatic beta cell regenerative Reg genes. Cell Regen. (Lond.) 2, 2.
- 174) Nakagawa, K., Takasawa, S., Nata, K., Yamauchi, A., Itaya-Hironaka, A., Ota, H. et al. (2013) Prevention of Reg I-induced β-cell apoptosis by IL-6/dexamethasone through activation of HGF gene regulation. Biochim. Biophys. Acta 1833, 2988–2995.
- 175) Yamauchi, A., Itaya-Hironaka, A., Sakuramoto-Tsuchida, S., Takeda, M., Yoshimoto, K., Miyaoka, T. et al. (2015) Synergistic activations of REG Iα and REG Iβ promoters by IL-6 and glucocorticoids through JAK/STAT pathway in human pancreatic β cells. J. Diabetes Res. 2015, 173058.
- 176) Nirodi, C., NagDas, S., Gygi, S.P., Olson, G., Aebersold, R. and Richmond, A. (2001) A role for poly(ADP-ribose) polymerase in the transcriptional regulation of the melanoma growth stimulatory activity (CXCL1) gene expression. J. Biol. Chem. 276, 9366–9374.
- 177) Chang, W.J. and Alvarez-Gonzalez, R. (2001) The sequence-specific DNA binding of NF-κB is reversibly regulated by automodification reaction of poly(ADP-ribose) polymerase 1. J. Biol. Chem. 276, 47664–47670.
- 178) Chiba-Falek, O., Kowalak, J.A., Smulson, M.E. and Nussbaum, R.L. (2005) Regulation of α-synuclein expression by poly (ADP Ribose) polymerase-1 (PARP-1) binding to the NACP-Rep1 polymorphic site upstream of the SNCA gene. Am. J. Hum. Genet. 76, 478–492.
- 179) Beranger, G.E., Momier, D., Guigonis, J.M., Samson, M., Carle, G.F. and Scimeca, J.C. (2007) Differential binding of poly(ADP-Ribose) polymerase-1 and JunD/Fra2 accounts for RANKL-induced Tcirg1 gene expression during osteoclastogenesis. J. Bone Miner. Res. 22, 975–983.
- 180) Ambrose, H.E., Papadopoulou, V., Beswick, R.W. and Wagner, S.D. (2007) Poly-(ADP-ribose) polymerase-1 (Parp-1) binds in a sequence-specific manner at the Bcl-6 locus and contributes to the regulation of Bcl-6 transcription. Oncogene 26, 6244–6252.
- 181) Beranger, G.E., Momier, D., Rochet, N., Carle, G.F. and Scimeca, J.C. (2008) Poly(adp-ribose) polymerase-1 regulates Tracp gene promoter activity during RANKL-induced osteoclastogenesis. J. Bone Miner. Res. 23, 564–571.
- 182) Chu, S., Xu, H., Ferro, T.J. and Rivera, P.X. (2007) Poly(ADP-ribose) polymerase-1 regulates vimentin expression in lung cancer cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L1127–L1134.
- 183) Lin, Y., Tang, X., Zhu, Y., Shu, T. and Han, X. (2011) Identification of PARP-1 as one of the transcription factors binding to the repressor element in the promoter region of COX-2. Arch. Biochem. Biophys. 505, 123–129.
- 184) Pelham, C., Jimenez, T., Rodova, M., Rudolph, A., Chipps, E. and Islam, M.R. (2013) Regulation of HFE expression by poly(ADP-ribose) polymerase-1 (PARP1) through an inverted repeat DNA sequence in the distal promoter. Biochim. Biophys. Acta 1829, 1257–1265.
- 185) Okada, H., Inoue, T., Kikuta, T., Kato, N., Kanno, Y., Hirosawa, N. et al. (2008) Poly(ADP-ribose) polymerase-1 enhances transcription of the profibrotic CCN2 gene. J. Am. Soc. Nephrol. 19, 933–942.
- 186) Ono, T., Kaneda, T., Muto, A. and Yoshida, T. (2009) Positive transcriptional regulation of the human micro opioid receptor gene by poly(ADP-ribose) polymerase-1 and increase of its DNA binding affinity based on polymorphism of G−172 → T. J. Biol. Chem. 284, 20175–20183.
- 187) Peng, H., Zhu, Q.S., Zhong, S. and Levy, D. (2015) Transcription of the human microsomal epoxide hydrolase gene (EPHX1) is regulated by PARP-1 and histone H1.2. association with sodium-dependent bile acid transport. PLoS One 10, e0125318.
- 188) Tian, Y.N., Chen, H.D., Tian, C.Q., Wang, Y.Q. and Miao, Z.H. (2020) Polymerase independent repression of FoxO1 transcription by sequence-specific PARP1 binding to FoxO1 promoter. Cell Death Dis. 11, 71.
- 189) Soldatenkov, V.A., Chasovskikh, S., Potaman, V.N., Trofimova, I., Smulson, M.E. and Dritschilo, A. (2002) Transcriptional repression by binding of poly(ADP-ribose) polymerase to promoter sequences. J. Biol. Chem. 277, 665–670.
- 190) Unno, M., Yonekura, H., Nakagawara, K., Watanabe, T., Miyashita, H., Moriizumi, S. et al. (1993) Structure, chromosomal localization, and expression of mouse reg genes, reg I and reg II. A novel type of reg gene, reg II, exists in the mouse genome. J. Biol. Chem. 268, 15974–15982.
- 191) Takasawa, S. (2016) Regenerating gene (REG) product and its potential clinical usage. Expert Opin. Ther. Targets 20, 541–550.
- 192) Moriizumi, S., Watanabe, T., Unno, M., Nakagawara, K., Suzuki, Y., Miyashita, H. et al. (1994) Isolation, structural determination and expression of a novel reg gene, human reg Iβ. Biochim. Biophys. Acta 1217, 199–202.
- 193) Lasserre, C., Simon, M.T., Ishikawa, H., Diriong, S., Nguyen, V.C., Christa, L. et al. (1994) Structural organization and chromosomal localization of a human gene (HIP/PAP) encoding a C-type lectin overexpressed in primary liver cancer. Eur. J. Biochem. 224, 29–38.
- 194) Dusetti, N.J., Frigerio, J.-M., Fox, M.F., Swallow, D.M., Dagon, J.-C. and Iovanna, J.L. (1994) Molecular cloning, genomic organization, and chromosomal localization of the human pancreatitis-associated protein (PAP) gene. Genomics 19, 108–114.
- 195) Nata, K., Liu, Y., Xu, L., Ikeda, T., Akiyama, T., Noguchi, N. et al. (2004) Molecular cloning, expression and chromosomal localization of a novel human REG family gene, REG III. Gene 340, 161–170.
- 196) Miyashita, H., Nakagawara, K., Mori, M., Narushima, Y., Noguchi, N., Moriizumi, S. et al. (1995) Human REG family genes are tandemly ordered in a 95-kilobase region of chromosome 2p12. FEBS Lett. 377, 429–433.
- 197) Hartupee, J.C., Zhang, H., Bonaldo, M.F., Soares, M.B. and Dieckgraefe, B.K. (2001) Isolation and characterization of a cDNA encoding a novel member of the human regenerating protein family: Reg IV. Biochim. Biophys. Acta 1518, 287–293.
- 198) Abe, M., Nata, K., Akiyama, T., Shervani, N.J., Kobayashi, S., Tomioka-Kumagai, T. et al. (2000) Identification of a novel Reg family gene, Reg IIIδ, and mapping of all three types of Reg family genes in a 75 kilobase mouse genomic region. Gene 246, 111–122.
- 199) Shervani, N.J., Takasawa, S., Uchigata, Y., Akiyama, T., Nakagawa, K., Noguchi, N. et al. (2004) Autoantibodies to REG, a beta-cell regeneration factor, in diabetic patients. Eur. J. Clin. Invest. 34, 752–758.
- 200) Omori, K., Matsuda, T., Ferreri, K., Todorov, I., Okamoto, H., Takasawa, S. et al. (2003) Induction of Reg gene expression, in vitro, in human islets. Transplantation 76, 126.
- 201) Lu, Y., Ponton, A., Okamoto, H., Takasawa, S., Herrera, P.L. and Liu, J.L. (2006) Activation of the Reg family genes by pancreatic-specific IGF-I gene deficiency and after streptozotocin-induced diabetes in mouse pancreas. Am. J. Physiol. Endocrinol. Metab. 291, E50–E58.
- 202) Planas, R., Alba, A., Carrillo, J., Puertas, M.C., Ampudia, R., Pastor, X. et al. (2006) Reg (Regenerating gene) overexpression in islets from NOD mice with accelerated diabetes: role of INFβ. Diabetologia 49, 2379–2387.
- 203) Huszarik, K., Wright, B., Keller, C., Nikoopour, E., Krougly, O., Lee-Chan, E. et al. (2010) Adjuvant immunotherapy increases β cell regenerative factor Reg2 in the pancreas of diabetic mice. J. Immunol. 185, 5120–5129.
- 204) Kapur, R., Højfeldt, T.W., Højfeldt, T.W., Rønn, S.G., Karlsen, A.E. and Heller, R.S. (2012) Short-term effects of INGAP and Reg family peptides on the appearance of small β-cells clusters in non-diabetic mice. Islets 4, 40–48.
- 205) Ota, H., Itaya-Hironaka, A., Yamauchi, A., Sakuramoto-Tsuchida, S., Miyaoka, T., Fujimura, T. et al. (2013) Pancreatic β cell proliferation by intermittent hypoxia via up-regulation of Reg family genes and HGF gene. Life Sci. 93, 664–672.
- 206) Aida, K., Saitoh, S., Nishida, Y., Yokota, S., Ohno, S., Mao, X. et al. (2014) Distinct cell clusters touching islet cells induce islet cell replication in association with over-expression of regenerating gene (REG) protein in fulminant Type 1 diabetes. PLoS One 9, e95110.
- 207) Calderari, S., Irminger, J.C., Giroix, M.H., Ehses, J.A., Gangnerau, M.N., Coulaud, J. et al. (2014) Regenerating 1 and 3b gene expression in the pancreas of Type 2 diabetic Goto-Kakizaki (GK) rats. PLoS One 9, e90045.
- 208) Aida, K., Kobayashi, T., Takeshita, A., Jimbo, E., Nishida, Y., Yagihashi, S. et al. (2018) Crucial role of Reg I from acinar-like cell cluster touching with islets (ATLASTIS) on mitogenesis of beta cells in EMC virus-induced diabetic mice. Biochem. Biophys. Res. Commun. 503, 963–969.
- 209) Satomura, Y., Sawabu, N., Ohta, H., Watanabe, H., Yamakawa, O., Motoo, Y. et al. (1993) The immunohistochemical evaluation of PSP/reg-protein in normal and diseased human pancreatic tissues. Int. J. Pancreatol. 13, 59–67.
- 210) Satomura, Y., Sawabu, N., Mouri, I., Yamakawa, O., Watanabe, H., Motoo, Y. et al. (1995) Measurement of serum PSP/reg-protein concentration in various diseases with a newly developed enzyme-linked immunosorbent assay. J. Gastroenterol. 30, 643–650.
- 211) Kadowaki, Y., Ishihara, S., Miyaoka, Y., Rumi, M., Sato, H., Kazumori, H. et al. (2002) Reg protein is overexpressed in gastric cancer cells, where it activates a signal transduction pathway that converges on ERK1/2 to stimulate growth. FEBS Lett. 530, 59–64.
- 212) Ogawa, H., Fukushima, K., Naito, H., Funayama, Y., Unno, M., Takahashi, K. et al. (2003) Increased expression of HIP/PAP and regenerating gene III in human inflammatory bowel disease and a murine bacterial reconstitution model. Inflamm. Bowel Dis. 9, 162–170.
- 213) Miyaoka, Y., Kadaowaki, Y., Ishihara, S., Ose, T., Fukuhara, H., Kazumori, H. et al. (2004) Transgenic overexpression of Reg protein caused gastric cell proliferation and differentiation along parietal cell and chief cell lineages. Oncogene 23, 3572–3579.
- 214) Sekikawa, A., Fukui, H., Fujii, S., Takeda, J., Nanakin, A., Hisatsune, H. et al. (2005) REG Iα protein may function as a trophic and/or anti-apoptotic factor in the development of gastric cancer. Gastroenterology 128, 642–653.
- 215) Ose, T., Kadowaki, Y., Fukuhara, H., Kazumori, H., Ishihara, S., Udagawa, J. et al. (2007) Reg I-knockout mice reveal its role in regulation of cell growth that is required in generation and maintenance of the villous structure of small intestine. Oncogene 26, 349–359.
- 216) Fukuhara, H., Kadowaki, Y., Ose, I., Ishihara, S., Takasawa, S. and Kinoshita, Y. (2010) In vivo evidence for the role of Reg I in gastric regeneration: Transgenic overexpression of Reg accelerates the healing of experimental gastric ulcers. Lab. Invest. 190, 555–565.
- 217) Vives-Pi, M., Takasawa, S., Pujol-Autonell, I., Planas, R., Cabré, E., Ojanguren, I. et al. (2013) Biomarkers for diagnosis and monitoring of Celiac disease. J. Clin. Gastroenterol. 47, 308–313.
- 218) Sun, C., Fukui, H., Hara, K., Kitayama, Y., Eda, H., Yang, M. et al. (2015) Expression of Reg family genes in the gastrointestinal tract of mice treated with indomethacin. Am. J. Physiol. Gastrointest. Liver Physiol. 308, G736–G744.
- 219) Kitayama, Y., Fukui, H., Hara, K., Eda, H., Kodani, M., Yang, M. et al. (2016) Role of regenerating gene I in claudin expression and barrier function in the small intestine. Transl. Res. 173, 92–100.
- 220) Tsuchida, C., Sakuramoto-Tsuchida, S., Takeda, M., Itaya-Hironaka, A., Yamauchi, A., Misu, M. et al. (2017) Expression of REG family genes in human inflammatory bowel diseases and its regulation. Biochem. Biophys. Rep. 12, 198–205.
- 221) Takasawa, S., Tsuchida, C., Sakuramoto-Tsuchida, S., Takeda, M., Itaya-Hironaka, A., Yamauchi, A. et al. (2018) Expression of human REG family genes in inflammatory bowel disease and their molecular mechanism. Immunol. Res. 66, 800–805.
- 222) Takasawa, S., Tsuchida, C., Sakuramoto-Tsuchida, S., Yamauchi, A., Makino, M., Takeda, Y. et al. (2021) Molecular biological analysis of Reg family genes in IBD patients/IBD models. Methods Mol. Biol. (in press).
- 223) Fukui, H., Franceschi, F., Penland, R.L., Sakai, T., Sepulveda, A.R., Fujimori, T. et al. (2003) Effects of Helicobacter pylori infection on the link between regenerating gene expression and serum gastrin levels in Mongolian gerbils. Lab. Invest. 83, 1777–1786.
- 224) Yoshino, N., Ishihara, S., Rumi, M.A., Ortega-Cava, C.F., Yuki, T., Kazumori, H. et al. (2005) Interleukin-8 regulates expression of Reg protein in Helicobacter pylori-infected gastric mucosa. Am. J. Gastroenterol. 100, 2157–2166.
- 225) Harada, K., Zen, Y., Kanemori, Y., Chen, T.C., Chen, M.F., Yeh, T.S. et al. (2001) Human REG I gene is up-regulated in intrahepatic cholangiocarcinoma and its precursor lesions. Hepatology 33, 1036–1042.
- 226) Simon, M.T., Pauloin, A., Normand, G., Lieu, H.T., Mouly, H., Pivert, G. et al. (2003) HIP/PAP stimulates liver regeneration after partial hepatectomy and combines mitogenic and anti-apoptotic functions through the PKA signaling pathway. FASEB J. 17, 1441–1450.
- 227) Wang, J., Koyota, S., Zhou, X., Ueno, Y., Ma, L., Kawagoe, M. et al. (2009) Expression and localization of Regenerating gene I in a rat liver regeneration model. Biochem. Biophys. Res. Commun. 380, 472–477.
- 228) Uchiyama, T., Ota, H., Itaya-Hironaka, A., Shobatake, R., Yamauchi, A., Sakuramoto-Tsuchida, S. et al. (2017) Up-regulation of selenoprotein P and HIP/PAP mRNAs in hepatocytes by intermittent hypoxia via down-regulation of miR-203. Biochem. Biophys. Rep. 11, 130–137.
- 229) Otsuka, N., Yoshioka, M., Abe, Y., Nakagawa, Y., Uchinami, H. and Yamamoto, Y. (2020) Reg3α and Reg3β expressions followed by JAK2/STAT3 activation play a pivotal role in the acceleration of liver hypertrophy in a rat ALPPS model. Int. J. Mol. Sci. 21, 4077.
- 230) Kiji, T., Dohi, Y., Nishizaki, K., Takasawa, S., Okamoto, H., Nagasaka, S. et al. (2003) Enhancement of cell viability in cryopreserved rat vascular grafts by administration of regenerating gene (Reg) inducers. J. Vasc. Res. 40, 132–139.
- 231) Kiji, T., Dohi, Y., Takasawa, S., Okamoto, H., Nonomura, A. and Taniguchi, S. (2005) Activation of regenerating gene in rat and human hearts in response to acute stress. Am. J. Physiol. Heart Circ. Physiol. 289, H277–H284.
- 232) Watanabe, R., Hanawa, H., Yoshida, T., Ito, M., Isoda, M., Chang, H. et al. (2008) Gene expression profiles of cardiomyocytes in rat autoimmune myocarditis by DNA microarray and increase of regenerating gene family. Transl. Res. 152, 119–127.
- 233) Yoshimoto, K., Fujimoto, T., Itaya-Hironaka, A., Miyaoka, T., Sakuramoto-Tsuchida, S., Yamauchi, A. et al. (2013) Involvement of autoimmunity to REG, a regenerating factor, in patients with primary Sjögren’s syndrome. Clin. Exp. Immunol. 174, 1–9.
- 234) Fujimura, T., Fujimoto, T., Itaya-Hironaka, A., Miyaoka, T., Yoshimoto, K., Yamauchi, A. et al. (2015) Interleukin-6/STAT pathway is responsible for the induction of gene expression of REG Iα, a new auto-antigen in Sjögren’s syndrome patients, in salivary duct epithelial cells. Biochem. Biophys. Rep. 2, 69–74.
- 235) Fujimura, T., Fujimoto, T., Itaya-Hironaka, A., Miyaoka, T., Yoshimoto, K., Sakuramoto-Tsuchida, S. et al. (2017) Significance of interleukin-6/STAT pathway for the gene expression of REG Iα, a new autoantigen in Sjögren’s syndrome patients, in salivary duct epithelial cells. Clin. Rev. Allergy Immunol. 52, 351–363.
- 236) Klasan, G.S., Ivanac, D., Erzen, D.J., Picard, A., Takasawa, S., Peharec, S. et al. (2014) Reg3G gene expression in regenerating skeletal muscle and corresponding nerve. Muscle Nerve 49, 61–68.
- 237) Tohma, Y., Dohi, Y., Shobatake, R., Uchiyama, T., Takeda, M., Takasawa, S. et al. (2017) Reg gene expression in periosteum after fracture and its in vitro induction triggered by IL-6. Int. J. Mol. Sci. 18, 2257.
- 238) Acquatella-Tran Van Ba, I., Marchal, S., François, F., Silhol, M., Lleres, C., Michel, B. et al. (2012) Regenerating islet-derived 1α (Reg-1α) protein is new neuronal secreted factor that stimulates neurite outgrowth via exostosin tumor-like 3 (EXTL3) receptor. J. Biol. Chem. 287, 4726–4739.
- 239) Livesey, J.F., O’Brien, A.J., Li, M., Smith, G.A., Murphy, J.L. and Hunt, P.S. (1997) A Schwann cell mitogen accompanying regeneration of motor neurons. Nature 390, 614–618.
- 240) Namikawa, K., Fukushima, M., Murakami, K., Suzuki, A., Takasawa, S., Okamoto, H. et al. (2005) Expression of Reg/PAP family members during motor nerve regeneration in rat. Biochem. Biophys. Res. Commun. 332, 126–134.
- 241) Nishimune, H., Vasseur, S., Wiese, S., Birling, M.C., Holtmann, B., Sendtner, M. et al. (2000) Reg-2 is a motoneuron neurotrophic factor and a signalling intermediate in the CNTF survival pathway. Nat. Cell Biol. 2, 906–914.
- 242) Fukui, H., Kinoshita, Y., Maekawa, T., Okada, A., Waki, S., Hassan, S. et al. (1998) Regenerating gene protein may mediate gastric mucosal proliferation induced by hypergastrinemia in rats. Gastroenterology 115, 1483–1493.
- 243) Kazumori, H., Ishirara, S., Hoshino, E., Kawashima, K., Moriyama, N., Suetsugu, H. et al. (2000) Neutrophil chemoattractant-2β regulates the expression of the Reg gene in injured gastric mucosa in rats. Gastroenterology 119, 1610–1622.
- 244) Macadam, R.C.A., Sarela, A.I., Farmery, S.M., Robinson, P.A., Markham, A.F. and Guillou, P.J. (2000) Death from early colorectal cancer is predicted by the presence of transcripts of the REG gene family. Br. J. Cancer 83, 188–195.
- 245) Yonemura, Y., Sakurai, S., Yamamoto, H., Endou, Y., Kawamura, T., Bandou, E. et al. (2003) REG gene expression is associated with the infiltrating growth of gastric carcinoma. Cancer 98, 1394–1400.
- 246) Dhar, D.K., Udagawa, J., Ishihara, S., Otani, H., Kinoshita, Y., Takasawa, S. et al. (2004) Expression of Regenerating gene I in gastric adenocarcinomas: Correlation with tumor differentiation status and patient survival. Cancer 100, 1130–1136.
- 247) Hayashi, K., Motoyama, S., Sugiyama, T., Izumi, J., Anbai, A., Nanjo, H. et al. (2008) REG Iα is a reliable marker of chemoradiosensitivity in squamous cell esophageal cancer patients. Ann. Surg. Oncol. 15, 1224–1231.
- 248) Zheng, H.C., Sugawara, A., Okamoto, H., Takasawa, S., Takahashi, H., Masuda, S. et al. (2011) Expression profiling of the REG gene family in colorectal carcinoma. J. Histochem. Cytochem. 59, 106–115.
- 249) Sasahira, T., Oue, N., Kirita, T., Luo, Y., Bhawal, U.K., Fujii, K. et al. (2008) Reg IV expression is associated with cell growth and prognosis of adenoid cystic carcinoma in the salivary gland. Histopathology 53, 667–675.
- 250) Ohara, S., Oue, N., Matsubara, A., Mita, K., Hasegawa, Y., Hayashi, T. et al. (2008) Reg IV is an independent prognostic factor for relapse in patients with clinically localized prostate cancer. Cancer Sci. 99, 1570–1577.
- 251) Wang, Q., Deng, J., Yuan, J., Wang, L., Zhao, Z., He, S. et al. (2012) Oncogenic Reg IV is a novel prognostic marker for glioma patient survival. Diagn. Pathol. 7, 69.
- 252) Kimura, M., Naito, H., Tojo, T., Itaya-Hironaka, A., Dohi, Y., Yoshimura, M. et al. (2013) REG Iα gene expression is linked with the poor prognosis of lung adenocarcinoma and squamous cell carcinoma patients via discrete mechanisms. Oncol. Rep. 30, 2625–2631.
- 253) Masui, T., Ota, I., Itaya-Hironaka, A., Takeda, M., Kasai, T., Yamauchi, A. et al. (2013) Expression of REG III and prognosis in head and neck cancer. Oncol. Rep. 30, 573–578.
- 254) Kaprio, T., Hagström, J., Mustonen, H., Koskensalo, S., Andersson, L.C. and Haglund, C. (2014) REG4 independently predicts better prognosis in non-mucinous colorectal cancer. PLoS One 9, e109600.
- 255) Mikami, S., Ota, I., Masui, T., Itaya-Hironaka, A., Shobatake, R., Okamoto, H. et al. (2016) Effect of resveratrol on cancer progression through the REG III expression pathway in head and neck cancer cells. Int. J. Oncol. 49, 1553–1560.
- 256) Sakaki, Y., Minamiya, Y., Takahashi, N., Nakagawa, T., Katayose, Y., Ito, S. et al. (2008) REGIA expression is an independent factor predictive of poor prognosis in patients with breast cancer. Ann. Surg. Oncol. 15, 3244–3251.
- 257) Rojas, E., Carroll, P.B., Ricordi, C., Boschero, S.C., Stojikovic, S.S. and Atwater, I. (1994) Control of cytosolic free calcium in cultured human pancreatic beta-cells occurs by external calcium-dependent and independent mechanisms. Endocrinology 134, 1771–1781.
- 258) Vague, P., Vialettes, B., Lassmann-Vague, V. and Vallo, I.J. (1987) Nicotinamide may extend remission phase in insulin-dependent diabetes. Lancet 330, 619–620.
- 259) Vialettes, B., Picq, R., du Rostu, M., Charbonnel, B., Rodier, M., Mirouze, J. et al. (1990) A preliminary multicentre study of the treatment of recently diagnosed Type 1 diabetes by combination nicotinamide-cyclosporin therapy. Diabet. Med. 7, 731–735.
- 260) Elliott, R.B., Pilcher, C.C., Fergusson, D.M. and Stewart, A.W. (1996) A population based strategy to prevent insulin-dependent diabetes using nicotinamide. J. Pediatr. Endocrinol. Metab. 9, 501–509.
- 261) Pozzili, P., Visalli, N., Cavallo, M.G., Signore, A., Baroni, M.G., Buzzetti, R. et al. (1997) Vitamin E and nicotinamide have similar effects in maintaining residual beta cell function in recent onset insulin-dependent diabetes (the IMDIAB IV study). Eur. J. Endocrinol. 137, 234–239.
- 262) Lampeter, E.F., Klinghammer, A., Scherbaum, W.A., Heinze, E., Haastert, B., Giani, G. et al. (1998) The Deutsche Nicotinamide Intervention Study. An attempt to prevent type 1 diabetes. Diabetes 47, 980–984.
- 263) Fernandez, I.C., del Carmen Camberos, M., Passicot, G.A., Martucci, L.C. and Crest, J.C. (2013) Children at risk of diabetes Type 1. Treatment with acetyl-L-carnitine plus nicotinamide — Case reports. J. Pediatr. Endocrinol. Metab. 26, 347–355.
- 264) Knip, M., Douek, I.F., Moore, W.P.T., Gillmor, H.A., McLean, A.E.M., Bingley, P.J. et al. (2000) Safety of high-dose nicotinamide: a review. Diabetologia 43, 1337–1345.
- 265) Gale, E.A., Bingley, P.J., Emmett, C.L. and Collier, T., European Nicotinamide Diabetes Intervention Trial (ENDIT) Group (2004) European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet 363, 925–931.
- 266) Akabane, A., Kato, I., Takasawa, S., Unno, M., Yonekura, H., Yoshimoto, T. et al. (1995) Nicotinamide inhibits IRF-1 mRNA induction and prevents IL-1β-induced nitric oxide synthase expression in pancreatic β cells. Biochem. Biophys. Res. Commun. 215, 524–530.
- 267) Okamoto, H. (1996) Okamoto model for B-cell damage: recent advances. In Lessons from Animal Diabetes VI: 75th Anniversary of the Insulin Discovery (ed. Shafrir, E.). Birkhäuser, Boston, Basel, Berlin, pp. 97–111.
- 268) Jung, D.Y., Park, J.B., Joo, S.Y., Joh, J.W., Kwon, C.H., Kwon, G.Y. et al. (2009) Effect of nicotinamide on early graft failure following intraportal islet transplantation. Exp. Mol. Med. 41, 782–792.
- 269) Ichii, H., Wang, X., Messinger, S., Alvarez, A., Fraker, C., Khan, A. et al. (2006) Improved human islet isolation using nicotinamide. Am. J. Transplant. 6, 2060–2068.
- 270) Zenilman, M.E., Chen, J. and Magnuson, T.H. (1998) Effect of reg protein on rat pancreatic ductal cells. Pancreas 17, 256–261.
- 271) Nair, G.G., Vincent, R.K. and Odorico, J.S. (2014) Ectopic Ptf1a expression in murine ESCs potentiates endocrine differentiation and models pancreas development in vitro. Stem Cells 32, 1195–1207.
- 272) Md. Shahjalal, H., Shiraki, N., Sakano, D., Kikawa, K., Ogaki, S., Baba, H. et al. (2014) Generation of insulin-producing β-like cells from human iPS cells in a defined and completely xeno-free culture system. J. Mol. Cell Biol. 6, 394–408.
- 273) Hwang, E.S. and Song, S.B. (2020) Possible adverse effects of high-dose nicotinamide: Mechanisms and safety assessment. Biomolecules 10, 687.
- 274) Uyeki, E.M., Doull, J., Cheng, C.C. and Misawa, M. (1982) Teratogenic and antiteratogenic effects of nicotinamide derivatives in chick embryos. J. Toxicol. Environ. Health 9, 963–973.
- 275) Handler, P. and Dann, W.J. (1942) The inhibition of rat growth by nicotinamide. J. Biol. Chem. 146, 357–368.
- 276) Yamagami, T., Miwa, A., Takasawa, S., Yamamoto, H. and Okamoto, H. (1985) Induction of rat pancreatic B-cell tumors by the combined administration of streptozotocin or alloxan and poly(adenosine diphosphate ribose) synthetase inhibitors. Cancer Res. 45, 1845–1849.
- 277) Hoorens, A. and Pipeleers, D. (1999) Nicotinamide protects human beta cells against chemically-induced necrosis, but not against cytokine-induced apoptosis. Diabetologia 42, 55–59.
- 278) Eizirik, D.L. and Darville, M.I. (2001) β-Cell apoptosis and defense mechanisms. Lessons from type 1 diabetes. Diabetes 50, S64–S69.
- 279) Germain, M., Scovassi, I. and Poirier, G.G. (2000) Role of poly(ADP-ribose) polymerase in apoptosis. In Cell Death: The Role of PARP (ed. Szabó, C.). CRC Press, Boca Raton, pp. 209–225.
- 280) Kaufmann, S.H., Desnoyers, S., Ottaviano, Y., Davidson, N.E. and Poirier, G.G. (1993) Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 53, 3976–3985.
- 281) Tewarim, M., Quan, L.T., O’Rourke, K., Desnoyers, S., Zeng, Z., Beidler, D.R. et al. (1995) Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 81, 801–809.
- 282) Chaitanya, G.V., Alexander, J.S. and Babu, P.P. (2010) PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 8, 31.
- 283) Sauter, B., Albert, M.L., Francisco, L., Larsson, M., Somersan, S. and Bhardwaj, N. (2000) Consequences of cell death: Exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 191, 423–433.
- 284) Cocco, R.E. and Ucker, D.S. (2001) Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol. Biol. Cell 12, 919–930.
- 285) Hallakou-Bozec, S., Kergoat, M., Fouqueray, P., Bolze, S., and Moller, D.E. (2021) Imeglimin amplifies glucose-stimulated insulin release from diabetic islets via a distinct mechanism of action. PLoS One 16, e0241651.
- 286) Dubourg, J., Fouqueray, P., Thang, C., Grouin, J.-M. and Ueki, K. (2021) Efficacy and safety of Imeglinmin monotherapy versus placebo in Japanese patients with Type 2 diabetes (TIMES 1): A double-blind, randomized, placebo-controlled. parallel-group, multicenter phase 3 trial. Diabetes Care 44, 952–959.
- 287) Okamoto, H., Miyamoto, S., Mabuchi, H. and Takeda, R. (1974) Inhibitory effect of quinaldic acid on glucose-induced insulin release from isolated Langerhans islets of the rat. Biochem. Biophys. Res. Commun. 59, 623–629.
- 288) Endo, M., Tanaka, M. and Ogawa, Y. (1970) Calcium induced release of calcium from the sarcoplasmic reticulum of skinned skeletal muscle fibres. Nature 228, 34–36.
- 289) Munshi, C., Aarhus, R., Graeff, R., Walseth, T.F., Levitt, D. and Lee, H.C. (2000) Identification of the enzymatic active site of CD38 by site-directed mutagenesis. J. Biol. Chem. 275, 21566–21571.
- 290) Kuemmerle, J.F. and Makhlouf, G.M. (1995) Agonist-stimulated cyclic ADP-ribose: Endogenous modulator of Ca2+-induced Ca2+ release in intestinal longitudinal muscle. J. Biol. Chem. 270, 25488–25494.
- 291) Campbell, R.K., Wells, R.W., Miller, D.V. and Paterson, W.G. (2007) Role of cADPR in sodium nitroprusside-induced opossum esophageal longitudinal smooth muscle contraction. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1543–G1548.
- 292) Takasawa, S., Itaya-Hironaka, A., Yamauchi, A., Ota, H., Takeda, M., Sakuramoto-Tsuchida, S. et al. (2016) Regulators of beta-cell death and regeneration. In Pancreatic Islet Biology (ed. Hardikar, A.A.). Springer, Cham, Switzerland, pp. 125–158.
- 293) Narushima, Y., Unno, M., Nakagawara, K., Mori, M., Miyashita, H., Suzuki, Y. et al. (1997) Structure, chromosomal localization and expression of mouse genes encoding type III Reg, RegIIIα, RegIIIβ, RegIIIγ. Gene 185, 159–168.
- 294) Inoue, C., Yamamoto, H., Nakamura, T., Ichihara, A. and Okamoto, H. (1989) Nicotinamide prolongs survival of primary cultured hepatocytes without involving loss of hepatocyte-specific functions. J. Biol. Chem. 264, 4747–4750.
Non-standard abbreviation list
A-kinasecyclic AMP-dependent protein kinase
alloxan2,4,5,6-tetraoxohexahydropyrimidine
ATF-2activating transcription factor 2
cADPRcyclic ADP-ribose
CaMcalmodulin
CaM kinase IIcalmodulin-dependent protein kinase II
CDKcyclin-dependent kinase
FK506Tacrolimus (Tsukuba macrolide immunosuppressant)
FKBPFK506-binding protein
FSBA5′-p-fluorosulfonylbenzoyladenosine
GLP-1glucagon-like peptide-1
IL-6interleukin-6
IP3inositol 1,4,5-trisphosphate
KOknockout
NOnitric oxide
NODnon-obese diabetic
NOS2type 2 nitric oxide synthase
NOS2type 2 nitric oxide synthase
PARPpoly(ADP-ribose) polymerase/synthetase
PI(3)Kphosphoinositide 3-kinase
RegReg protein
RegRegenerating gene
RyRryanodine receptor
streptozotocin2-deoxy-2-(3-methyl-3-nitrosoureido)-D-glucopyranose
Profile

Hiroshi Okamoto is Professor Emeritus of Tohoku University and of Toyama University, and holds Honorary Doctorate from Kanazawa University. He was born in Kanazawa (1939) and graduated from Kanazawa University School of Medicine in 1964. After internship in St. Luke's International Hospital in Tokyo, he studied on the tryptophan-NAD+ metabolism in rat liver at the Medical Chemistry Department (late Prof. Osamu Hayaishi) of the graduate school of Kyoto University. He was promoted to Assistant Professor in the department in 1969. In 1972, he returned to Kanazawa University and started studies using pancreatic islets of Langerhans. He was offered a Professorship at Toyama Medical and Pharmaceutical University School of Medicine (currently University of Toyama, Faculty of Medicine) to become Professor and Chairman of the Department of Biochemistry in 1976. In Toyama, his group revealed translational control of proinsulin synthesis by glucose and proposed the Okamoto model for β-cell damage and its prevention. In 1984, he moved to Tohoku University School of Medicine as Professor and Chairman of the Department of Biochemistry. At this university, studies of the CD38-cyclic ADP-ribose signal system and Reg-Reg receptor system were performed as an extension of the Okamoto model. In 2003, he established a new department, the Department of Advanced Biological Sciences for Regeneration (Kotobiken Medical Laboratories) in the Tohoku University Graduate School of Medicine to further these studies and became Professor at Tohoku University. He worked as an auditor and senior president advisor in Tohoku University.
For his important contributions to the field of biochemistry and diabetology, he was honored to receive the Japanese Biochemistry Society Young Investigator Award in 1973, the 36th Chunichi Award for Science and Culture in 1983, the Special Lecture at the 20th Anniversary of the Insulin Crystal Structure Determination (University of York, York, U.K.) in 1988, the 13th International Diabetes Federation Plenary Lecture Medal in 1988 (Sydney, Australia), the 3rd Krock Lecture Award (University of Pennsylvania) in 1988, the 1st Roerig-Pfizer Lecture Award (University of Michigan) in 1990, the Special Lecture at the 14th International Diabetes Federation (Washington, D.C.) in 1991, Hagedorn Award of the Japan Diabetes Society in 1993, Takeda Award for Medical Science in 1993, the 33rd Erwin von Bälz Award for Medical Science in 1996, Special Lectures at American Diabetes Association (San Antonio, Texas) in 2000, European Association for the Study of Diabetes (Paris, France in 2000 & Prien am Chiemsee, Germany in 2004), and the 2nd World Congress on Regenerative Medicine (Leipzig, Germany) in 2005. He was awarded the Medal with Purple Ribbon (Shiju-ho-sho) in 2002, the Japan Academy Prize in 2003, and Japan Endocrine Society Meister Award in 2019. He is an Academic Editor of the Journal of Diabetes Research (Hindawi, London).

Shin Takasawa was born in 1958 and graduated from Yamagata University School of Medicine in 1983. He carried out further studies in the Department of Biochemistry (Chairman Prof. Hiroshi Okamoto) of the Graduate School of Toyama Medical and Pharmaceutical University (currently University of Toyama, Faculty of Medicine). He was promoted to Assistant Professor in 1986, to Lecturer in 1993, and to Associate Professor in 1996 at the Department of Biochemistry, Tohoku University School of Medicine. In 2006, he was promoted to Professor of the Department of Advanced Biological Sciences for Regeneration (Kotobiken Medical Laboratories), Tohoku University Graduate School of Medicine. During this research studies, he performed research on the CD38-cyclic ADP-ribose signal system and Reg-Reg receptor system. In 2007, he moved to Nara Medical University as Professor and Chairman in the Department of Biochemistry. At this university, he has extended his studies concerning the CD38-cyclic ADP-ribose signal system and Reg-Reg receptor system.
For his achievements in the field of biochemistry, he was honored to receive the 33rd Erwin von Bälz Award for Medical Science and the Japanese Biochemistry Society Young Investigator Award in 1996. He is as an editorial board member of Life Sciences and International Journal of Molecular Sciences.


