Reviews
Possibilities and challenges of small molecule organic compounds for the treatment of repeat diseases
2022 年 98 巻 1 号 p. 30-48
詳細
2022 年 98 巻 1 号 p. 30-48
The instability of repeat sequences in the human genome results in the onset of many neurological diseases if the repeats expand above a certain threshold. The transcripts containing long repeats sequester RNA binding proteins. The mechanism of repeat instability involves metastable slip-out hairpin DNA structures. Synthetic organic chemists have focused on the development of small organic molecules targeting repeat DNA and RNA sequences to treat neurological diseases with repeat-binding molecules. Our laboratory has studied a series of small molecules binding to mismatched base pairs and found molecules capable of binding CAG repeat DNA, which causes Huntington’s disease upon expansion, CUG repeat RNA, a typical toxic RNA causing myotonic dystrophy type 1, and UGGAA repeat RNA causing spinocerebellar ataxia type 31. These molecules exhibited significant beneficial effects on disease models in vivo, suggesting the possibilities for small molecules as drugs for treating these neurological diseases.
The human genome contains many repeat sequences related to human genetic diseases.1) Telomere repeats (TTAGGG) at the ends of human chromosomes shorten as the genome replicates,2)–4) and when the repeat length falls below a certain threshold, cell death is induced. In cancer cells that have acquired immortality, the enzyme telomerase is activated to elongate the shortened telomere repeat.5) Depending on their genomic location, sequence, and the length of repeats, repeat sequences are associated with the development of various neurodegenerative diseases.6) Repetitions of aberrantly expanded trinucleotide sequences (trinucleotide repeats) are known to be the cause of trinucleotide repeat diseases.7),8)
Specifically, the CGG repeat sequence in exon 1 of the FMR1 gene on the X chromosome is approximately 40 repeats in healthy individuals, but when it exceeds 200 repeats due to aberrant expansion, Fragile X Syndrome develops.9)–11) (Table 1) Similarly, the CCG repeat sequence on the FMR2 gene on the X chromosome is approximately 40 repeats in normal individuals but exceeds 200 repeats in patients with Fragile XE syndrome.12)–14) The CGG repeat sequence in exon 1 of the FMR1 gene and the CCG repeat sequence in the FMR2 gene are both located in the 5′-untranslated region (5′-UTR) of their respective genes. Huntington’s disease (HD) is caused by an expansion of CAG repeats in the coding region of the first exon of the HTT gene, which encodes the huntingtin protein on chromosome 4.15)–17) The length of the CAG repeat is approximately 35 repeats or less in healthy individuals, but when it exceeds 40 repeats, the probability of developing HD increases. The CAG repeats are translated into glutamine, so the aberrantly expanded CAG repeats form a long continuous region of glutamine in the huntingtin protein. Aggregation of the glutamine region was thought to be the cause of the disease onset,18)–20) HD is also called polyglutamine disease. Myotonic dystrophy type 1 (DM1) is a systemic disease characterized by muscle atrophy, myotonia, testicular atrophy, cataracts, and glucose intolerance.21)–23) The gene responsible for DM1 is the DMPK gene on chromosome 19, and the direct cause of the disease is the aberrant expansion of CTG repeats in its 3′-UTR. The number of repeats in normal individuals ranges from 5 to 37, and DM1 is diagnosed when the number of repeats exceeds 50.
| Repeat sequence | Gene (location) | Gene disorder | Wild-type (n =) | Mutant type (n =) |
|---|---|---|---|---|
| (CGG)n | FMR1 (Chr. X, exon 1, 5′-UTR) |
Fragile X syndrome | 5–40 | 60–200 |
| (CCG)n | FMR2 (Chr. X, exon 1, 5′-UTR) |
Fragile XE syndrome | 4–39 | >200 |
| (CAG)n | HTT (Chr. 4, exon 1, ORF) |
Huntington disease | 6–39 | 36–121 |
| (GAA)n | X25 (frataxin) (Chr. 9, intron 1) |
Friedreich ataxia | 6–34 | 112–1700 |
| (CTG)n | DMPK (Chr. 19, exon 15, 3′-UTR) |
Myotonic dystrophy type 1 |
5–37 | 50–6500 |
| (TGGAA)n | BEAN, TK2 (Chr. 19, intron) |
Spinocerebellar ataxia type 31 |
0 | 500–760 |
| (GGGGCC)n | C9orf72 (Chr. 9, intron) |
Amyotrophic lateral sclerosis |
2–19 | 250–1600 |
Disease-causing repeat sequences are not limited to trinucleotide sequences; diseases involving penta and hexanucleotide repeat sequences are also known. Spinocerebellar ataxia type 31 (SCA31)24) is caused by the insertion of a TGGAA pentanucleotide repeat extending 2.5 to 3.8 kilobases into an intron shared by the BEAN and TK2 genes on chromosome 16.25) In addition, approximately 40% of familial amyotrophic lateral sclerosis is reported to be caused by aberrant expansion of the GGGGCC hexanucleotide repeat sequence in intron 1 of C9orf72.26),27) Expansion or insertion of these repeats directly affects the amino acid sequence of the protein if the repeats are in the translated region, as in the HTT gene. In addition, many repeat diseases are now thought to be caused by aberrantly long repeat sequences in transcripts, whether translated or not, that acquire the ability to capture RNA-binding proteins. The term “toxic RNA” is used to describe RNA that has acquired toxicity.28)–35)
Repeat sequences in the genome can expand or shorten during biological reactions such as replication and transcription, where the double strands dissociate.36),37) Aberrantly expanded repeat sequences are known to form metastable slipped strand DNA (S-DNA) with a hairpin structure in each repeat region on both strands when the double strand is dissociated, and the stability of the S-DNA structure is thought to be related to the ease of repeat expansion and shortening.38)–40) (Fig. 1a) Sequestration of RNA-binding proteins by toxic RNA also involved the hairpin structure of the expanded RNA repeat. For example, in the aberrantly expanded CAG repeats that cause HD, a hairpin structure consisting of CAG repeats is formed on one strand and a hairpin structure consisting of CTG repeats is formed on the complementary strand. The CAG hairpin consists of a CAG/CAG unit comprising of an A-A mismatch flanked by C-G base pairs, and the CTG hairpin consists of a CTG/CTG unit with a T-T mismatch sandwiched between C-G base pairs. (Fig. 1b) We were fortunate to discover a CAG repeat-binding molecule, naphthyridine-azaquinolone (NA),41) which binds to the CAG/CAG unit.42)–44) (Fig. 2a) When this NA was administered to the brain striatum of a mouse model of HD, we found that the CAG repeat length was contracted45) (Figs. 2b and 2c). This suggested the possibility that the administration of small organic compounds may inhibit the onset of the disease by contracting the repeat length, alleviate the symptoms, and even cure the disease by shortening the repeat length below the threshold for onset. In this review, we will first show the in vivo effects of our molecules on repeat diseases models, and then explain the binding mode of these molecules to repeat hairpins, and the ideas and concepts from the chemistry viewpoint that led to the development of these molecules, from the last part of the research to the first, as opposed to the flow of the research, to reiterate the real appeal and importance of organic chemistry and synthetic organic chemistry, which deals with molecules at the atomic level. Finally, the challenges and perspective for innovative academic questions derived from this research will be discussed.

(a) Illustration of the formation of slipped strand DNA (S-DNA) structures in the CTG/CAG repeat. (b) The secondary structures of CTG and CAG hairpins. The dotted line indicated the repeated units of T-T and A-A mismatches flanked by C-G base pairs.

(a) Chemical structure of CAG repeat-binding molecule NA (shown in bold) and illustration of the formation of hydrogen bond pairs with guanine and adenine. (b) Direct injection of NA into the striatum of an HD mouse model. (c) Comparison of CAG repeats treated with NA (left striatum, solid line) and those treated with saline (right striatum, dotted line) after four injections at 1-week intervals.
The specificity of NA, a molecule that binds to the CAG hairpin structure, for binding to the CAG S-DNA structure was examined by our collaborator Dr. Christopher Pearson (University of Toronto and Sick Kids Institute, Toronto, Canada) using the CAG/CTG S-DNA with an electrophoretic mobility shift assay (EMSA).45) As a result, it was confirmed that NA binds to slip-out with longer CAG repeats and hardly binds to CTG slip-out. In vitro repair assays using human cell extracts showed that NA inhibited CAG slip-out repair but not CTG slip-out repair and had no effect on G-T mismatch repair, which plays an important role in DNA damage recovery in vivo.46) To verify the effect of NA on CAG repeat length, Dr. Masayuki Nakamori of Osaka University, a co-investigator of the study, analyzed the expanded CAG repeat length in fibroblasts derived from patients with HD.47) HD fibroblasts were cultured in the presence and absence of NA for 1 month, and DNA was collected for detailed analysis of the variation in CAG repeat length. The results showed that NA treatment clearly increased the number of cells with shortened CAG repeats, with an average shortening of 9.2 repeats. It was also confirmed that NA had no effect on normal length CAG repeats on other genes. To investigate the effect of NA on the shortening of CAG repeats in vivo, we directly administered NA by stereotactic neurosurgery into the striatal tissue of the R6/2 mouse model of HD48) (Figs. 2b and 2c). We found that the CAG repeat length in the left striatum treated with NA was shorter than that in the right striatum treated with saline. The shortening of CAG repeats in an HD mouse model by NA was also demonstrated to reduce aberrant protein aggregates in the brain striatum, which underlies the pathology of the disease. There was no variation in repeat length in brain tissue not at the site of administration, nor was there any effect on normal length CAG repeats. The mutagenic effect of NA on the entire gene was analyzed using a next-generation sequencer for the Hprt1 gene, and no significant mutagenic effect was observed. The mechanism of NA-induced shortening of CAG repeats has been investigated, and it has been shown that it involves the formation of CAG slip-outs that occur during DNA transcription, not during DNA replication, and that it promotes the processing of DNA-RNA hybrids (R-loop) during transcription.49) The reason for contracting the repeat but not expanding the repeat by stabilizing S-DNA structure in this case remains to be clarified.
SCA31 is a neurodegenerative genetic disease caused by insertions of aberrantly long TGGAA repeat sequences, and the long UGGAA repeats produced by transcription have been reported to exhibit RNA toxicity.24),25) The pathogenic mechanism of SCA31 is similar to that of trinucleotide repeat diseases, including the formation of RNA foci and the sequestration of RNA-binding proteins (RBPs) in addition to the repeat assisted non-AUG translation.50)–53) We have started a collaborative study on SCA31 with Dr. Yoshitaka Nagai of Osaka University (currently Kinki University) and Dr. Kinya Ishikawa of Tokyo Medical and Dental University. Because we had no information on small molecules that are likely bind to UGGAA repeats, we screened our own chemical library of repeat-binding molecules using an surface plasmon resonance (SPR) assay with a sensor chip containing immobilized UGGAA repeats. We found naphthyridine carbamate dimer (NCD)54)–58) as a UGGAA repeat binding molecule59) (Fig. 3). Details of the screening and binding of UGGAA repeats to NCD will be discussed later.

(a) Chemical structures of NCD (X = N, Y = CH3 shown in bold) and its control compound QCD (X = CH, Y = H) and the mode of hydrogen bonding to the two guanines. (b) Alleviation of compound eye degeneration for Drosophila model of SCA31 using NCD and QCD (top row) and Drosophila expressing non-toxic repeat RNA (bottom row). (c) Quantitative analysis of the effects of NCD and QCD on the area of compound eyes expressing (left) r(UGGAA)exp and (right) non-toxic repeat RNAs. (Modified from figures in Ref. 59.)
UGGAA repeats capture RBPs, including splicing factors and form RNA foci in the cell nucleus. It is important to note that the UGGAA repeat sequence is coincidentally found in a long non-coding RNA called HSATIII.60) HSATIII also sequesters multiple RBPs to form RNA foci. We collaborated with Dr. Tetsuro Hirose of Osaka University to study the effect of NCD binding on the UGGAA repeat RNA-RBP interactions. The results showed that the binding of RBPs, including splicing factors, to the UGGAA repeat RNA was inhibited upon NCD treatment as evaluated using an in vitro pull-down assay.59) In contrast, quinoline carbamate dimer (QCD), a control compound for NCD, did not inhibit RNA-RBP interactions. In addition, the formation of RNA foci from the expression of UGGAA repeats was evaluated by RNA fluorescence in situ hybridization; QCD did not affect the formation of RNA foci, but NCD treatment reduced RNA foci in the cell nucleus. In collaboration with Dr. Nagai, we examined the effect of NCDs on RNA toxicity using a Drosophila model of SCA31, which exhibits compound eye degeneration due to the expression of UGGAA repeats in the compound eye. We found that feeding NCD to SCA31 Drosophila model larvae alleviated compound eye degeneration such as reduced compound eye area and lack of pigmentation in adult Drosophila. When QCD was fed as a control compound, the compound did not show any significant effects, suggesting that NCD alleviates RNA toxicity by inhibiting RNA-RBP interactions and RNA foci formation. These results demonstrated that NCD alleviates RNA toxicity caused by UGGAA repeats in vivo.
In DM1, aberrantly expanded CUG repeats trap mRNA metabolism-related proteins such as MBNL1 and induce selective splicing, which is important for the pathogenesis of the disease.21)–23) In our laboratory, we have been working to develop compounds that exhibit binding activity to G-T and G-U mismatches.61)–64) Although we have found some effective compounds such as diaminophenanthrene derivatives,62),63) their binding selectivity remains an issue because they show affinity for other RNA sequences, such as CCG repeats, in addition to CUG repeats. From structure-activity relationship studies using computer simulations and SPR binding data of synthetic molecules, we found a CUG repeat-selective binding molecule JM642 with a diaminoisoquinoline structure64) (Fig. 4). Using a DM1 mouse model, we administered JM642 intraperitoneally (20 mg/kg) for 5 days to investigate the effect of releasing splicing factors trapped by toxic RNA to reduce splicing defects leading to pathogenicity. Only the isoform containing exon 22 (+ex22) is expressed from the Atp2a1 gene in control WT mice. On the other hand, the expression level of +ex22 isoform was 15.8% in DM1 model mice, but it was greatly restored to 73.6% in JM642-treated mice (Fig. 4b). The reduction in splicing defects was also observed in the Clcn1 gene (Fig. 4c). Apart from this, RNA aggregate formation, which is frequently observed in myoblasts derived from DM1 patients, was greatly reduced from 41% of 255 cells to 6.7% of 286 cells in JM642-treated cells. These results supported the mechanism of action in which JM642 binds to CUG repeats and inhibits RNA aggregate formation, resulting in the release of trapped splicing factors, suggesting that the selective and high affinity binding properties of JM642 to CUG repeats are important for the splicing aberration recovery effect. By optimizing the substituent at position 5 of 1,3-diaminoisoquinoline, the parent nucleus of JM642, we can expect to create a binding molecule with higher efficacy and lower toxicity.

(a) Chemical structure of JM642 (shown in bold) and the proposed hydrogen bonding scheme to uracil. Rescue of mis-splicing of the pre-mRNAs of the (b) Atp2a1 and (c) Clcn1 genes by intraperitoneal injection of JM642. Exon 22 is included in the wild-type for the splicing of pre-mRNA of Atp2a1, whereas exon 7a is excluded from the pre-mRNA of Clcn1. (Modified from figures in Ref. 64.)
NA, NCD, and JM642 showed remarkable activity in vivo. NA shortened CAG repeats in the striatum of the brain in an HD mouse model,45) NCD alleviated the degeneration of compound eyes in a SCA31 Drosophila model,59) and JM642 alleviated splicing defects in a DM1 mouse model.64) Although more research is needed, all these compounds are expected to be important leads in the development of effective drugs for alleviating symptoms and even inhibiting the onset of neurodegenerative diseases. Each of these compounds has been shown to bind to a target repeat sequence. In other words, compounds that bind to the target repeat sequence are likely to be active in vivo, indicating that the evaluation of repeat binding is effective in screening for potential therapeutic agents for repeat diseases. In the following sections, through the use of techniques such as SPR and UV-melting measurements, we will show that these compounds bind to their target repeats.
In our research, we start by designing and synthesizing molecules that are likely to bind to the target DNAs or RNAs and then evaluate the binding to determine whether to proceed to further biological evaluation.65)–68) Therefore, binding evaluation and screening are major milestones in research, and we used SPR and melting temperature measurement of relatively short DNA or RNA as simple and informative methods. In our SPR assay, compounds flowed continuously onto a sensor chip with target DNA or RNA immobilized on a thin gold film via dextran, and the response was recorded. In practice, dielectric constant changes on the thin gold film surface were observed. Because single-stranded repeat DNA or RNA is immobilized on the sensor, it is expected that the repeat DNA or RNA changes into a hairpin structure, i.e., an S-DNA structure, when the compound binds. Upon a structural change from the single-stranded state to the S-DNA hairpin, the distance of the negative charge of the nucleic acid from the gold surface changes significantly, and, therefore, a large response is expected.42)
Figure 5 shows the results of evaluation with a sensor coated with immobilized d(CAG)9 repeats with different concentrations of NA.44) In the experiment, the solution containing NA was allowed to flow for 60 seconds from the background condition adjusted with flowing buffer, and only buffer was supplied after 60 seconds. The binding of NA to the CAG repeat DNA on the sensor surface was observed between 0 and 60 seconds, and the dissociation of NA from the NA-CAG repeat complex was observed after 60 seconds. The binding of NA to CAG repeats is characterized by a very slow binding and dissociation response, i.e., slow binding and dissociation rates. This is qualitative, but it can be ratioalized that the single-stranded repeat DNA shows a large structural change.

(a) SPR analysis of NA interaction with the d(CAG)9-immobilized surface. NA was added stepwise at 0, 0.13, 0.25, 0.5, 1.0, and 2.0 µM (from bottom to top). The sensors were exposed to the ligand for 0–60 s. d(CAG)9 was immobilized on the streptavidin-coated (SA) sensor chip for 497 RU. (b) Thermal stabilization of A-A mismatches by NA-binding. Melting temperature (Tm) of 11-mer duplexes containing A-A mismatches in the vAw-xAy sequence in the presence of NA is shown above the bar. v-y and x-w denote the Watson-Crick base pairs. Error in Tm measurements was ±0.8 °C and is reported as the standard error of three independent measurements.
Another method for the evaluation of binding is change in melting temperature (ΔTm). This is a technique to observe how the temperature at which a double strand of DNA or RNA melts into a single strand changes depending on whether a test compound is present or not. In double-stranded DNAs and RNAs, the two strands are bound together by hydrogen bonds, and when the temperature is increased, the hydrogen bonds break, and the two strands melt apart to give a single-stranded form. The absorbance at 260 nm increases upon melting of the double strand. Therefore, when the temperature is increased continuously and the absorbance at 260 nm is recorded, the change in absorbance against the temperature gives a sigmoidal graph. The inflection point of this graph is defined as the melting temperature (Tm) of the duplex. Molecules that bind to DNA or RNA stabilize the double-stranded state, and thus the melting temperature increases. In other words, the degree of binding of a compound can be estimated from the melting temperature that has increased in the presence of the compound. For the same DNA or RNA, the degree of binding can be ranked unambiguously by ΔTm, but for different DNA or RNA, careful evaluation is required, for example, when there is a marked difference in the original Tm.
Looking at the actual data, we used double-stranded DNA containing an A-A mismatch (CAG/CAG) sandwiched between C-G base pairs (cf. Fig. 1), which appears in the hairpin structure formed by CAG repeats.42) When NA was added to this DNA, Tm changed from 18.1 °C to 50.9 °C with an increase of 34.8 °C. When the base pairs flanking the A-A mismatch were changed from C-G base pairs to other base pairs, the increase in Tm was drastically reduced. For CAC/GAG DNA, in which one C-G base pair was replaced by a G-C base pair, the Tm change was almost halved to 17.0 °C (from 20.2 °C to 37.2 °C). The most striking result was the Tm of GAC/GAC DNA where the A-A mismatch was sandwiched between G-C base pairs. The ΔTm was −0.6 °C (from 22.0 °C to 21.4 °C). Because the measurement error was around ±0.5 °C, it was shown that there was no temperature change due to the addition of NA, i.e., NA did not bind to this sequence. The CAG/CAG sequence, which binds NA, and the GAC/GAC sequence, which does not, have the same number and type of bases, and the only difference is the order of the bases. DNA is a polymer consisting of deoxyribose units with asymmetric carbon atoms and has a right-handed double-helix structure. DNA is a chiral molecule. Therefore, DNA with a CAG/CAG sequence and DNA with a GAC/GAC sequence are completely different compounds, and it is not surprising that the binding of NA is different. This series of DNA binding analyses revealed that NA binds not only to the A-A mismatch, but also to the flanking C-G base pairs. A similar sequence selectivity was observed for NCD, which favorably binds to the CGG/CGG (that is 5′-CGG-3′/5′-CGG-3′) sequence DNA but only weakly to the GGC/GGC (5′-GGC-3′/5′-GGC-3′).55) It is important to note that NCD was originally found to bind to CGG/CGG DNA and CGG repeat DNA, as discussed in the next chapter NCD was found to bind to UGGAA/UGGAA RNA and UGGAA repeat RNA, too.
We screened for compounds that bind to UGGAA repeat RNA using SPR.59) Because there was no information about compounds that bind to this sequence, we decided to choose from compounds that we have synthesized in our laboratory. If the UGGAA repeats adopted an S-DNA structure, the hairpin structure would have a series of GGA/GGA units (Fig. 6a). Because of the high content of guanine, we thought that a compound that forms complementary hydrogen bonds with guanine might bind to it, and we selected 20 compounds containing NCD.56),69) On the other hand, as explained by the sequence selectivity of the NA molecule binding to CAG repeats, DNA is a chiral molecule; therefore, there is a directionality (5′→3′ or 3′→5′) in the sequence. That is, CGG/CGG (5′-CGG-3′/5′-CGG-3′) DNA to which NCD binds has the 5′-XGG-3′ sequence, but the GGA/GGA (5′-GGA-3′/5′-GGA-3′) repeat unit of the SCA31 RNA has the 5′-GGX-3′ sequence, which is the reverse of the sequence direction. Therefore, we were not confident whether our library of compounds bound to DNA repeats can bind to RNA repeats. We immobilized r(UGGAA)9 on an SPR sensor, evaluated the binding of 20 compounds, and found seven binding compounds. Among the bound compounds, LC-1 and LC-2 were first-generation G-G mismatch binding molecules and their derivatives. LC-3, LC-5, and LC-9 were NCD and their derivatives, and LC-8 and LC-11 were dimers of NCD. These results indicated that LC-9 (NCD) is the smallest structural unit that binds to UGGAA repeat RNA.
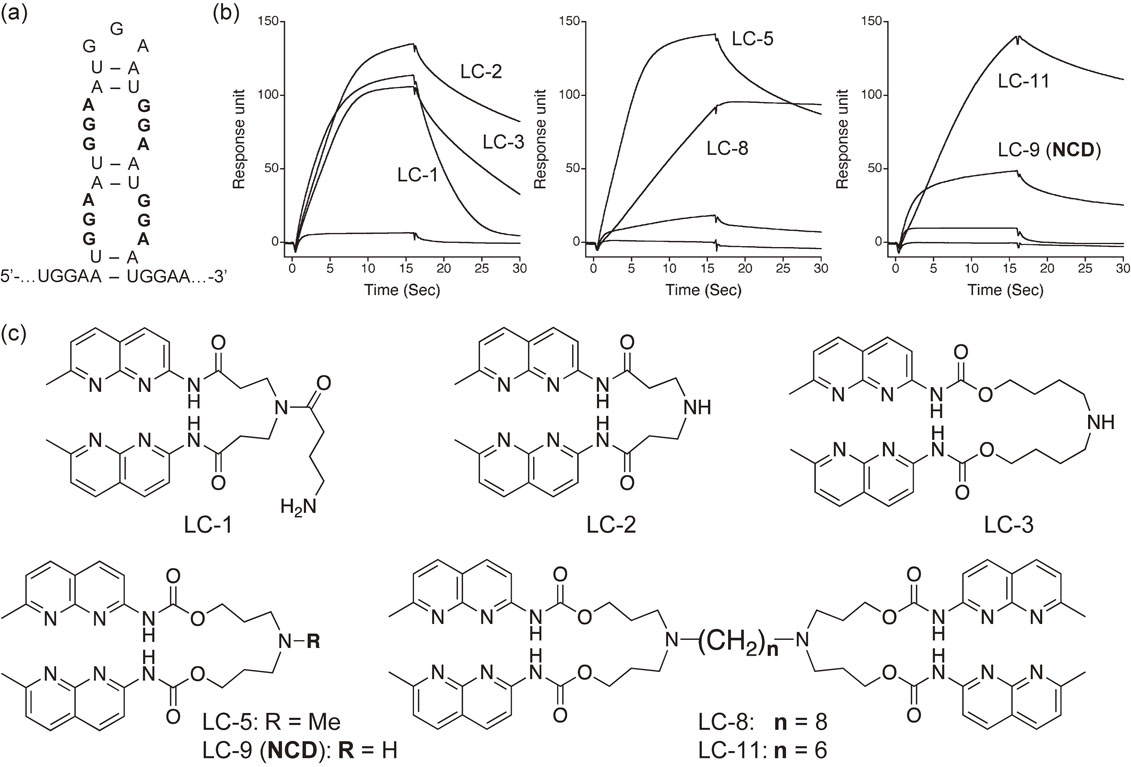
(a) Proposed secondary structure of UGGAA repeat involving a GGA/GGA internal loop. (b) SPR profiles of compounds applied to the r(UGGAA)9 RNA immobilized on the sensor surface. (c) Structures of compounds exhibited affinity to UGGAA repeat RNA.
The sequence selectivity of NCD in binding to UGGAA repeat RNA was investigated using Tm measurement. We prepared a control RNA in which guanine in the GGA/GGA sequence was replaced with adenine in a stepwise manner. For these RNAs, NCD was added, and the Tm was measured. When NCD was added to UGGAA/UGGAA double-stranded RNA (Tm 31.1 °C), the Tm increased to 52.6 °C (ΔTm of 21.5 °C). (Fig. 7a) In the case of the GGA/AGA sequence, in which the 5′-side G of GGA was replaced with A, the ΔTm by the addition of NCD was markedly reduced to 5.2 °C. Furthermore, in the AGA/AGA sequence in which the 5′-side G of both strands was replaced with A, the effect of NCD was further reduced to 2.7 °C in ΔTm, and when the G in the center of the sequence was replaced with A, no increase in Tm was observed.
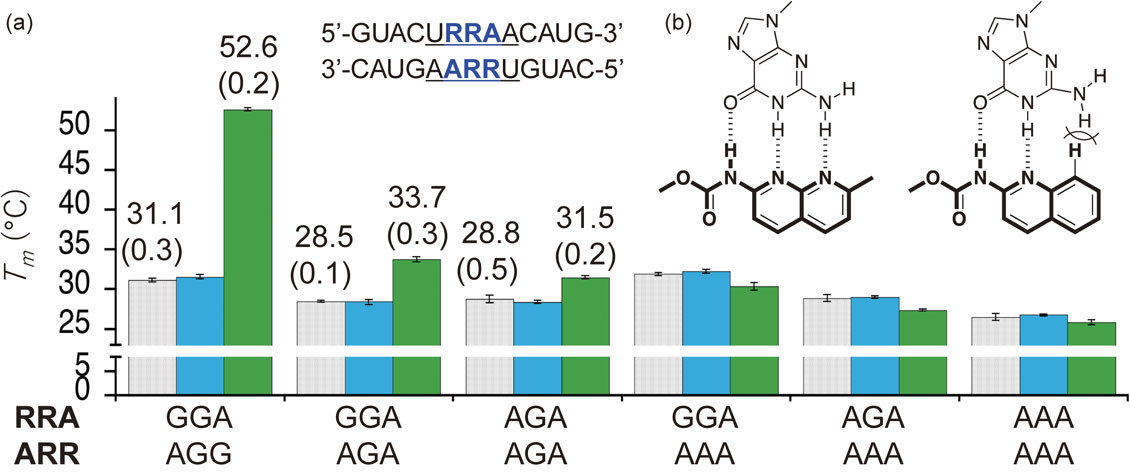
(a) Thermal stabilization of RNA duplexes containing URRAA/URRAA pentad in the absence (gray) and presence of NCD (green) and QCD (blue). The data from Tm measurements are presented as the mean ± SD (n = 3 independent experiments). (b) The decisive effect of quinoline ring on the hydrogen bonding with guanine.
QCD, a control compound of NCD, has a structure in which one of the two 2-amino-1,8-naphthyridine heterocycles of NCD is replaced by a 2-aminoquinoline ring. (Fig. 7b) The methyl group at position 7 was excluded for simplicity of the synthesis. The substitution of a naphthyridine ring with a quinoline ring had a decisive effect on the hydrogen bonding with guanine. The substitution of one nitrogen atom with a carbon (C-H) caused steric repulsion with the amino group hydrogen at position 2 of guanine and prevented hydrogen bonding with guanine. No increase in Tm was observed at all for the UGGAA/UGGAA sequence with QCD, suggesting that the hydrogen bond between NCD and guanine plays a decisive role in the binding of NCD to the UGGAA/UGGAA sequence.
In our research on the molecular design of organic compounds targeting nucleic acids, it is of course important to know whether they bind or not, but it is of utmost importance to see the complex structure. If we can see the complex structure, we can understand how a small change in functional group or substituent may affect the complex formation based on the structure. Structural information of the complexes is essential for more precise molecular design. Fortunately, in our study, our collaborators were able to analyze the complex structures of NA with CAG/CAG double-stranded DNA42) and NCD with UGGAA/UGGAA double-stranded RNA59) using nuclear magnetic resonance (NMR) spectroscopy and complexes of CAG repeat RNA by X-ray analysis.70)
We asked Dr. Chojiro Kojima (currently a professor at Yokohama National University) to analyze the structure of the NA-CAG/CAG double-stranded DNA complex.42) The results were clear, and we were able to immediately understand a mystery that we had not understood, namely, why NA binds to the CAG/CAG sequence. NA was designed based on the prediction that each heterocycle consisting of NA, 2-amino-1,8-naphthyridine and 8-azaquinolone, can bind to guanine and adenine, respectively, through hydrogen bonding. In other words, NA is a designed molecule targeting G-A mismatch.41) However, there is no G-A mismatch in the CAG/CAG sequence, and we did not understand how NA binds to this sequence at all. There was one remaining mystery. From mass spectrometry of the complex, it was known that two molecules of NA bound to the CAG/CAG sequence, but it was not clear why. The structure of the NA-CAG/CAG complex revealed by Dr. Kojima solved both mysteries at once. (Fig. 8a) First, the naphthyridine and azaquinolone rings formed hydrogen bonds with guanine and adenine, respectively. In this respect, the molecular design of NA aimed at binding to the G-A mismatch was found to be successful. The naphthyridine ring deprived the guanine from the hydrogen-bonded C-G base pair. As a result, cytosine, which had no hydrogen-bonding partner, was pushed out of the duplex and flipped out. Two molecules of NA were needed to bind the A of one strand of the central A-A mismatch and the G of the other strand. The naphthyridine-G pairs and azaquinolone-A pairs produced well-stacked structure in the interior of DNA (Fig. 8b).

(a) NMR-determined structure of the NA-CAG/CAG complex. DNA is shown as a stick model, and the flipped out cytidine residues are shown in red. Two NA molecules are shown in a CPK model. The view is from the major groove. (b) Illustration of a mode of NA-binding to the CAG/CAG site. Rectangular boxes colored in red and blue indicate naphthyridine and azaquinolone heterocycles, respectively. Two cytosines are in a flipped-out position.
The structure of the complex between NCD and UGGAA/UGGAA RNA was analyzed by Dr. Gota Kawai of Chiba Institute of Technology.59) Mass spectral analysis of the complex indicated that two molecules of NCD bound to UGGAA/UGGAA, and the circular dichroism (CD) spectral measurement also suggested that a large structural change occurred on the RNA side upon binding of NCD. The structure of the complex analyzed is shown in Fig. 9. The naphthyridine ring of NCD formed a hydrogen bond with guanine, which was in good agreement with the importance of guanine indicated by the Tm measurement. Two molecules of NCD bound to the guanine of both strands, as in the NA-CAG/CAG complex.42) Four guanine-naphthyridine hydrogen bond pairs were stacked well with each other in the interior of the RNA duplex. The fourth adenine from the 5′ end of UGGAA/UGGAA was flipped out in the complex (indicated by an asterisk in the figure). The complex structure shown in Fig. 9 is viewed from the major groove side, and the complex of NA-CAG/CAG shown in Fig. 8 is also viewed from the major groove side, but there is a marked difference between the two complex structures. In the NA-CAG/CAG complex structure, the linker connecting the two heterocycles can be seen on the major groove side, whereas it is not the case in the NCD-UGGAA/UGGAA complex structure, indicating that NCD bound from the minor groove side of the UGGAA/UGGAA RNA double strand. The guanine recognition heterocycle, 2-amino-1,8-naphthyridine, formed a hydrogen bond with guanine. Therefore, for the naphthyridine to bind from the major or minor groove side, the order of the hydrogen-bonding groups on the guanine must change accordingly.

(a) NMR-determined structure of the NCD-UGGAA complex viewed toward the major groove side. Nucleotides are colored differently; A (Green), C (sky blue), G (orange), and U (yellow). Two As in the flipped-out position are marked with an asterisk. (b) Illustration of a mode of NCD-binding to the UGGAA/UGGAA site. Rectangular boxes colored in green and orange indicate naphthyridine. Two adenines are in a flipped-out position. (Modified from figures in Ref. 59.)
Figure 10 shows the structural features of the NCD-UGGAA/UGGAA complex. First, the stoichiometry of the binding is 1 RNA to 2 small molecules, i.e., a binding ratio of 2:1. This is also a characteristic of mismatch binding molecules, as seen in the NA-CAG/CAG and NCD-CGG/CGG complexes. The second feature is the flipping out of nucleobases by binding. In the NCD-UGGAA/UGGAA complex, the fourth adenine base counting from 5′ was flipped out. This base flip out is also a common feature in the binding mode of mismatch-bound molecules.42),55) The last feature, which is unique to the NCD-UGGAA/UGGAA structure, concerns the orientation of the glycosidic bond that connects the base and ribose portions of the nucleobase. In normal nucleic acids, the glycosidic bond is anti-type, with the base and sugar portions in an s-trans configuration.71) In this case, in guanine, the amino group at position 2, which is the hydrogen bond donor, is located on the minor groove side, and the carbonyl group at position 6, which is the hydrogen bond acceptor, is located on the major groove side. For the formation of a complete hydrogen bond between guanine and 2-amino-1,8-naphthyridine, the amino group at position 2 of the naphthyridine ring must be on the major groove side (see Fig. 3a). In the NCD-UGGAA/UGGAA complex, the linker of the NCD was not on the major groove side, indicating that the glycosidic bond of the guanine to which NCD was bound is not anti-type. A closer look at the NMR structure showed that the glycosidic bonds of the four guanines were of the syn type. When the glycosidic bond was syn-type, the order of hydrogen bonding groups was switched, so the carbonyl group at position 6 was located on the minor groove side and the amino group at position 2 was located on the major groove side. Therefore, there was no question that 2-amino-1,8-naphthyridine binds from the minor groove side. On the other hand, a new question emerged. Does the NCD bind to RNA in which four guanine bases are all syn-type? Or did the configuration of the glycosidic bond change from anti-type to syn-type with the binding of NCD? These questions will be discussed one last time.

Characteristic features of NCD-UGGAA/UGGAA RNA binding. (a) Stoichiometry, two NCD molecules bound to the UGGAA/UGGAA site. (b) Flipping out of adenines. (c) Conformation of glycosidic bond.
NA, which binds to CAG/CAG DNA, and NCD, which binds to CGG/CGG DNA and UGGAA/UGGAA RNA, discussed so far are compounds that were designed as mismatch binding ligands (MBLs)65),72) and showed better-than-expected effects in vivo. At the same time, NMR structural analysis of the complexes revealed that they bind to their respective target sequences in a manner different from the designed binding mode. In particular, it has also become clear that factors that are difficult to incorporate in the design are more important. For example, MBLs were designed based on a 1:1 stoichiometry at the design stage, but in reality they formed a 2:1 complex. By flipping out bases that are not involved in the binding, the MBL acquires stabilization through a stacking effect in the interior of duplex. In this section, we will discuss the concept of molecular design of MBLs in detail.
The basis for the molecular design of MBLs was the function of uracil DNA glycosylase (UDG),73) which removes uracil bases from DNA, and the mode of its functional expression. Uracil in DNA is mainly caused by mis-incorporation or the deamination of cytosine resulting in the formation of G-U mismatched base pair in duplex DNA (Fig. 11a), and if left unrepaired, adenine is incorporated opposite the uracil during replication, resulting in a G to A mutation. To prevent this mutation, it is necessary to repair the DNA by removing U from the DNA and replacing it with C. The first step in this repair process is the recognition of uracil in the DNA.

Illustration of the concept of mismatch binding ligand. (a) DNA duplex containing a G-U mismatch. (b) Mode of recognition of U by uracil DNA glycosylase (UDG). (c) Hypothetical structure of G-U mismatch holding space opposite each nucleotide. (d) Binding of two heterocycles (oval and rectangular shapes) in the space opposite G and U.
The co-crystal structure of UDG and U-containing DNA in the protein data bank (ID 4SKN) shows that UDG inserts an amino acid side chain (Arg272) into the duplex to find a weak G-U base pair by flipping out U and recognizes it in the pocket of UDG (Fig. 11b). We initially planned to reproduce this function in a small molecule, but the mechanism for recognizing the flipped-out U was too complex. Instead, we considered recognizing U in the internal region of the nucleobase stack without flipping out uracil.65) We considered a hypothetical structure in which the G-U mismatch base pairs are displaced (Fig. 11c), so that neither G nor U has a hydrogen bonding partner and hypothesized that a molecule consisting of a heterocyclic ring that fills the space in front of G and U and forms hydrogen bonds with G and U would be able to stabilize this hypothetical structure.
Molecules that recognize and form hydrogen bonds with nucleobases have been studied for a long time.74)–86) A 2-amino-1,8-naphthyridine derivative that forms hydrogen bonds with guanine has been studied.87)–89) However, the studies focused exclusively on the interaction in organic solvents and did not evaluate the interaction with DNA in water. When we started our research in the late 1990s, we were inexperienced in molecular recognition research, so we did not know how difficult it was to form hydrogen bond complexes in water.90) As a result, the competition with water molecules likely had a positive effect on suppressing the non-specific binding of molecules, because the interior of the nucleic acid is a hydrophobic space. The basic idea behind the MBL design was as follows (Fig. 11d): (1) For the hypothetical structure of a target mismatched base pair, a heterocyclic compound with hydrogen-bonding groups complementary to each nucleobase can be designed. (2) The two heterocyclic compounds are connected by a linker in order to favor the binding entropy. (3) The hydrogen-bonding pair between the heterocyclic ring and the nucleobase is stabilized by stacking in the nucleic acid duplex.65)
The other important point in MBL design is the charge that the molecule carries. Nucleic acids exist as polyanions due to the negative charge of the phosphate diester moiety, and thus are clothed with positively charged ions in the vicinity. In addition, the presence of divalent cations such as magnesium ions is important for the expression of functions such as ribozymes.91),92) We intentionally introduced an amino group in the center of the linker connecting the two heterocycles in the hope that the molecule would carry a positive charge and undergo electrostatic interaction with the nucleic acid, thereby promoting binding to the nucleic acid. This amino group undergoes protonation at neutral pH, bringing a positive charge to the molecule. By extending the linker through this amino group, the MBL can be fixed to a solid phase surface such as an SPR sensor.41),66),93),94) The length of the linker depends on the estimated degree of freedom of the two heterocycles, but the longer it is, the greater the degree of freedom of the heterocycles and the more entropically unfavorable the binding will be. Because there was no specific index, the length was chosen to be convenient for synthesis and considering the stability of the compound. The first-generation MBL, naphthyridine dimer (LC-2) shown in Fig. 6, which binds to the G-G mismatch sequence in CGG/CGG was reported in 2001.66),67) In parallel with this study, we also searched for molecules that bind to bulge structures in DNA double strands.67) The bulge structure was considered to be part of the hypothetical structure shown in Fig. 11c. Figure 12 shows the binding motif to the cytosine bulge and the structure of the 2,7-diamino-1,8-naphthyridine derivative (DANP) actually bound to the cytosine bulge.95)–99) The dimer of 2-amino-1,8-naphthyridine derivative, named ANP77, was found to bind to the stem-loop composed of CC/T and CC/C in DNA.100),101) Both DANP and ANP77 have high fluorescence intensity and can be used as fluorescent probes and indicators to screen out molecules binding to nucleic acids.102)–105)

Illustration of the concept of the bulge binding. (a) Binding of a heterocycle in the space opposite C. (b) Hydrogen bonding scheme of protonated 2,7-diamino-1,8-naphthyridine (DANP) with cytosine. (c) A proposed binding mode of dimeric 2-amino-1,8-naphthyridine derivative (ANP77) to the CC/T and CC/C bulge loops. (d) The structure of ANP77.
An important factor in the molecular design of MBLs is the formation of hydrogen-bond pairs with the target nucleobases. So far, we have used 2-amino-1,8-naphthyridine, 8-azaquinolone, and protonated 2-amino-1,8-naphthyridine as heterocycles that form hydrogen-bond pairs with adenine, guanine, and cytosine, respectively. Other heterocycles developed by other researchers are also known.76),77),82) On the other hand, the development of heterocycles that bind to thymine (uracil) has not yet been successful in our group. Although JM642 is a characteristic molecule that binds to CUG repeats,64) its binding mode has not been clarified. In thymine, the sequence of hydrogen bonding groups is Acceptor (A), Donor (D), and Acceptor (A). It is well known that even if the number of hydrogen bonds increases, the intermolecular interaction does not become stronger in such a sequence of hydrogen bonding groups in which D and A alternate.106)–108) Specifically, 2,6-diaminopyridine, which has a complementary hydrogen-bonding group to thymine, can bind to thymine with three hydrogen bonds, but its binding constant is two orders of magnitude smaller than that of guanine-cytosine, which also has three hydrogen bonds. The design of heterocycles that recognize thymine remains a major challenge.
2) Stoichiometry.Despite the increasing popularity of molecular modeling software and the accuracy of the force field used in the calculations, most MBLs do not bind to the target nucleic acid in the designed binding mode. One of the reasons for this is stoichiometry. Usually, when designing molecules, we assume a 1:1 complex between the target and the molecule, and it is difficult to predict 2:1 complex formation. As already mentioned, MBLs prefer a 2:1 binding ratio instead of 1:1. It may be necessary to develop a molecular design method that incorporates variable binding ratios.
3) Structural change.In the NA-CAG/CAG and NCD-UGGAA/UGGAA complexes, large structural changes occur in both the nucleic acid and the MBL. Such binding might involve induced fit and/or conformational selection mechanisms.109),110) Is it possible to predict such large conformational changes in the interaction of small molecules with nucleic acids? The success or failure of the prediction depends on the ability to correctly estimate the stability of the predicted structure. Recently, artificial intelligence that can predict the folding of proteins with high accuracy has been developed and has become a hot topic.111) This success was probably due in large part to the abundance of data on protein structures. Unfortunately, data on nucleic acid structures, especially complex structures of nucleic acids and small molecules, are scarce, and the accumulation of data on such complexes will probably be necessary first to be able to predict structural changes involving binding to small molecules.
In the NA-CAG/CAG complex, there are two naphthyridine-guanine hydrogen-bond pairs, two azaquinolone-adenine hydrogen-bond pairs, and the cytosine that formed the C-G base pair is flipped out. Similarly, in the NCD-UGGAA/UGGAA complex, four naphthyridine-guanine hydrogen bond pairs are formed. Furthermore, in the latter case, all the guanines form syn-type glycosidic bonds. How is this major structural change occurring? We have already mentioned that QCD, in which one of the two naphthyridine rings of NCD is replaced by a quinoline ring, cannot form a hydrogen bond pair with guanine. However, if QCD bound to the nucleic acid from the naphthyridine ring, would it not be at the last stage of the binding process that the 2-aminoquinoline ring is found to be unable to make hydrogen bond pairs? If this hypothesis is correct, the rate-limiting step in the complex formation between NCD and UGGAA/UGGAA would be in the latter half of the complex formation. Furthermore, does NCD bind only to RNAs with guanine whose glycosidic bonds are in the syn form? Or does the glycosidic bond change from the anti-type to the syn-type during the NCD binding process? In addition, why did NCD not bind to guanine with the anti-type of glycosidic bond from the major groove side? There are many unanswered questions about the binding of MBLs to mismatched base pairs. Although it is unlikely that we will be able to answer all these questions any time soon, the question of how small molecules bind to nucleic acid targets that flexibly change their structure is mysterious and presents a very important scientific challenge.
In recent years, small molecule drug discovery targeting nucleic acids, especially RNA, has shown great promise.112)–118) As a result of the encyclopedia of DNA elements (ENCODE; https://www.encodeproject.org)119) project after the completion of the sequencing of the human genome, it is known that much of the human genome is involved in the maintenance of life as non-coding RNA. If we can find molecules that bind to non-coding RNAs, we can regulate the expression and function of a wide variety of RNAs in the cell using small molecules. The fact that NA-CAG/CAG complex formation shortened CAG repeats in striatal DNA of an HD mouse model and that NCD-UGGAA/UGGAA complex formation alleviated compound eye degeneration in an SCA31 Drosophila model strongly suggests that nucleic acid-targeted small molecule drug discovery is not hypothetical but will be a reality in the near future. In fact, Risdiplam, which was thought to bind to the RNA-protein interface, was approved by the FDA in October 2020 as a treatment for spinal muscular atrophy (SMA).120) Although antibody drugs and nucleic acid drugs with high drug prices are attracting attention, pharmaceutical companies have accumulated know-how in small molecule drug discovery and a diverse library of organic small molecule compounds. I am convinced that these resources will be quite important and useful for the discovery of small molecules that regulate nucleic acid functions and further development. In order to strongly promote nucleic acid-targeted small molecule drug discovery, it is necessary to gain a deeper chemical understanding of the interactions between nucleic acids and small molecules. At the same time, we need to accelerate our research by making full use of new information science technologies and artificial intelligence.
This study is a part of my 20-year research on mismatch-binding ligands, and I would like to express my sincere gratitude to my many collaborators, including those who were not named in the references. I would also like to express my sincere gratitude to the Ministry of Education, Culture, Sports, Science and Technology (MEXT), the Japan Science and Technology Foundation (JST), the Japan Society for the Promotion of Science (JSPS), and the Ministry of Health, Labor and Welfare (MHLW) for support through a series of research funding programs.
Edited by Keisuke SUZUKI, M.J.A.
Correspondence should be addressed: K. Nakatani, SANKEN, The Institute of Scientific and Industrial Research, Osaka University, 8-1 Mihogaoka, Ibaraki, Osaka 567-0074, Japan (e-mail: nakatani@sanken.osaka-u.ac.jp).
5′-untranslated region
ΔTmchange in melting temperature
CDcircular dichroism
DANP2,7-diamino-1,8-naphthyridine
DM1myotonic dystrophy type 1
EMSAelectrophoretic mobility shift assay
HDHuntington’s disease
MBLmismatch binding ligand
NAnaphthyridine-azaquinolone
NCDnaphthyridine carbamate dimer
NMRnuclear magnetic resonance
QCDquinoline carbamate dimer
R-loopDNA-RNA hybrids
RBPRNA-binding protein
S-DNAslipped strand DNA
SCA31spinocerebellar ataxia type 31
SPRsurface plasmon resonance
Tmmelting temperature
UDGuracil DNA glycosylase

Kazuhiko Nakatani was born in Nara Prefecture in 1959. He graduated from the Department of Chemistry, Faculty of Science, Osaka City University in 1982, and went on to graduate school. In 1985, after completing his first year of the doctoral program, he left the university to study under Professor Gilbert Stork at the Department of Chemistry, Columbia University, U.S.A. Three years later, in 1988, he returned to Japan to complete his doctoral degree at Osaka City University and to become a postdoctoral researcher at the Sagami Chemical Research Center in Sagamihara City, Kanagawa Prefecture. In 1991, he started his academic career as a research associate at the Faculty of Science, Osaka City University. Since his student days, he has continued his research on the synthesis of natural products. In 1993, he was hired as a research associate by Professor Isao Saito in the Department of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, and since then, he has been working on nucleic acid chemistry and the interaction of nucleic acids with small organic molecules. He was promoted to Assistant Professor at Kyoto University in 1997, and then to Professor at Osaka University in 2005, where he established the Regulatory Bioorganic Chemistry Laboratory at the Institute of Scientific and Industrial Research, SANKEN. He served as the Director of SANKEN from August 2015 to March 2018, and since August 2019, he has been serving as the Executive Vice President for Finance and Facilities at Osaka University.