Abstract
Metal-catalyzed asymmetric synthesis is one of the most important methods for the economical and environmentally benign production of useful optically active compounds. The success of the asymmetric transformations is significantly dependent on the structure and electronic properties of the chiral ligands coordinating to the center metals, and hence the development of highly efficient ligands, especially chiral phosphine ligands, has long been an important research subject in this field. This review article describes the synthesis and applications of P-chiral phosphine ligands possessing chiral centers at the phosphorus atoms. Rationally designed P-chiral phosphine ligands are synthesized by the use of phosphine–boranes as the intermediates. Conformationally rigid and electron-rich P-chiral phosphine ligands exhibit excellent enantioselectivity and high catalytic activity in various transition-metal-catalyzed asymmetric reactions. Recent mechanistic studies of rhodium-catalyzed asymmetric hydrogenation are also described.
1. Introduction
Catalytic asymmetric synthesis is one of the most efficient methods for the production of optically active compounds used in pharmaceuticals, agrochemicals, fragrances, and so on.1)–5) Among available methodologies for asymmetric catalysis, transition-metal-catalyzed reactions have been studied for more than six decades, and a variety of reactions of this class have been developed. The enantioselectivities and catalytic activities of the reactions are generally largely affected by the chiral ligands as well as the center metals.
Among various types of chiral ligands, phosphine ligands play an outstanding role in asymmetric catalysis owing to their intrinsic property of coordinating strongly to the transition metals and to promoting the catalytic reactions. A great number of chiral phosphine ligands have been synthesized and used in this class of asymmetric reactions.6)–20)
Chiral phosphine ligands are divided into two classes. One class includes backbone chirality ligands that possess their stereogenic centers on the linking carbon chain. Representative examples of this class are shown in Fig. 1. Most of the hitherto reported chiral phosphine ligands belong to this class, and some of them are used as benchmark ligands not only for the synthesis of various chiral compounds but also for the development of new catalytic asymmetric reactions.
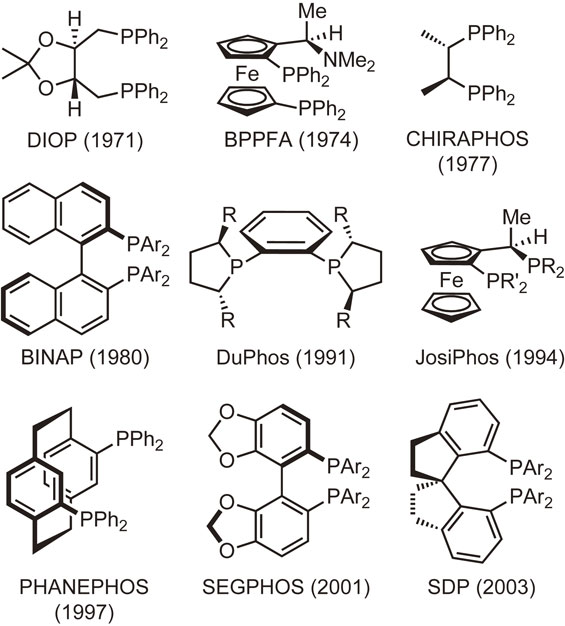
Another class is composed of P-chiral (P-chirogenic or P-stereogenic) phosphine ligands possessing their stereogenic centers at the phosphorus atoms. Figure 2 shows the representative examples of this class. Although P-chiral phosphine ligands are small in number compared with backbone chirality ligands, some of the ligands play a critical role in the early stage of the study on Rh-catalyzed homogeneous asymmetric hydrogenation.21)–23) In particular, the use of DIPAMP ligand, which was developed by Knowles and coworkers at Monsanto Company, provided high enantioselectivities of up to 96% in 1975, the highest at that time.24)–26) The same ligand was successfully employed in the manufacture of (S)-3,4-dihydroxyphenylalanine (l-DOPA) that is used to treat Parkinson’s disease.27),28) However, despite those landmark achievements, P-chiral phosphine ligands including DIPAMP have not been widely used for more than 20 years. This is mainly attributed to the synthetic difficulties in generating optically pure P-chiral phosphines. In addition, the fact that some phosphines bearing electron-withdrawing groups are stereochemically unstable and gradually racemize via pyramidal inversion even at room temperature may have discouraged researchers. Another reason is that many backbone chirality phosphine ligands such as BINAP have been synthesized and their superior catalytic performance has been proven in various asymmetric syntheses.

On the other hand, in our studies of the synthesis and reactions of phosphine–boranes, we found that P-chiral phosphine ligands could be synthesized via the stereospecific removal of the BH3 group of chiral phosphine–boranes. This finding inspired us to study the design and synthesis of new P-chiral phosphine ligands and their applications in catalytic asymmetric synthesis. This review article contains our studies on phosphine–boranes and P-chiral phosphine ligands. It also mentions how we discovered new research themes and developed them, taking on the many challenges that came with the work and overcoming the difficulties along the way.
2. Prologue to the study of P-chiral phosphine ligands
2.1. Use of cerium element in organic synthesis.
After receiving my Ph.D. degree in physical organic chemistry in 1972, I went on to work at four universities to pursue further studies in organic synthesis. For 8 years, I worked as a postdoctoral fellow or an assistant professor, seeking tenure. Fortunately, in 1980, I was appointed assistant professor at Chiba University. I was 37, and that appointment gave me an excellent opportunity to begin my own research work. However, at that time, my research group was quite small, and it was almost impossible to undertake a big project. I decided to seek out new research subjects in the largely unexplored areas. Perusing the periodic table of elements in an effort to find elements that had not yet been utilized in organic synthesis, I arrived at the conclusion that lanthanide elements were the perfect choice. Although Professors Kagan and Luche had already reported their pioneering and outstanding investigations at that time,29)–32) I believed that this area offered the exciting possibility of discovering new and useful synthetic methods. Among 15 lanthanides, I focused on cerium element because cerium exists in high natural abundance, and its major salts are commercially available at affordable prices.
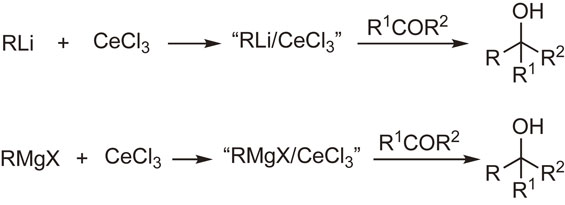
After many attempts using metallic cerium or cerium(III) salts, we succeeded in preparing new reagents called “organocerium reagents” or “cerium(III)-modified organometallic reagents”. These reagents are readily prepared by the reaction of organolithiums or Grignard reagents with anhydrous cerium(III) chloride in tetrahydrofuran. The cerium reagents undergo nucleophilic addition reactions with carbonyl groups more efficiently than the parent organometallic reagents. Thus, the reagents react with various carbonyl compounds to afford the corresponding normal addition products in high yields, even though the substrates are susceptible to so-called abnormal reactions, such as enolization, reduction, conjugate addition, condensation, and metal–halogen exchange reaction (Scheme 1).33)–35) Several reaction examples are shown in Scheme 2. This method, together with Knochel’s improved method,36),37) is widely used in organic synthesis.
2.2. Working with phosphine–boranes.
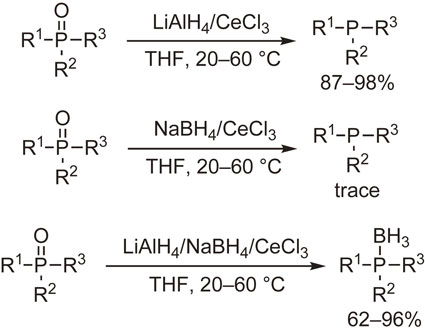
The above-mentioned cerium chloride methodology was extended to a LiAlH4/CeCl3 system to examine the enhanced or modified reducing ability of LiAlH4. Our initial experiments on the reduction of phosphine oxides and organic halides including fluorides demonstrated the powerful reducing ability of LiAlH4 under mild conditions.38) Notable is that various phosphine oxides including sterically congested ones were readily converted into the corresponding phosphines in high yields. In contrast, the use of NaBH4 instead of LiAlH4 afforded only trace amounts of the products. Taking one step further, we conducted the reaction with a three-component reagent, LiAlH4–NaBH4–CeCl3, to find that phosphine–boranes were produced in good to high yields (Scheme 3).39)
We were surprised to find that phosphine–borane compounds, including secondary ones having a P–H bond, were virtually inert to air and hardly decomposed even on contact with an acid or a base. Phosphine–boranes possess formal charges of +1 and −1 on the phosphorus and boron atoms, respectively, and are a kind of phosphorus ylide and boron ate complex. I was fascinated by these structural features and decided to study phosphine–boranes from the perspective of organic synthesis. I emphasized the significance of this research to my collaborator students by mentioning that two Nobel Prize Laureates, G. Wittig and H.C. Brown, were involved in our phosphine–borane research. The following studies were conducted in parallel with the synthesis and application of P-chiral phosphine ligands. The main purpose of these studies was to create interesting and fundamentally important chemical species and to find unprecedented reactions by utilizing the characteristic properties of phosphine–boranes.
2.2.1. Generation and reactions of tricoordinate boron dianions.
Tricoordinate boron dianions have an isoelectronic relationship with tricoordinate carbanions, which are one of the most important chemical species in organic chemistry. We were interested in whether or not the boron dianions exhibit reactivities similar to the carbanions and intended to generate such chemical species by using phosphine–boranes. After various trials, we succeeded in the generation of the desired chemical species possessing a formal −2 charge on the boron atom, and demonstrated their high nucleophilicity and strong basicity, which are similar to those of carbanions. Thus, tricyclohexylphosphine–monoiodoborane (1) was reacted with lithium 4,4′-di-tert-butylbiphenylide (LDBB) to generate boron dianion 2, which was allowed to react with various electrophiles to give the boron-functionalized phosphine–boranes (3) (Scheme 4).40) Similarly, the reduction of tri-tert-butylphosphine–monoiodoborane (4) with LDBB, followed by the reaction with electrophiles, furnished compounds 7. It is reasonable to consider that the generated boron dianion 5 underwent a pericyclic reaction at −78 °C and the resulting phosphide anion 6 was trapped by the electrophiles. This reaction was compared with the corresponding carbanion species, tri-tert-butylphosphonium methylide, which was reported to undergo the same type of pericyclic reaction at a much higher temperature (20 °C).41) These results clearly indicate that the basicity of the boron dianion is greater than that of the corresponding carbanion.

Reports of nucleophilic substitution reactions at the tetracoordinate boron atom abound, but little attention has been given to the stereochemistry of the reactions. We initially succeeded in synthesizing enantiopure B-stereogenic tetracoordinate boron compound 8 containing a bromo substituent as the leaving group and examined the stereochemistry of both the nucleophilic and the electrophilic substitution reactions (Scheme 5).42)
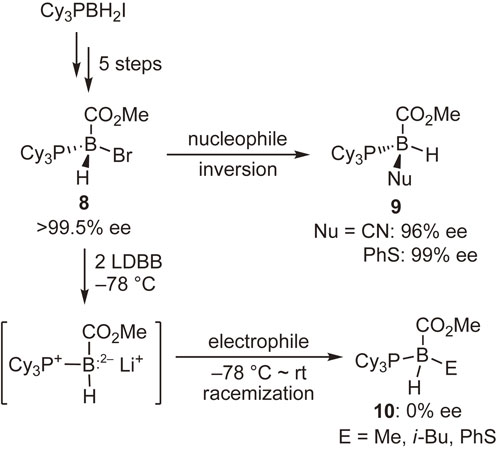
The reactions of 8 with nucleophiles afforded the substitution products 9 with excellent enantiomeric excesses. These results apparently demonstrate that the SN2 reaction at the sp3 boron atom proceeded with inversion of configuration. On the other hand, the reduction of 8 with LDBB, followed by the reactions with electrophiles, provided completely racemized products 10. These experimental facts can be explained by assuming that the intermediate boranyl radical or the boron dianion species would be stereochemically unstable and undergo rapid stereomutation under the reaction conditions.
2.2.3. Preparation and reactions of boranophosphorylation reagents.
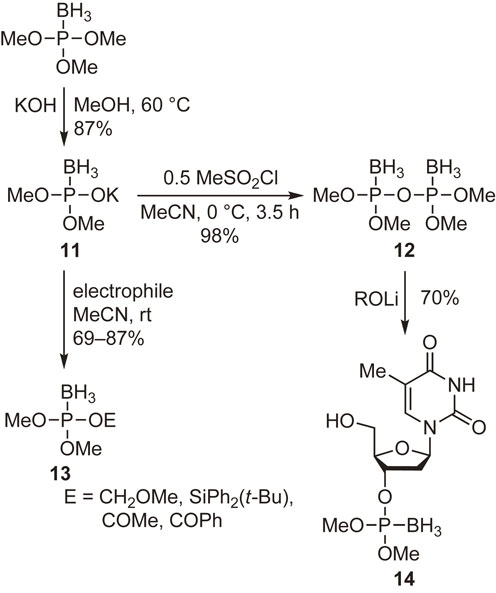
Boranophosphates, which have an isoelectronic relationship with phosphates, are useful in biochemical investigations. They also have potential utility as carriers of 10B in boron neutron capture therapy for cancer treatment. We tried to develop new reagents for the synthesis of similar compounds. As shown in Scheme 6, dimethyl boranophosphate monopotassium salt 11 and tetramethyl boranopyrophosphate 12 were prepared from the borane adduct of trimethylphosphite. The former compound underwent substitution reactions with various electrophilic reagents to generate compounds 13, whereas the latter reacted with metal alkoxides to give the corresponding boranophosphate derivatives such as compound 14.43) These are simple synthetic routes to various boranophosphate derivatives, including the borano analogs of naturally occurring phosphates.
2.2.4. Synthesis and reactions of various phosphine–borane derivatives.
We examined the functionalization of the phosphine moiety of phosphine–boranes. As shown in Scheme 7, tertiary phosphine–boranes 15 bearing a methyl group at the phosphorus atom were subjected to deprotonation, followed by the reaction with alkyl halides or carbonyl compounds to give various phosphine–borane derivatives 16. Oxidative dimerization proceeded smoothly to afford compounds 17 with the boranato group intact. Secondary phosphine–boranes 18 reacted with a variety of electrophiles in the presence of a base to yield corresponding phosphine–borane derivatives 19, similarly to the reactions of secondary phosphine oxides.39),44)

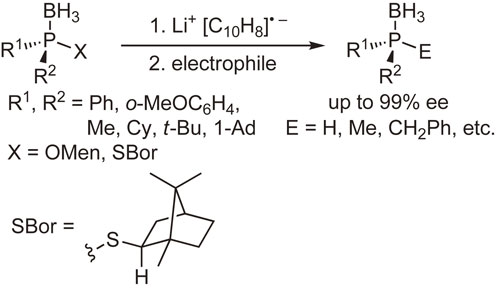
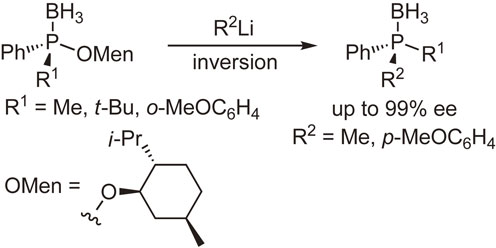
Our subsequent interest was the synthesis and reactions of optically active phosphine–boranes. Several phosphine–boranes bearing l-menthoxy or bornylthio group were prepared in diastereomerically pure form, and their reactions with nucleophiles or one-electron reducing agents were examined. The results shown in Schemes 8–10 indicate that chiral phosphine–boranes can be obtained in excellent enantiopurity, most of which are limited to compounds bearing aryl groups at the phosphorus atom.45)–51)
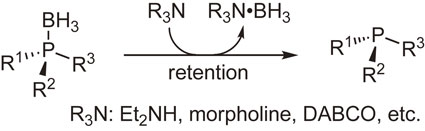
Our study of the reactivities of phosphine–boranes was extended to the transformation of the boranato group moiety. After many trials, we found that phosphine–boranes reacted with amines such as diethylamine and 1,4-diazabicyclo[2.2.2]octane to produce parent tertiary phosphines and amine–boranes. Further trials using optically active phosphine–boranes indicated that this deboranation proceeded with complete retention of configuration (Scheme 11).39),44) We were delighted with the finding of this simple and unprecedented reaction and were convinced that various phosphines including optically active ones can be synthesized via this deboranation process. It is noteworthy that the BH3 group plays the role of protecting group of the labile phosphine moieties.
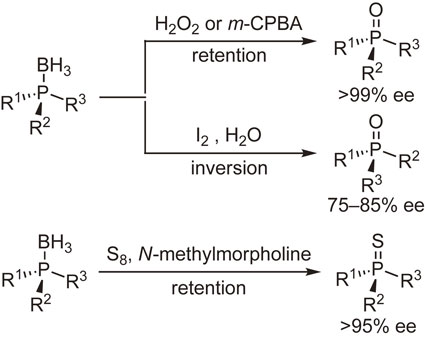
The conversion of the boranato group of phosphine–boranes into oxygen or sulfur atom was examined. The reaction with hydrogen peroxide or m-chloroperbenzoic acid proceeded at 0 °C with complete retention of configuration to provide the corresponding phosphine oxides. On the contrary, the reaction with iodine in the presence of water occurred at room temperature with inversion of configuration, while it was accompanied by partial racemization. The reaction with sulfur in the presence of N-methylmorpholine proceeded at 110 °C while retaining stereochemical integrity to give the corresponding phosphine sulfides in almost quantitative yields (Scheme 12).52)
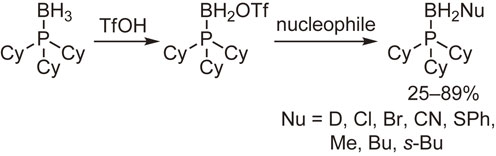
The functionalization of phosphine–boranes at the boron atom was then examined. It was found that one of the hydrogen atoms of the boranato group was readily replaced with a trifluoromethanesulfonyloxy or methanesulfonyloxy group by the reaction with trifluoromethanesulfonic acid or methanesulfonic acid. The resulting triflate and mesylate were subjected to a nucleophilic substitution reaction to afford the functionalized phosphine–boranes (Schemes 13 and 14).53),54)
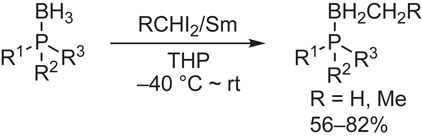
Direct boron–carbon bond formation was also examined through the reactions with metal carbenoids. Scheme 15 shows our observation that samarium carbenoids undergo methylene insertion into the B–H bond to afford B-alkylated compounds.55)
3. Synthesis of P-chiral phosphine ligands
3.1. Synthesis of P-chiral bisphosphine ligands with aryl groups at phosphorus atoms.
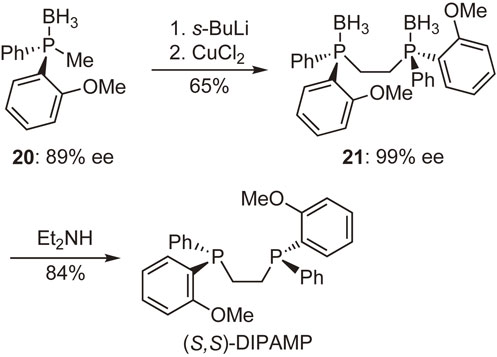
Motivated by the results mentioned above, we tried to synthesize bidentate P-chiral phosphine ligands using phosphine–boranes as the intermediates. Our initial attempt commenced with the preparation of DIPAMP, as shown in Scheme 16. Compound 20 with 89% ee was successively reacted with s-BuLi and CuCl2 to give compound 21 with 99% ee after removal of the concomitantly formed meso-isomer. The subsequent reaction of 21 with diethylamine furnished enantiopure (S,S)-DIPAMP, demonstrating the potential utility of this phosphine–borane methodology.39)
In a similar manner, new P-chiral phosphine ligands 22a–d and their rhodium complexes 23a–d were prepared from dichlorophenylphosphine (Scheme 17), and their enantioinduction abilities were evaluated in the hydrogenation of a typical probing substrate, methyl (Z)-α-acetamidocinnamate (MAC). The hydrogenation results are shown in Scheme 18 together with that obtained by the use of a Rh-DIPAMP complex.

It is noted that complex 23b bearing an o-ethylphenyl group provided remarkably high enantioselectivity (97%) comparable to that (96%) of [Rh((S,S)-DIPAMP)(cod)]BF4. Complexes 23c and 23d with larger ortho-alkyl substituents afforded almost perfect enantioselectivities (>99%), exceeding that of DIPAMP after 20 years.56),57) Another significant fact is that the sense of the enantioselectivity of the newly synthesized ligands with (S,S)-configuration is the same as that of (S,S)-DIPAMP in the Rh-catalyzed asymmetric hydrogenation. These results clearly indicate that the coordinative interaction between methoxy oxygen atom and rhodium atom in the Rh-DIPAMP complex is not significant in the stereoregulation and the enantioselection is determined by the steric effects of the ligands.
3.2. Synthesis of electron-rich P-chiral phosphine ligands and their enantioinduction ability.
Although the above-mentioned ligands 22c and 22d exhibited higher enantioselectivity than DIPAMP, their molecular structures closely resembled DIPAMP, and hence we next tasked ourselves with the development of structurally new P-chiral phosphine ligands. In 1990–1991, Burk and coworkers reported that enantiopure 1,2-bis(trans-2,5-dialkylphospholano)ethanes (BPE) and 1,2-(trans-2,5-dialkylphospholano)benzenes (DuPhos), both of which are electron-rich bisphosphine ligands, exhibited exceedingly high enantioinduction abilities and catalytic efficiencies in Rh- and Ru-catalyzed asymmetric hydrogenations.58)–61) Inspired by their outstanding achievements, we concentrated on the design and synthesis of new P-chiral phosphine ligands.
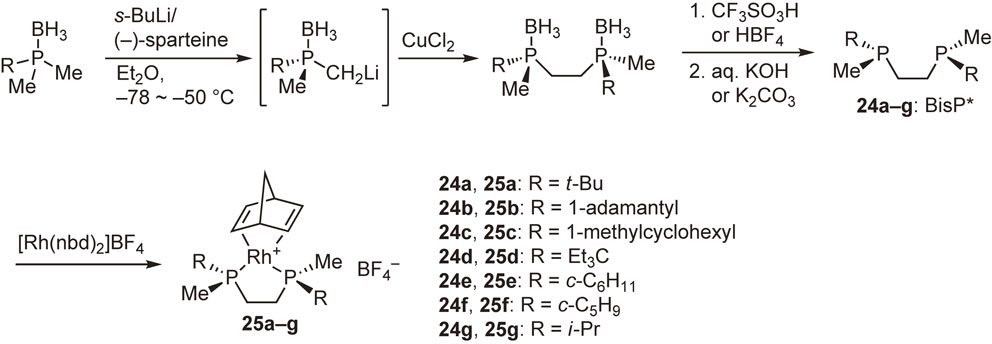
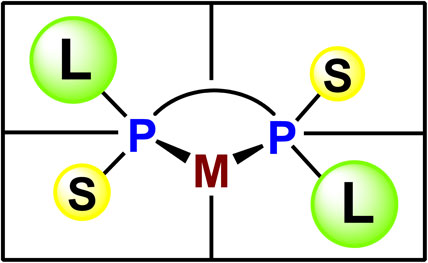
Our ligand design concept was based on the quadrant diagram proposed by Knowles.26) The newly designed ligands are C2-symmetric bisphosphines, in which a large alkyl group and a small alkyl group are bonded to each phosphorus atom, and the asymmetric environment around the center metal is quite clear (Fig. 3). This idea was immediately implemented in experiments of the synthesis of ethylene-bridged ligands, (S,S)-1,2-bis(alkylmethylphosphino)ethanes named BisP* (24a–g), possessing a tertiary or secondary alkyl group as the large group and a methyl group as the small group. The ligands were air-sensitive semi-solids or oils, and hence they were converted into air-stable rhodium complexes 25a–g (Scheme 19).62)
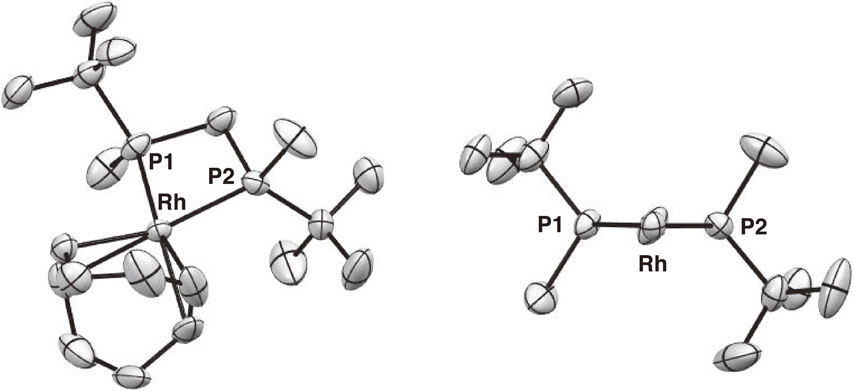
The structure of complex 25a having a t-butyl group as the large group was determined by single-crystal X-ray analysis. The ORTEP drawing shown in Fig. 4 clearly indicates the expected C2-symmetric environment, where the bulky t-butyl groups occupy the quasi-equatorial positions and the methyl groups are located at the quasi-axial positions to form a λ-chelate structure. This evidently defined asymmetric environment led us to anticipate the high utility of t-Bu-BisP* ligand in asymmetric catalysis.

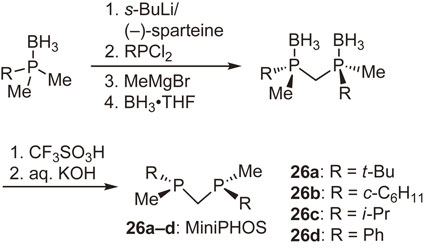
Subsequent to the above-mentioned work, we synthesized structurally simpler methylene-bridged bisphosphine ligands, (R,R)-bis(alkylmethylphosphino)methanes (MiniPHOS) (26a–d) (Scheme 20).63) The complexation of t-Bu-MiniPHOS (26a) with Rh-diene complexes afforded two different four-membered Rh complexes depending on the diene ligand. Thus, the reaction of 26a with norbornadiene complexes [Rh(nbd)2]X (X = BF4, PF6) provided bischelated complexes [Rh((R,R)-t-Bu-MiniPHOS)2]X,63) whereas the reaction with a cyclooctadiene complex [Rh(cod)2]SbF6 afforded monochelated complex [Rh((R,R)-t-Bu-MiniPHOS)(cod)]SbF6.64) Figure 5 shows an ORTEP drawing of the monochelated complex. It is noted that the four-membered chelate ring is almost entirely flat, the bulky t-butyl groups effectively shield the diagonal quadrants, and the two methyl groups locate on the other diagonal quadrants, constructing the expected asymmetric environment just as we designed. This C2-symmetric Rh-t-Bu-MiniPHOS complex is one of my favorite compounds because of its very simple structure.
The prepared Rh complexes of BisP* and MiniPHOS ligands were used in the asymmetric hydrogenation of α- and β-dehydroamino acid derivatives and enamides to evaluate their enantioinduction abilities. In most cases, the ligands possessing a t-butyl group exhibited very high enantioselectivities of up to 99.9%.62),63),65)–67) We were pleased to find that the two ligands, t-Bu-BisP* and t-Bu-MiniPHOS, were applicable to other representative catalytic asymmetric reactions, such as the Ir-catalyzed hydrogenation of imines and the Rh-catalyzed hydrosilylation of ketones.68),69)
Our next work was to develop more efficient chiral bisphosphine ligands. Day in and day out, we designed new P-chiral phosphine ligands using molecular models, finally designing ligand 27 consisting of two phospholane rings as our most promising candidate. The ligand was expected to form metal complex 28 having three fused five-membered rings, and the complex would be quite rigid to effectively block the two diagonal quadrants, eventually creating an ideal asymmetric environment around the metal center (Fig. 6).
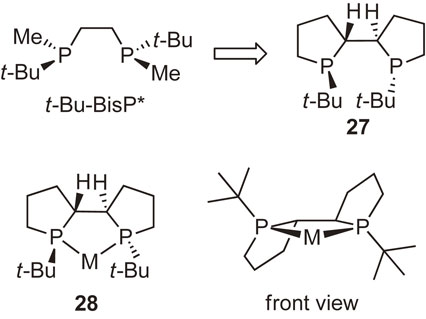
Initially, we presumed that ligand 27 could be readily synthesized from 1-tert-butylphospholane–borane 29 via C2-symmetric precursor 30. However, the oxidative coupling proceeded sluggishly to form undesired meso-isomer 31 in 10% yield (Scheme 21).70) To obtain the desired compound 30, we further examined the coupling reaction under various conditions and at the same time sought other different synthetic routes; however, we were unable to isolate 30. Meanwhile, I was astonished to find the paper by Tang and Zhang describing the synthesis and exceedingly high enantioinduction ability of ligand 27 (Scheme 22).71) They used phosphine sulfide 32 as the starting material to obtain oxidative coupling product 33 as the major product. Actually, we had also carried out the experiments using the same phosphine sulfide 32, but were unsuccessful in isolating the desired 33. To this day, I very much regret not being more persistent in performing the experiments to achieve the first synthesis of ligand 27.


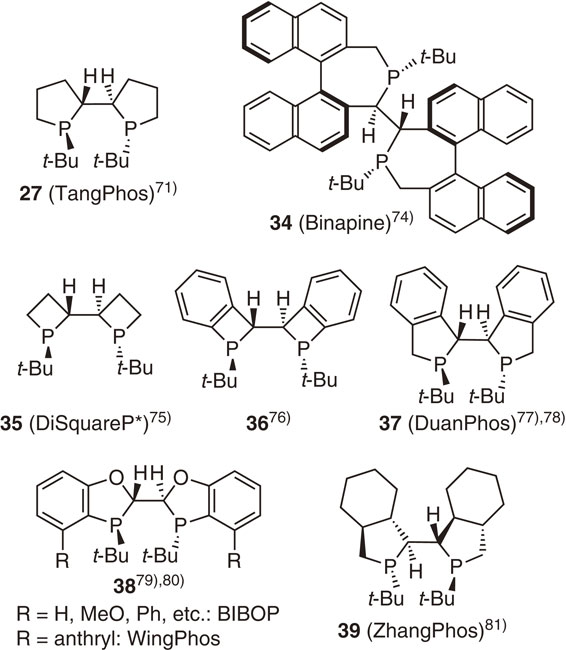
The superior performance of TangPhos in not only Rh-catalyzed asymmetric hydrogenation reactions but also many other catalytic asymmetric reactions was demonstrated through extensive investigations by Zhang and Tang.71)–73) Following to TangPhos, analogous P-chiral bisphosphacycle ligands (34–39) bearing t-butyl groups at the phosphorus atom were reported (Fig. 7).74)–81) We also prepared ligands 35 and 36 consisting of more rigid four-membered phosphacycles, expecting that they would exhibit high enantioselectivity.75),76) Among these ligands, Binapine (34),74) DuanPhos (37),77),78) BIBOP (38),79),80) and WingPhos (38)79),80) are widely used in both academia and industry for the production of many useful optically active compounds.82) I am amazed at the powerful research activities of Professors Zhang and Tang and their coworkers. At the same time, I am delighted that these remarkable developments originated from the discovery of the t-Bu-BisP* ligand.
Enantiopure 1,2-bis(tert-butylmethylphosphino)ethane (t-Bu-BisP*), a representative P-chiral phosphine ligand developed in our laboratory, exhibits very high enantioselectivities in several catalytic asymmetric reactions. However, this ligand is an extremely air-sensitive semi-solid, and this property has hampered its widespread application in catalytic asymmetric synthesis. In this context, I pursued a new ligand that is not an oily material but an air-stable crystalline solid to exhibit excellent enantioinduction ability similar to t-Bu-BisP*. Newly designed ligand 2,3-bis(tert-butylmethylphosphino)quinoxaline (40) (QuinoxP*) possessing the quinoxaline backbone was prepared from enantiopure (R)-tert-butyl(hydroxymethyl)methylphosphine–borane (41) (Scheme 23).83) Compound 41 was transformed by a ruthenium-catalyzed stereospecific oxidative one-carbon degradation into (S)-tert-butylmethylphosphine–borane (42),84) which was deprotonated with n-BuLi and allowed to react with 2,3-dichloroquinoxaline, followed by treatment with TMEDA to furnish the desired ligand 40 in an enantiopure form. We were overjoyed to obtain the ligand as an air-stable crystalline solid and to confirm its high performance in representative transition-metal-catalyzed asymmetric reactions.83),85)
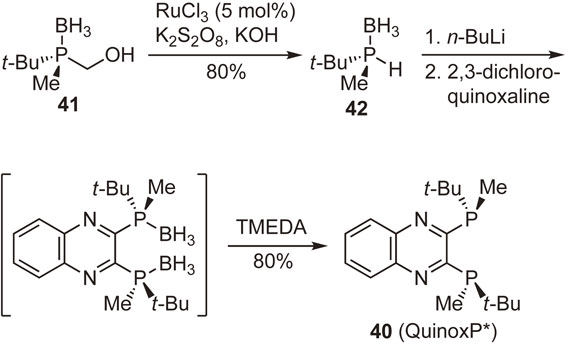
In a similar manner, we prepared analogous C2-symmetric ligands, (R,R)-2,3-bis((1′-adamantyl)methylphosphino)quinoxaline ((R,R)-Ad-QuinoxP*) (43) and (R,R)-2,3-bis((1′,1′,3′,3′-tetramethylbutyl)methylphosphino)quinoxaline ((R,R)-TMB-QuinoxP*) (44) (Fig. 8). The former ligand, unfortunately, did not crystallize, whereas the latter one was obtained as orange plates. Both ligands showed excellent performance as chiral ligands in a few representative catalytic asymmetric reactions.64),86)
3.3.2. 1,2-Bis(tert-butylmethylphosphino)benzene (BenzP*) and related ligands.
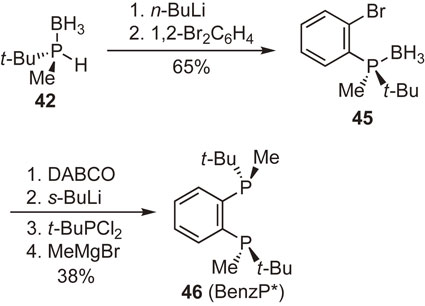
For years, we were extremely interested in the synthesis of 1,2-bis(tert-butylmethylphosphino)benzene (BenzP*), an ortho-phenylene-bridged P-chiral bisphosphine ligand. Although the ligand has a simple molecular structure, our attempts to synthesize it took approximately 10 years. Finally, in 2010, we found a method that enabled gram-scale synthesis.87)–89) We owe our success to our private communication with Professor Sylvain Jugé of the University of Bourgogne. When I visited Professor Jugé’s laboratory, he kindly taught me the reaction of lithiated secondary phosphine–boranes with o-dibromobenzene to produce (2-bromophenyl)(dialkyl)phosphine–boranes.90) By applying his protocol to the reaction of optically pure 42 with 1,2-dibromobenzene, we were able to obtain enantiopure 45 in good yield (Scheme 24). The conversion of 45 into BenzP* (46) in four steps was accomplished in one pot.89) During the reaction sequence, the introduction of another t-butylmethylphosphino group at the ortho-position failed to proceed stereoselectively, resulting in the formation of a considerably large amount of the undesired meso-isomer as the co-product. Fortunately, however, only the desired (R,R)-BenzP* (46) was isolated as a colorless crystalline solid from the reaction mixture. It was also fortunate that the enantiopure BenzP* was considerably air-stable, in contrast to the mixture of its stereoisomers, which was reported to be an air-sensitive oil.91) This air-stable property of the ligand in conjunction with its high enantioinduction ability makes it potentially useful in various catalytic asymmetric syntheses.
As has been described above, enantiopure tert-butylmethylphosphine–borane (42) plays a key role in the synthesis of QuinoxP* and BenzP*. I find this structurally simple secondary phosphine–borane very attractive, because it consists of a stereogenic phosphorus atom, a boranato group, a hydrogen atom, a methyl group, and a t-butyl group, all of which play respective roles in ligand synthesis or in the construction of an effective asymmetric environment. Whereas this compound was initially prepared on a laboratory scale as described in Schemes 9 and 23, its enantiomers are currently manufactured on a large scale through the optical resolution of the racemate at Nippon Chemical Industrial Co., Ltd.
With both enantiomers in hand, we prepared various P-chiral phosphine ligands by the reactions with electrophiles, followed by the removal of the boranato groups. The ligands prepared by this method are illustrated in Fig. 9. It should be noted that both enantiomers of t-Bu-BisP* could be conveniently prepared from 1,2-dichloroethane.64) Among these ligands, AlkynylP*,92) BulkyP*,93) 5,8-TMS-QuinoxP*,94) and 3H-QuinoxP*95)–97) have shown unique and remarkably high enantioselectivities in some transition-metal-catalyzed asymmetric transformations.
4. Use of QuinoxP* and related air-stable phosphine ligands in transition-metal-catalyzed asymmetric reactions
Since the commercialization of QuinoxP* and BenzP* by reagent dealers, these ligands have been widely used in both academia and industry for the development of new transition-metal-catalyzed asymmetric reactions as well as for the synthesis of the desired optically active compounds. The successful results obtained by the use of these ligands are many, and hence, only representative examples are described herein.
4.1. Asymmetric hydrogenation.
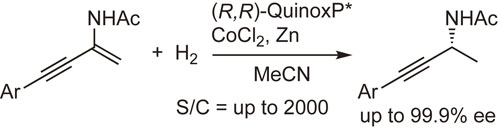
The rhodium-catalyzed asymmetric hydrogenation of functionalized alkenes is one of the most important catalytic asymmetric syntheses.98),99) Even though plentiful examples have been reported, the further development of the method by using new ligands is still a popular research subject. We have examined the asymmetric hydrogenation of a variety of functionalized alkenes by the use of QuinoxP* and analogous ligands and found that the reaction resulted in excellent enantioselectivities and had a broad substrate scope. The hydrogenation reactions of α- or β-dehydroamino acid derivatives are shown in Schemes 25 and 26 as typical examples.85),93) It is worth noting that the ligands could be employed in the hydrogenation of not only (E)- but also (Z)-β-dehydroamino acid derivatives (Scheme 26). These results indicate that the ligands, particularly QuinoxP*, are useful for the production of chiral ingredients containing an amino acid or amine moiety.85)

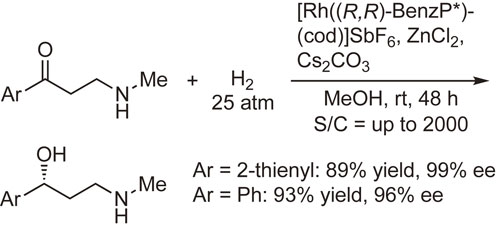
High enantioselectivities have also been observed in the asymmetric hydrogenation of ketones using QuinoxP* or BenzP*. For example, the asymmetric hydrogenation of β-secondary-amino ketones by a Rh-BenzP* catalyst was significantly promoted by ZnCl2 to afford the corresponding hydrogenation products with excellent enantiomeric excesses in high yields (Scheme 27).100) This procedure is potentially useful for the production of synthetic intermediates of (S)-duloxetine, (R)-fluoxetine, and (R)-atomoxetine, which are used as antidepressant drugs.
Recently, the use of non-precious metals, such as cobalt, nickel, and iron, in asymmetric hydrogenation has attracted increasing attention as an economical and environmentally friendly methodology. One reaction example using a cobalt-QuinoxP* catalyst is shown in Scheme 28.101) In this reaction, the alkene moiety was hydrogenated with very high enantioselectivity while keeping the alkyne intact.
Another example is the nickel-catalyzed asymmetric hydrogenation (Scheme 29).102) The use of a much lower catalyst loading (0.0095 mol%, S/C = 10500) has resulted in the highest catalytic activity for the Ni-catalyzed asymmetric hydrogenation reactions reported to date.
4.2. Carbon–carbon and carbon–heteroatom bond-forming reactions.
QuinoxP* and BenzP* have also been used in metal-catalyzed asymmetric carbon–carbon and carbon–heteroatom bond-forming reactions. Scheme 30 shows an example of a carbon–carbon bond-forming reaction reported by Buchwald and coworkers.103) This allylation reaction of ketones with allene, an underutilized hydrocarbon feedstock, proceeds without specialized equipment or pressurization to give allylation products with high enantiomeric excesses.
Some air-stable P-chiral phosphine ligands have found use in the enantioselective synthesis of optically active pharmaceutical ingredients. Scheme 31 shows an example reaction that was established by the research group of Merck & Co., Inc. for the production of the HCV drug candidate elbasvir.104) It should be noted that the C–N coupling reaction with the formation of the optically active hemiaminal ether is catalyzed by the Pd(0)–QuinoxP* mono-oxide complex produced by the reaction of Pd(OAc)2 with QuinoxP*.
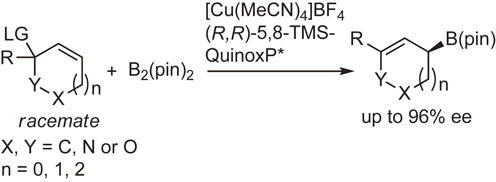
In 2010, Ito et al. disclosed a direct enantio-convergent transformation of racemic substrates without racemization or symmetrization by the use of Cu(I)–QuinoxP* complex as the chiral catalyst.105) By employing the modified ligand (R,R)-5,8-bis(trimethylsilyl)quinoxaline ((R,R)-5,8-TMS-QuinoxP*), the substrate scope was significantly expanded from five-membered compounds to six- and seven-membered ones (Scheme 32).94)
5. Mechanistic study of rhodium-catalyzed asymmetric hydrogenation of functionalized alkenes: A new approach for predicting the sense of enantioselectivity
As has been described in Section 3.2, we found that the asymmetric hydrogenation of α-dehydroamino acid derivatives with rhodium complex [Rh((S,S)-t-Bu-BisP*)(nbd)]BF4 (25a) provided (R)-configuration products with up to 99.9% ee.62) With the results in hand, we at first tried to explain the stereochemical outcome with respect to the hitherto reported empirical rule regarding the correlation between the catalyst δ or λ conformer and the absolute configuration of the products.26),106)–108) The rule predicts that the catalyst with δ-conformation provides (R)-enantiomer products and the catalyst with λ-conformation furnishes (S)-enantiomers. Therefore, according to the rule, our catalyst 25a with λ-conformation should provide (S)-configuration products. However, it turned out that the use of 25a afforded (R)-configuration products, which are the opposite enantiomers of those predicted by the empirical rule. On the other hand, in the case of the (R,R)-t-Bu-MiniPHOS (26a)–Rh complex, the δ,λ-empirical rule was not applicable to the entirely flat four-membered chelate cycle with no distinct quasi-axial and equatorial groups, although the complex afforded (R)-configuration products with excellent enantiomeric excesses. Another attempted explanation was made by applying the Halpern–Brown mechanism, which is also known as the unsaturated mechanism, the major/minor concept, or the anti-lock-and-key mechanism.109)–117) However, not only the origin and the sense of the enantioselection but also the observed very high enantioselectivities could not be reasonably explained by the mechanism. These discrepancies are the main reasons why we initiated our mechanistic studies of the rhodium-catalyzed asymmetric hydrogenation.
To detect the reaction intermediates and to examine their reactivities, we began our multinuclear NMR study with the use of complex 25a. Fortunately, complex 25a was sufficiently soluble in deuteriomethanol even at a very low temperature, and the spectral data obtained could be analyzed without difficulty owing to its simple molecular structure.
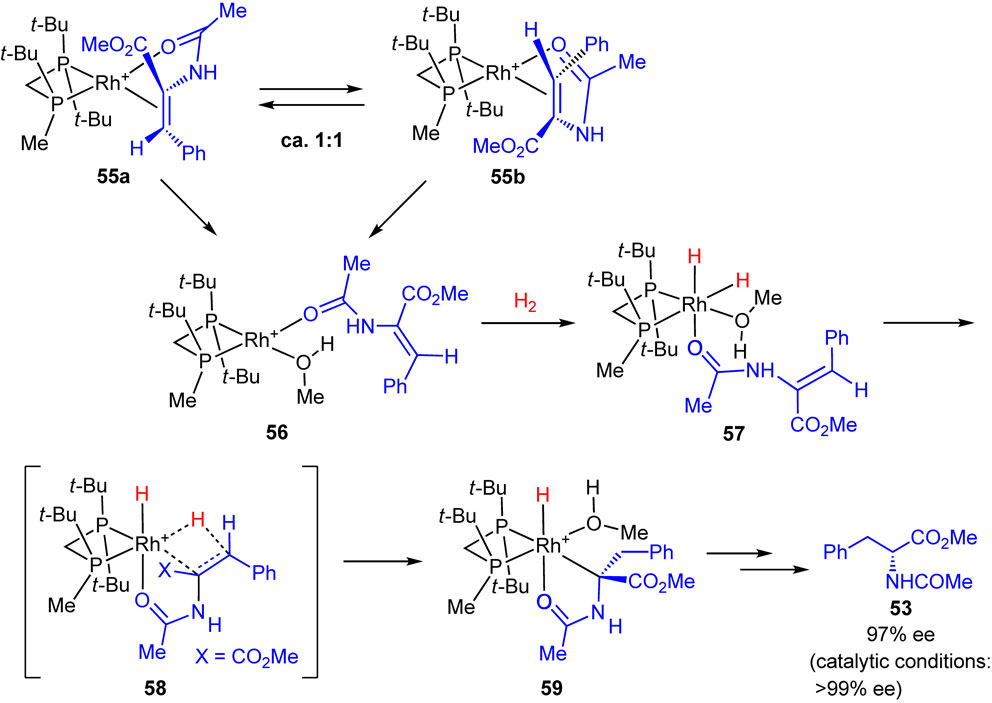
Our experimental findings and a plausible enantioselection mechanism including the catalytic cycle are shown in Scheme 33.118) One of the important findings is that solvate complex 47 generated from catalyst precursor 25a reacted reversibly with H2 at a low temperature to give diastereomeric dihydride intermediates 48a and 48b in approximately 10:1 ratio; these are the first observable Rh dihydrides with a bisphosphine ligand. These dihydrides reacted with probing substrate MAC very rapidly at −90 °C to give monohydride complex 52 detected by NMR measurement. The reaction was considered to proceed through 49, 50, and 51. Thus, the amide oxygen atom of MAC coordinated to the rhodium atom at the trans to the Rh–H bond to form complex 49, and subsequently, the carbon–carbon double bond moved to the Rh–H bond, leading to the transition state 50. The migratory insertion of the alkene into the Rh–H bond afforded monohydride complex 51, which in turn isomerized into a more stable monohydride 52. At temperatures above −50 °C, 52 underwent reductive elimination to afford hydrogenation product 53 with 99% ee and solvate complex 47.

On the other hand, solvate complex 47 reacted with MAC to form Rh-alkene complexes 54a and 54b in the ratio of approximately 10:1. These diastereomeric complexes were allowed to react with H2 (2 atm) at −80 °C for 1 h to give monohydride 52, which was detected by NMR measurement along with the signals of solvate complex 47 and dihydrides 48a and 48b. At the temperatures higher than −50 °C, it was converted into the hydrogenation product 53 with 97% ee (R). The absolute configuration of the product corresponded to that of si-coordinated minor complex 54b, and this hydrogenation followed seemingly the Halpern–Brown mechanism. However, this transformation proceeded at a relatively higher temperature and required a longer reaction time than the reaction of the dihydride complexes 48a and 48b with MAC. Therefore, it is reasonable to consider that the alkene complexes 54a and 54b exist in a resting state and are not directly subjected to hydrogenation; they reversibly dissociate into the solvate complex 47 and substrate MAC and eventually furnished 52 via the dihydride pathway.
We consider that the enantioselection would be determined at the association step to form hexacoordinated Rh(III) dihydride complex 50. Among the eight possible stereoisomers of the transition state structure, only 50 as the most favorable transition state satisfies the following steric and electronic requirements:
-
1.
A chelate ring is formed, avoiding steric repulsion with the bulky t-butyl group of the ligand.
-
2.
The carbon–carbon double bond is parallel to the Rh–H bond trans to the Rh–P bond.
-
3.
The α-carbon of the ester binds to the Rh atom during the migratory insertion.
The origin of the very high enantioselectivity is responsible for the multiple factors for lowering the transition state energy. This enantioselection mechanism is analogous to that of enzyme reactions, even though the catalyst is not a macromolecule similar to an enzyme. It is also worth mentioning that the relationship between the catalyst structure and the product chirality can be reasonably explained by considering transition state structure 50.
To confirm the dihydride mechanism, we studied the reactivities of tetracoordinated Rh(I)-alkene chelating complexes toward H2 using other electron-rich P-chiral phosphine ligands (R,R)-t-Bu-MiniPHOS, (R)-(tert-butylmethylphosphino)(di-tert-butylphosphino)methane (Trichikenfootphos: TCFP), (R,R)-BenzP*, etc. Scheme 34 shows an example of the studies based on low-temperature NMR measurements and DFT computations.119) In this case, TCFP–Rh solvate complex reacted with MAC to provide alkene complexes 55a and 55b in approximately 1:1 ratio, and no distinct major/minor concentrations were observed. A significant finding is that both the re- and si-coordinated alkene complexes 55a and 55b reacted with H2 at −78 °C to give the same (R)-enantiomer product in 97% ee. The NMR monitoring and extensive DFT computations led us to conclude that the hydrogenation of 55a and 55b does not occur directly, but is preceded by the dissociation of the double bond to result in the more reactive species 56. Semi-dissociated species 56 reacts with H2 to give non-chelating Rh(III)-dihydride complex 57, which undergoes migratory insertion via transition state 58 to generate monohydride 59. Finally, 59 is converted into product 53 via reductive elimination. It is noted that transition state 58 closely resembles 50 and the observed very high enantioselectivities would be responsible for the above-described steric and electronic effects.
In addition to the example mentioned above, the mechanisms of the Rh-catalyzed asymmetric hydrogenation of many functionalized alkenes, such as β-dehydroamino acid derivatives, enamides, and α,β-unsaturated phosphonates, were studied using not only (S,S)-t-Bu-BisP* but also other several P-chiral bisphosphine ligands. In all cases, the resulting high enantioselectivity and stereochemical outcome (R or S) were reasonably explained by considering the dihydride pathway. In addition, the absolute configuration of the product could be predicted by considering the transition structure in the association and migratory insertion step. Furthermore, the proposed catalytic cycle and the enantioselection mechanism would be crucial for the design of new chiral ligands and catalysts. More details are described in original papers65)–67),85),120)–126) and reviews.127)–129)
6. Conclusions
A new method for the synthesis of enantiopure P-chiral phosphine ligands has been established by utilizing the characteristic properties of phosphine–boranes. A rationally designed P-chiral phosphine ligand, t-Bu-BisP*, was synthesized in 1998, and its very high enantioinduction ability was demonstrated in the Rh-catalyzed asymmetric hydrogenation of functionalized alkenes. The discovery of the t-Bu-BisP* ligand ushered in a revival of P-chiral phosphine ligands in asymmetric catalysis, and many P-chiral ligands have been synthesized and successfully applied in a variety of transition-metal-catalyzed asymmetric reactions. In particular, TangPhos, DuanPhos, BIBOP, and QuinoxP* have found widespread use in both academia and industry owing to their exceedingly high enantioinduction ability and superior catalytic efficiency.
Structurally simple P-chiral phosphine ligands, such as BisP*, TCFP, and BenzP*, have been employed in the mechanistic studies of the Rh-catalyzed asymmetric hydrogenation of enamides and related substrates. It has been suggested that the hydrogenation proceeds through the dihydride pathway and the enantioselection is determined by the formation of the octahedral Rh(III) dihydride complex and the subsequent migratory insertion step. Furthermore, the absolute configuration of the products can be predicted by considering the multiple stereoregulating factors at the transition state.
Asymmetric catalysis is undoubtedly one of the most environmentally benign and economical methods for the production of enantiomerically pure or enriched high value-added compounds. I believe that P-chiral phosphine ligands along with many other backbone chirality ligands will play a vital role in the further development of the sophisticated and truly useful catalytic synthetic transformations.
Acknowledgments
The work described in this review was carried out at Chiba University and Nippon Chemical Industrial Co., Ltd., where many coworkers were engaged in these studies. I deeply thank all collaborators, whose names appear in the References section. My sincere appreciation also goes to the Ministry of Education, Culture, Sports, Science and Technology of Japan for the financial support in the form of a Grant-in-Aid for Scientific Research.
Notes
Edited by Ryoji NOYORI, M.J.A.
Correspondence should be addressed: T. Imamoto, Department of Chemistry, Graduate School of Science, Chiba University, 1-33, Yayoicho, Inage-ku, Chiba 263-8522, Japan (e-mail: imamoto@faculty.chiba-u.jp).
References
- 1) Noyori, R. (1994) Asymmetric Catalysis in Organic Synthesis. Wiley, New York.
- 2) Jacobsen, E.N., Pfaltz, A. and Yamamoto, H. (eds.) (1999) Comprehensive Asymmetric Catalysis, Vols. I–III. Springer-Verlag, Berlin.
- 3) Ojima, I. (ed.) (2010) Catalytic Asymmetric Synthesis, 3rd ed. John Wiley, Hoboken.
- 4) Blaser, H.-U. and Federsel, H.-J. (eds.) (2010) Asymmetric Catalysis on Industrial Scale, 2nd ed. Wiley-VCH, Weinheim.
- 5) Zhou, Q.-L. (ed.) (2011) Privileged Chiral Ligands and Catalysis. Wiley-VCH, Weinheim.
- 6) Tang, W. and Xumu, X. (2003) New chiral phosphorus ligands for enantioselective hydrogenation. Chem. Rev. 103, 3029–3070.
- 7) Börner, A. (ed.) (2008) Phosphorus Ligands in Asymmetric Catalysis: Synthesis and Application. Vols. 1–3, Wiley-VCH, Weinheim.
- 8) Grabulosa, A. (2011) P-Stereogenic Ligands in Enantioselective Catalysis. RSC Publishing, Cambridge.
- 9) Li, W. and Zhang, X. (2013) Chiral phosphines and diphosphines. In Phosphorus(III) Ligands in Homogeneous Catalysis (eds. Kamer, P.C.J. and van Leewen, P.W.N.M.). Wiley, Chichester, pp. 27–80.
- 10) Bayardon, J. and Jugé, S. (2013) P-Chiral ligands. In Phosphorus(III) Ligands in Homogeneous Catalysis (eds. Kamer, P.C.J. and van Leewen, P.W.N.M.). Wiley, Chichester, pp. 355–389.
- 11) Kolodiazhnyl, O.I. (2014) Recent advances in asymmetric synthesis of P-stereogenic phosphorus compounds. Top. Curr. Chem. 360, 161–236.
- 12) Dutartre, M., Bayardon, J. and Jugé, S. (2016) Applications and stereoselective syntheses of P-chirogenic phosphorus compounds. Chem. Soc. Rev. 45, 5771–5794.
- 13) Kolodiazhny, O.I. (2016) Asymmetric Synthesis in Organophosphorus Chemistry: Synthetic Methods, Catalysis, and Applications. Wiley-VCH, Weinheim.
- 14) Imamoto, T. (2016) Searching for practically useful P-chiral phosphine ligands. Chem. Rec. 16, 2655–2669.
- 15) Xu, G., Senanayake, C.H. and Tang, W. (2019) P-Chiral phosphorus ligands based on a 2,3-dihydrobenzo[d][1,3]oxaphosphole motif for asymmetric catalysis. Acc. Chem. Res. 52, 1101–1112.
- 16) Carbré, A., Riera, A. and Verdaguer, X. (2020) P-Stereogenic amino-phosphines as chiral ligands: From privileged intermediates to asymmetric catalysis. Acc. Chem. Res. 53, 676–689.
- 17) Zhao, Q., Chen, C., Wen, J., Dong, X.-Q. and Zhang, X. (2020) Noncovalent interaction-assisted ferrocenylphosphine ligands in asymmetric catalysis. Acc. Chem. Res. 53, 1905–1921.
- 18) Lemouzy, S., Giordano, L., Hérault, D. and Buono, G. (2020) Introducing chirality at phosphorus atoms: An update on the recent synthetic strategies for the preparation of optically pure P-stereogenic molecules. Eur. J. Org. Chem. 2020, 3351–3366.
- 19) Ye, F., Xu, Z. and Xu, L.-W. (2021) The discovery of multifunctional chiral P ligands for the catalytic construction of quarternary carbon/silicon and multiple stereogenic centers. Acc. Chem. Res. 54, 452–470.
- 20) Glueck, D.S. (2021) Catalytic asymmetric synthesis of P-stereogenic phosphines: Beyond precious metals. Synlett 32, 875–884.
- 21) Knowles, W.S. and Sabacky, M.J. (1968) Catalytic asymmetric hydrogenation employing a soluble optically active rhodium complex. Chem. Commun. 1445–1446.
- 22) Knowles, W.S., Sabacky, M.J. and Vineyard, B.D. (1972) Catalytic asymmetric hydrogenation. J. Chem. Soc. Chem. Commun. 10–11.
- 23) Horner, L., Siegel, H. and Büthe, H. (1968) Asymmetric catalytic hydrogenation with an optically active phosphinerhodium complex in homogeneous solution. Angew. Chem. Int. Ed. Engl. 7, 942.
- 24) Knowles, W.S., Sabacky, M.J., Vineyard, B.D. and Weinkauff, D.J. (1975) Asymmetric hydrogenation with a complex of rhodium and a chiral bisphosphine. J. Am. Chem. Soc. 97, 2567–2569.
- 25) Vineyard, B.D., Knowles, W.S., Sabacky, M.J., Bachman, G.L. and Weinkauff, D.J. (1977) Asymmetric hydrogenation. Rhodium chiral bisphosphine catalyst. J. Am. Chem. Soc. 99, 5946–5952.
- 26) Knowles, W.S. (1983) Asymmetric hydrogenation. Acc. Chem. Res. 16, 106–1012.
- 27) Knowles, W.S. (2002) Asymmetric hydrogenation (Nobel Lecture). Angew. Chem. Int. Ed. 41, 1998–2007.
- 28) Knowles, W.S. (2004) Asymmetric hydrogenation — The Monsanto l-dopa process. In Asymmetric Catalysis on Industrial Scale (eds. Blaser, H.-U. and Schmidt, E.). Wiley–VCH, Weinheim, pp. 23–38.
- 29) Namy, J.L., Girard, P. and Kagan, H.B. (1977) A new preparation of some divalent lanthanides and their usefulness in organic synthesis. Nouv. J. Chim. 1, 5–7.
- 30) Girard, P., Namy, J.L. and Kagan, H.B. (1980) Divalent lanthanide derivatives in organic synthesis. 1. Mild preparation of samarium iodide and ytterbium iodide and their use as reducing or coupling agents. J. Am. Chem. Soc. 102, 2693–2698.
- 31) Luche, J.L. (1978) Lanthanides in organic chemistry. 1. Selective 1,2 reductions of conjugated ketones. J. Am. Chem. Soc. 100, 2226–2227.
- 32) Gemal, A.L. and Luche, J.L. (1981) Lanthanoids in organic synthesis. 6. Reduction of α-enones by sodium borohydride in the presence of lanthanoid chlorides: Synthetic and mechanistic aspects. J. Am. Chem. Soc. 103, 5454–5459.
- 33) Imamoto, T., Kusumoto, T., Tawarayama, Y., Sugiura, Y., Mita, T., Hatanaka, Y. et al. (1984) Carbon–carbon bond-forming reactions using cerium metal or organocerium(III) reagents. J. Org. Chem. 49, 3904–3912.
- 34) Imamoto, T., Takiyama, N., Nakamura, K., Hatajima, T. and Kamiya, Y. (1989) Reactions of carbonyl compounds with Grignard reagents in the presence of cerium chloride. J. Am. Chem. Soc. 111, 4392–4398.
- 35) Takeda, N. and Imamoto, T. (1999) Use of cerium(III) chloride in the reactions of carbonyl compounds with organolithiums or Grignard reagents for the suppression of abnormal reactions: 1-Butyl-1,2,3,4-tetrahydro-1-naththol. Org. Synth. 76, 228–238.
- 36) Krasovskiy, A., Kopp, F. and Knochel, P. (2006) Soluble lanthanide salts (LnCl3•2LiCl) for the improved addition of organomagnesium reagents to carbonyl compounds. Angew. Chem. Int. Ed. 45, 497–500.
- 37) Krasovskiy, A., Gavryushin, A. and Knochel, P. (2009) LaCl3•2LiCl-catalyzed addition of Grignard reagents to ketones. Synlett 1433–1436.
- 38) Imamoto, T., Takeyama, T. and Kusumoto, T. (1985) Facile reduction of organic halides and phosphine oxides with LiAlH4–CeCl3. Chem. Lett. 1491–1492.
- 39) Imamoto, T., Kusumoto, T., Suzuki, N. and Sato, K. (1985) Phosphine oxides and LiAlH4–NaBH4–CeCl3: Synthesis and reactions of phosphine–boranes. J. Am. Chem. Soc. 107, 5301–5302.
- 40) Imamoto, T. and Hikosaka, T. (1994) Reactions of phosphine–monoiodoboranes with 4,4′-di-tert-butylbiphenylide and electrophiles. Trials of generation of tricoordinate boron anions and synthesis of B-functionalized phosphine–boranes. J. Org. Chem. 59, 6753–6759.
- 41) Schmidbaur, H., Blaschke, G. and Köhler, F.H. (1977) Tri(tert-butyl)methylenephosphorane: Consequences of steric hindrance for intramolecular motion and the thermal decomposition mechanism. Z. Naturforsch. 32, 757–761.
- 42) Imamoto, T. and Morishita, H. (2000) An enantiomerically pure tetracoordinate boron compound: Stereochemistry of substitution reactions at the chirogenic boron atom. J. Am. Chem. Soc. 122, 6329–6330.
- 43) Imamoto, T., Nagato, E., Wada, Y., Masuda, H., Yamaguchi, K. and Uchimaru, T. (1997) New boranophosphorylation reagents, dimethyl boranophosphate monopotassium salt and tetramethyl boranopyrophosphate. J. Am. Chem. Soc. 119, 9925–9926.
- 44) Imamoto, T., Oshiki, T., Onozawa, T., Kusumoto, T. and Sato, K. (1990) Synthesis and reactions of phosphine–boranes. Synthesis of new bidentate ligands with homochiral phosphine centers via optically pure phosphine–boranes. J. Am. Chem. Soc. 112, 5244–5252.
- 45) Oshiki, T. and Imamoto, T. (1990) The reactions of optically pure menthyloxymethylphenylphosphine–borane with organolithium reagents. Bull. Chem. Soc. Jpn. 63, 3719–3721.
- 46) Imamoto, T. and Tsuruta, H. (1996) Remarkable rate enhancement effect of the o-methoxy group in the nucleophilic substitution reactions at the phosphorus of phosphine-boranes. Chem. Lett. 25, 707–708.
- 47) Oshiki, T., Hikosaka, T. and Imamoto, T. (1991) Stereospecific reduction of menthyloxyphosphine–boranes with one-electron reducing agents. Tetrahedron Lett. 32, 3371–3374.
- 48) Imamoto, T., Matsuo, M., Nonomura, T., Kishikawa, K. and Yanagawa, M. (1993) Synthesis and reactions of optically pure cyclohexyl(o-methoxyphenyl)phosphine–borane and t-butyl(o-methoxyphenyl)phosphine–borane. Heteroatom Chem. 4, 475–486.
- 49) Miura, T., Yamada, H., Kikuchi, S. and Imamoto, T. (2000) Synthesis and reactions of optically active secondary dialkylphosphine–boranes. J. Org. Chem. 65, 1877–1880.
- 50) Oshiki, T. and Imamoto, T. (1992) Unprecedented stereochemistry of the electrophilic arylation at chiral phosphorus. J. Am. Chem. Soc. 114, 3975–3977.
- 51) Imamoto, T., Oshiki, T., Onozawa, T., Matsuo, M., Hikosaka, T. and Yanagawa, M. (1992) Synthesis and reactions of optically active phosphine–boranes. Heteroatom Chem. 3, 563–575.
- 52) Imamoto, T., Hirose, K., Amano, H. and Seki, H. (1996) Stereospecific replacement of the boranato group of phosphine–boranes by oxygen or sulfur. Synthesis of optically active P-chiral phosphine oxides, phosphine sulfides, phosphinates, and phosphinothioates. Main Group Chem. 1, 331–338.
- 53) Imamoto, T., Asakura, K., Tsuruta, H., Kishikawa, K. and Yamaguchi, K. (1996) Synthesis and properties of trifluoromethanesulfonyloxy derivatives of tricyclohexylphosphine–borane. Tetrahedron Lett. 37, 503–504.
- 54) Imamoto, T. and Oshiki, T. (1989) Synthesis of organic phosphorus compounds containing a linear P–B bond chain. Tetrahedron Lett. 30, 383–384.
- 55) Imamoto, T. and Yamanoi, Y. (1996) Methylene insertion reactions of samarium carbenoids into boron–hydrogen and phosphorus–hydrogen bonds. Chem. Lett. 25, 705–706.
- 56) Imamoto, T., Tsuruta, H., Wada, Y., Masuda, H. and Yamaguchi, K. (1995) A new P-chiral bisphosphine, (S,S)-1,2-bis[(o-ethylphenyl)phenylphosphino]ethane, as an effective ligand in catalytic asymmetric hydrogenation of α-(acylamino)acrylic acids. Tetrahedron Lett. 36, 8271–8274.
- 57) Wada, Y., Imamoto, T., Tsuruta, H., Yamaguchi, K. and Gridnev, I.D. (2004) Optically pure 1,2-bis[(o-alkylphenyl)phenylphosphino]ethanes and their use in rhodium-catalyzed asymmetric hydrogenation of α-(acylamino)acrylic derivatives. Adv. Synth. Catal. 346, 777–788.
- 58) Burk, M.J., Feaster, J.E. and Harlow, R.L. (1990) New electron-rich chiral phosphines for asymmetric catalysis. Organometallics 9, 2653–2655.
- 59) Burk, M.J. (1991) C2-Symmetric bis(phospholanes) and their use in highly enantioselective hydrogenation reactions. J. Am. Chem. Soc. 113, 8518–8519.
- 60) Burk, M.J., Feaster, J.E. and Nugent, R.L. (1993) Preparation and use of C2-symmetric bis(phospholanes): Production of α-amino acid derivatives via highly enantioselective hydrogenation reactions. J. Am. Chem. Soc. 115, 10125–10138.
- 61) Burk, M.J. (2000) Modular phospholane ligands in asymmetric catalysis. Acc. Chem. Res. 33, 363–370.
- 62) Imamoto, T., Watanabe, J., Wada, Y., Masuda, H., Yamada, H., Tsuruta, H. et al. (1998) P-Chiral bis(trialkylphosphine) ligands and their use in highly enantioselective hydrogenation reactions. J. Am. Chem. Soc. 120, 1635–1636.
- 63) Yamanoi, Y. and Imamoto, T. (1999) Methylene-bridged P-chiral diphosphines in highly enantioselective reactions. J. Org. Chem. 64, 2988–2989.
- 64) Imamoto, T., Horiuchi, Y., Hamanishi, E., Takeshita, S., Tamura, K., Sugiya, M. et al. (2015) Utilization of optically active secondary phosphine–boranes: Synthesis of P-chiral diphosphines and their enantioinduction ability in rhodium-catalyzed asymmetric hydrogenation. Tetrahedron 71, 6471–6480.
- 65) Gridnev, I.D., Yamanoi, Y., Higashi, N., Tsuruta, H., Yasutake, M. and Imamoto, T. (2001) Asymmetric hydrogenation catalyzed by (S,S)-R-BisP*-Rh and (R,R)-R-MiniPHOS complexes: Scope, limitations, and mechanism. Adv. Synth. Catal. 343, 118–136.
- 66) Gridnev, I.D., Yasutake, N., Higashi, N. and Imamoto, T. (2001) Asymmetric hydrogenation of enamides with Rh-BisP* and Rh-MiniPHOS catalysts. Scope, limitation, and mechanism. J. Am. Chem. Soc. 123, 5268–5276.
- 67) Gridnev, I.D., Yasutake, M., Imamoto, T. and Beletskaya, I.P. (2004) Asymmetric hydrogenation of α,β-unsaturated phosphonates with Rh-BisP* and Rh-MiniPHOS catalysts: Scope and mechanism of the reaction. Proc. Natl. Acad. Sci. U.S.A. 101, 5385–5390.
- 68) Imamoto, T., Iwadate, N. and Yoshida, K. (2006) Enantioselective hydrogenation of acyclic aromatic N-aryl imines catalyzed by an iridium complex of (S,S)-1,2-bis(tert-butylmethylphosphino)ethane. Org. Lett. 8, 2289–2292.
- 69) Imamoto, T., Itoh, T., Yamanoi, Y., Narui, R. and Yoshida, K. (2006) Highly enantioselective hydrosilylation of simple ketones catalyzed by rhodium complexes of P-chiral diphosphine ligands bearing tert-butyl groups. Tetrahedron Asymmetry 17, 560–565.
- 70) Ohashi, A. and Imamoto, T. (2000) 1-tert-Butyl-2-methylphospholane–borane and its coupling product 2,2′-bis(1-tert-butylphospholane–borane). Acta Crystallogr. C 56, 723–725.
- 71) Tang, W. and Zhang, X. (2002) A chiral 1,2-bisphospholane ligand with a novel structural motif: Applications in highly enantioselective Rh-catalyzed hydrogenation. Angew. Chem. Int. Ed. 41, 1612–1614.
- 72) Tang, W. and Xumu, X. (2002) Highly efficient synthesis of chiral beta-amino acid derivatives via asymmetric hydrogenation. Org. Lett. 4, 4159–4161.
- 73) Min, G., Meng, J.-J., Lv, H. and Zhang, X. (2015) Highly regio- and enantioselective synthesis of γ,δ-unsaturated amido esters by catalytic hydrogenation of conjugated enamides. Angew. Chem. Int. Ed. 54, 1885–1887 and references cited therein.
- 74) Tang, W., Wang, W., Chi, Y. and Zhang, X. (2003) A bisphosphepine ligand with stereogenic phosphorus centers for the practical synthesis of β-aryl-β-amino acids by asymmetric hydrogenation. Angew. Chem. Int. Ed. 42, 3509–3511.
- 75) Imamoto, T., Oohara, N. and Takahashi, H. (2004) Optically active 1,1-di-tert-butyl-2,2-diphosphetanyl and its application in rhodium-catalyzed asymmetric hydrogenations. Synthesis 1353–1358.
- 76) Imamoto, T., Crépy, K.V.L. and Katagiri, K. (2004) Optically active 1,1′-di-tert-butyl-2,2′-dibenzophosphetanyl: A highly strained P-stereogenic diphosphine ligand. Tetrahedron Asymmetry 15, 2213–2218.
- 77) Liu, D. and Zhang, X. (2005) Practical P-chiral phosphane ligand for Rh-catalyzed asymmetric hydrogenation. Eur. J. Org. Chem. 2005, 646–649.
- 78) Yang, H., Wang, E., Yang, P., Lv, H. and Zhang, X. (2017) Pyridine-directed asymmetric hydrogenation of 1,1-diarylalkenes. Org. Lett. 19, 5062–5065 and references cited therein.
- 79) Tang, W., Qu, B., Capacci, A.G., Rodriguez, S., Wei, X., Haddad, N. et al. (2010) Novel, tunable, and efficient chiral bisdihydrobenzooxaphosphole ligands for asymmetric hydrogenation. Org. Lett. 12, 176–179.
- 80) Xu, G., Senanayake, C.H. and Tang, W. (2019) P-Chiral phosphorus ligands based on a 2,3-dihydrobenzo[d][1,3]oxaphosphole motif for asymmetric catalysis. Acc. Chem. Res. 52, 1101–1112 and references cited therein.
- 81) Zhang, X., Huang, K., Hou, G., Cao, B. and Zhang, X. (2010) Electron-donating and rigid P-stereogenic bisphospholane ligands for highly enantioselective rhodium-catalyzed asymmetric hydrogenations. Angew. Chem. Int. Ed. 49, 6421–6424.
- 82) Wang, F. and Tang, W. (2021) Phosphorus ligands from the Zhang lab: Design, asymmetric hydrogenation, and industrial applications. Chin. J. Chem. 39, 954–968.
- 83) Imamoto, T., Sugita, K. and Yoshida, K. (2005) An air-stable P-chiral phosphine ligand for highly enantioselective transition-metal-catalyzed reactions. J. Am. Chem. Soc. 127, 11934–11935.
- 84) Nagata, K., Matsukawa, S. and Imamoto, T. (2000) Stereoselective, oxidative one-carbon degradation of alkyl(dimethyl)phosphine–boranes. Synthesis of enantiomerically enriched secondary phosphine–boranes. J. Org. Chem. 65, 4185–4188.
- 85) Imamoto, T., Tamura, K., Zhang, Z., Horiuchi, Y., Sugiya, M., Yoshida, K. et al. (2012) Rigid P-chiral phosphine ligands with tert-butylmethylphosphino groups for rhodium-catalyzed asymmetric hydrogenation of functionalized alkenes. J. Am. Chem. Soc. 134, 1754–1769.
- 86) Imamoto, T., Kumada, A. and Yoshida, K. (2007) Air-stable P-chiral bidentate phosphine ligand with (1-adamantyl)methylphosphino group. Chem. Lett. 36, 500–501.
- 87) Miura, T. and Imamoto, T. (1999) Enantiomerically pure 1,2-bis(isopropylmethylphosphino)benzene and its use in highly enantioselective Rh-catalyzed asymmetric hydrogenation. Tetrahedron Lett. 40, 4833–4836.
- 88) Yamamoto, Y., Koizumi, T., Katagiri, K., Furuya, Y., Danjo, H., Imamoto, T. et al. (2006) Facile synthesis of highly congested 1,2-diphosphinobenzenes from bis(phosphine)boronium salts. Org. Lett. 8, 6103–6106.
- 89) Tamura, K., Sugiya, M., Yoshida, K., Yanagisawa, A. and Imamoto, T. (2010) Enantiopure 1,2-bis(tert-butylmethylphosphino)benzene as a highly efficient ligand in rhodium-catalyzed asymmetric hydrogenation. Org. Lett. 12, 4400–4403.
- 90) Bayardon, J., Laureano, H., Diemer, V., Dutartre, M., Das, U., Rousselin, Y. et al. (2012) Stereoselective synthesis of o-bromo (or iodo)aryl P-chirogenic phosphines based on aryne chemistry. J. Org. Chem. 77, 5759–5769.
- 91) Kyba, E.P., Kerby, M.C. and Rines, S.P. (1986) A convenient synthesis of symmetrical and unsymmetrical 1,2-bis(phosphino)benzenes as ligands for transition metals. Organometallics 5, 1189–1194.
- 92) Imamoto, T., Saitoh, Y., Koide, A., Ogura, T. and Yoshida, K. (2007) Synthesis and enantioselectivity of P-chiral phosphine ligands with alkynyl groups. Angew. Chem. Int. Ed. 46, 8636–8639.
- 93) Sawatsugawa, Y., Tamura, K., Sano, N. and Imamoto, T. (2019) A bulky three-hindered quadrant bisphosphine ligand: Synthesis and application in rhodium-catalyzed asymmetric hydrogenation of functionalized alkenes. Org. Lett. 21, 8874–8876.
- 94) Iwamoto, H., Ozawa, Y., Takenouchi, Y., Imamoto, T. and Ito, H. (2021) Backbone-modified C2-symmetrical chiral bisphosphine TMS-QuinoxP*: Asymmetric borylation of racemic allyl electrophiles. J. Am. Chem. Soc. 143, 6413–6422.
- 95) Zhang, Z., Tamura, K., Mayama, D., Sugiya, M. and Imamoto, T. (2012) Three-hindered quadrant phosphine ligands with an aromatic ring backbone for the rhodium-catalyzed asymmetric hydrogenation of functionalized alkenes. J. Org. Chem. 77, 4184–4188.
- 96) Iwamoto, H., Imamoto, T. and Ito, H. (2018) Computational design of high performance ligand for enantioselective Markovnikov hydroboration of aliphatic terminal alkenes. Nat. Commun. 9, 2290.
- 97) Iwamoto, H., Endo, K., Ozawa, Y., Watanabe, Y., Kobota, K., Imamoto, T. et al. (2019) Copper(I)-catalyzed enantioconvergent borylation of racemic benzyl chlorides enabled by quadrant-by-quadrant structure modulation of chiral bisphosphine ligands. Angew. Chem. Int. Ed. 58, 11112–11117.
- 98) Shang, G., Li, W. and Zhang, X. (2010) Transition metal-catalyzed homogeneous asymmetric hydrogenation. In Catalytic Asymmetric Synthesis (ed. Ojima, I.). Wiley, New Jersey, pp. 343–436.
- 99) Imamoto, T. (2019) Rhodium(I)-catalyzed asymmetric hydrogenation. In Rhodium Catalysis in Organic Synthesis (ed. Tanaka, K.). Wiley-VCH, Weinheim, pp. 3–37.
- 100) Hu, Q., Zhang, Z., Liu, Y., Imamoto, T. and Zhang, W. (2015) ZnCl2-promoted asymmetric hydrogenation of β-secondary-amino ketones catalyzed by a P-chiral bisphosphine–Rh complex. Angew. Chem. Int. Ed. 54, 2260–2264.
- 101) Hu, Y., Zhang, Z., Liu, Y. and Zhang, W. (2021) Cobalt-catalyzed chem- and enantioselective hydrogenation of conjugated enynes. Angew. Chem. Int. Ed. 60, 16989–16993.
- 102) Li, B., Chen, J., Zhang, Z., Gridnev, I.D. and Zhang, W. (2019) Ni-catalyzed asymmetric hydrogenation of N-sulfonyl imines. Angew. Chem. Int. Ed. 58, 7329–7334.
- 103) Liu, R.Y., Zhou, Y., Yang, Y. and Buchwald, S.L. (2019) Enantioselective allylation using allene, a petroleum cracking byproduct. J. Am. Chem. Soc. 141, 2251–2256.
- 104) Li, H., Belyk, K.M., Yin, J., Chen, Q., Hyde, A., Ji, Y. et al. (2015) Enantioselective synthesis of hemiaminals via Pd-catalyzed C–N coupling with chiral bisphosphine mono-oxide. J. Am. Chem. Soc. 137, 13728–13731.
- 105) Ito, H., Kunii, M. and Sawamura, M. (2010) Direct enantio-convergent transformation of racemic substrates without racemization or symmetrization. Nat. Chem. 2, 972–976.
- 106) Frizuk, M.D. and Bosnich, B. (1977) Asymmetric synthesis. Production of optically active amino acids by catalytic hydrogenation. J. Am. Chem. Soc. 99, 6262–6267.
- 107) Samuel, O., Couffignal, R., Lauer, M., Zhang, S.Y. and Kagan, H.B. (1981) Phellanphos and nopaphos. New diphosphines for asymmetric catalysis. Nouv. J. Chim. 5, 15–20.
- 108) Pavlov, V.A., Klabunovskii, E.I., Struchkov, Y.T., Voloboev, A.A. and Yanovsky, A.I. (1988) Asymmetric reduction of C=C and C=O bonds: Stereochemical approach. J. Mol. Catal. 44, 217–243.
- 109) Chan, A.C.S. and Halpern, J. (1980) Interception and characterization of a hydridoalkylrhodium intermediate in a homogeneous catalytic hydrogenation reaction. J. Am. Chem. Soc. 102, 838–840.
- 110) Chan, A.C.S., Pluth, J.J. and Halpern, J. (1980) Identification of the enantioselection step in the asymmetric catalytic hydrogenation of a prochiral olefin. J. Am. Chem. Soc. 102, 5952–5954.
- 111) Halpern, J. (1982) Mechanism and stereoselectivity of asymmetric hydrogenation. Science 217, 401–407.
- 112) Halpern, J. (1985) Asymmetric catalytic hydrogenation: Mechanism and origin of enantioselection. In Asymmetric Synthesis (ed. Morrison, J.D.). Academic Press, Orlando, Vol. 5, pp. 41–69.
- 113) Landis, C.R. and Halpern, J. (1987) Asymmetric hydrogenation of methyl (Z)-α-acetamidocinnamate catalyzed by [1,2-bis((phenyl-o-anisoyl)phosphino]ethane]rhodium(I): Kinetics, mechanism and origin of enantioselection. J. Am. Chem. Soc. 109, 1746–1754.
- 114) Brown, J.M. and Chalonger, P.A. (1980) Structural characterization of a transient intermediate in rhodium-catalyzed asymmetric hydrogenation reaction. J. Chem. Soc. Chem. Commun. 344–346.
- 115) Brown, J.M. and Chalonger, P.A. (1980) The mechanism of asymmetric homogeneous hydrogenation. Rhodium(I) complexes of dehydroamino acids containing asymmetric ligands related to bis(1,2-diphenylphosphino)ethane. J. Am. Chem. Soc. 102, 3040–3048.
- 116) Brown, J.M. (1999) Hydrogenation of functionalized carbon–carbon double bonds. In Comprehensive Asymmetric Catalysis (eds. Jacobsen, E.N., Pfaltz, A. and Yamamoto, H.). Springer-Verlag, Berlin, Vol. 1, pp. 121–182.
- 117) Brown, J.M. (2014) Rhodium asymmetric hydrogenation observed during its experimental growth phase. Organometallics 33, 5912–5923.
- 118) Gridnev, I.D., Higashi, N., Asakura, K. and Imamoto, T. (2000) Mechanism of asymmetric hydrogenation catalyzed by a rhodium complex of (S,S)-1,2-bis(tert-butylmethylphosphino)ethane. Dihydride mechanism of asymmetric hydrogenation. J. Am. Chem. Soc. 122, 7183–7194.
- 119) Gridnev, I.D., Imamoto, T., Hoge, G., Kouchi, M. and Takahashi, H. (2008) Asymmetric hydrogenation catalyzed by a rhodium complex of (R)-(tert-butylmethylphosphino)(di-tert-bututylphosphino)methane: Scope of enantioselectivity and mechanistic study. J. Am. Chem. Soc. 130, 2560–2572.
- 120) Gridnev, I.D., Higashi, N. and Imamoto, T. (2000) On the origin of opposite stereoselection of the asymmetric hydrogenation of phenyl- and tert-butyl-substituted enamides. J. Am. Chem. Soc. 122, 10486–10487.
- 121) Gridnev, I.D., Higashi, N. and Imamoto, T. (2001) Interconversion of monohydride intermediates in Rh(I)-catalyzed asymmetric hydrogenation of dimethyl 1-benzoyloxyethenephosphonates. J. Am. Chem. Soc. 123, 4631–4632.
- 122) Gridnev, I.D., Higashi, N. and Imamoto, T. (2001) Formation of a stable rhodium(III) dihydride complex and its reaction with prochiral substrates of asymmetric hydrogenation. Organometallics 20, 4542–4553.
- 123) Imamoto, T., Yashio, K., Crépy, K.V.L., Katagiri, K., Takahashi, H., Kouchi, M. et al. (2006) P-Chiral tetraphosphine dirhodium complex as a catalyst for asymmetric hydrogenation: Synthesis, structure, enantioselectivity, and mechanism. Stereoselective formation of a dirhodium tetrahydride complex and its reaction with methyl (Z)-α-acetamidocinnamate. Organometallics 25, 908–914.
- 124) Imamoto, T., Ito, T., Yoshida, K. and Gridnev, I.D. (2008) Marked deuterium isotope effects on the enantioselectivity in rhodium-catalyzed asymmetric hydrogenation of enamides. Chem. Asian J. 3, 1636–1641.
- 125) Gridnev, I.D., Liu, Y. and Imamoto, T. (2014) Mechanism of asymmetric hydrogenation of β-dehydroamino acids catalyzed by rhodium complexes: Large-scale experimental and computational study. ACS Catal. 4, 203–219.
- 126) Gridnev, I.D. and Imamoto, T. (2015) Challenging the major/minor concept in Rh-catalyzed asymmetric hydrogenation of enamides. ACS Catal. 5, 2911–2915.
- 127) Gridnev, I.D. and Imamoto, T. (2004) On the mechanism of stereoselection in Rh-catalyzed asymmetric hydrogenation: A general approach for predicting the sense of enantioselectivity. Acc. Chem. Res. 37, 633–644.
- 128) Gridnev, I.D. and Imamoto, T. (2009) Mechanism of enantioselection in Rh-catalyzed asymmetric hydrogenation. The origin of utmost catalytic performance. Chem. Commun. 7447–7464.
- 129) Gridnev, I.D. and Imamoto, T. (2016) Enantioselection mechanism in Rh-catalyzed asymmetric hydrogenation. Russ. Chem. Bull. Int. Ed. 65, 1514–1534.
Profile

Tsuneo Imamoto was born in 1942 in Shizuoka Prefecture, Japan. After graduating from Shizuoka University, he proceeded to study physical organic chemistry under the supervision of Professor Yasuhide Yukawa at Osaka University and obtained his Ph.D. degree in 1972. After postdoctoral work under the direction of Professor Teruaki Mukaiyama at Tokyo Institute of Technology, he was appointed an assistant professor at Osaka University in 1973. In 1975, he moved to Wayne State University in Detroit, Michigan, where he studied organophosphorus chemistry and asymmetric synthesis as a postdoctoral fellow under Professor Carl R. Johnson. In 1978, he joined the research group of Professor Mukaiyama at the University of Tokyo, and in 1980, he moved to Chiba University where he worked as an assistant professor. He was promoted to associate professor in 1987 and professor in 1993, and he retired from Chiba University in 2008. Currently, he is a professor emeritus and a grand fellow of Chiba University. He worked as a research consultant of Nippon Chemical Industrial Co., Ltd. (2008–2020) and was a visiting professor of Shanghai Jiao Tong University (2009–2018). Since 2020, he has been working as a visiting professor of Hokkaido University. His research interests lie in the areas of synthetic methodology, organoelement chemistry, asymmetric catalysis, and organic reaction mechanism. He pioneered the use of phosphine–boranes for the synthesis of phosphine ligands and invented cerium(III)-modified organometallic reagents, which have found widespread use in the efficient addition reactions of carbonyl compounds. He is a recipient of the Synthetic Organic Chemistry Award (Academic Division), Japan (1997); the Rare Earth Society Award, Japan (2001); the Prize for Science and Technology by the Ministry of Education, Culture, Sports, Science and Technology (2008); the Synthetic Organic Chemistry Award (Technology Division), Japan (2013); and the Ryoji Noyori Prize (2020).