Abstract
Extracellular fluids, including blood, lymphatic fluid, and cerebrospinal fluid, are collectively called body fluids. The Na+ concentration ([Na+]) in body fluids is maintained at 135–145 mM and is broadly conserved among terrestrial animals. Homeostatic osmoregulation by Na+ is vital for life because severe hyper- or hypotonicity elicits irreversible organ damage and lethal neurological trauma. To achieve “body fluid homeostasis” or “Na homeostasis”, the brain continuously monitors [Na+] in body fluids and controls water/salt intake and water/salt excretion by the kidneys. These physiological functions are primarily regulated based on information on [Na+] and relevant circulating hormones, such as angiotensin II, aldosterone, and vasopressin. In this review, we discuss sensing mechanisms for [Na+] and hormones in the brain that control water/salt intake behaviors, together with the responsible sensors (receptors) and relevant neural pathways. We also describe mechanisms in the brain by which [Na+] increases in body fluids activate the sympathetic neural activity leading to hypertension.
1. Introduction
The sensing site of body fluid conditions was postulated to be circumventricular organs (CVOs) in the periventricular region of the brain.1)–3) CVOs, which are midline structures in the brains of all vertebrates,4) are so named because of their proximity to the ventricles of the brain. Their specialized common features are extensive vascularization lacking a blood-brain barrier and atypical ependymal cells. Among CVOs, the subfornical organ (SFO), organum vasculosum of the lamina terminalis (OVLT), and area postrema (AP) are called sensory CVOs (sCVOs) because they contain neuronal cell bodies, whereas the neurohypophysis (NH), median eminence (ME), and pineal gland are termed secretory CVOs.4)–7) sCVOs harboring neurons with efferent neural connections to many other areas of the brain are exposed to the chemical environment, such as electrolytes and hormones in the general circulation, including blood and cerebrospinal fluid (CSF).4),5) This review summarizes advances in this scientific field in the past 20 years by highlighting the critical roles of sCVOs as sensing loci of sodium ion (Na+) concentration ([Na+]), osmolality, and relevant hormones in circulation, such as angiotensin II (Ang II) and aldosterone, to the regulation of water/salt intake behaviors and blood pressure (BP).
2. [Na+] sensors and osmosensors
Na is one of the main electrolytes present in extracellular fluids and is the main ion affecting osmolality. [Na+] of the plasma and CSF is strictly controlled at ∼145 mM in mammals including humans.8) Dehydration is associated with increases in both [Na+] and osmolality in body fluids. Because Na homeostasis is essential for life, [Na+] in plasma and CSF has been postulated to be continuously monitored within the brain to maintain physiological [Na+] in body fluids.9) Andersson hypothesized the existence of a putative [Na+] sensor in the brain that is distinct from osmosensors nearly four decades ago,10),11) based on the findings that drinking and antidiuretic responses in conscious sheep were increased more by an injection of hypertonic sodium chloride (NaCl) solution than that of an equiosmolar hypertonic sucrose solution into the 3rd ventricle.12) Thus, [Na+] sensors and osmosensors were both proposed to be present in the brain for the control of water/Na intake as well as excretion by the kidneys.1),3),13)
2.1. [Na+] sensors.
2.1.1. Nax channel.
[Na+]-sensitive Nax channel expressed in glial cells.
Nax belongs to the voltage-gated Na channel family; however, its primary structure is largely divergent (∼50% identical) from other members, including the voltage sensor region.14) Nax has long remained as an enigmatic channel because of its loss of voltage sensitivity. Its expression has been detected in a number of CVOs in mice, including the SFO, OVLT, ME, and NH (posterior pituitary gland).15) The expression of Nax in mice has also been detected in peripheral regions, including dorsal root ganglion neurons, the myometrium of the pregnant uterus, non-myelinating Schwann cells, and alveolar type II cells in the lungs.15),16)
Our previous study revealed a marked elevation of intracellular [Na+] ([Na+]i) in dissociated SFO and OVLT cells from wild-type (WT) mice after the extracellular [Na+] ([Na+]o) had been increased from 145 mM (a physiological level) to 170 mM in vitro.17) These [Na+]-sensitive cells were all immunoreactive for Nax. The introduction of a Nax cDNA-harboring expression plasmid into dissociated SFO cells from Nax-knockout (KO) mice led to a similar increase in [Na+]i to that in Nax-positive cells from WT mice.18) The expression of Nax cDNA in homologous and heterologous cells induced a similar Na current to that observed in native cells.17)–19)
In a two-bottle test using pure water and 0.3 M NaCl solution, WT and Nax-KO mice fully satiated with water and salt showed no marked preference for either15),20); however, extensive water intake and an aversion to saline were observed in dehydrated WT mice, whereas dehydrated Nax-KO mice did not show an aversion to saline.20) Consistent with these findings, the intracerebroventricular (i.c.v.) infusion of a hypertonic saline did not induce an aversion to saline in Nax-KO mice, in contrast to WT animals.20) Importantly, an aversion to salt was not induced by the i.c.v. infusion of a hyperosmotic mannitol solution with physiological [Na+] in either genotype of mice, suggesting the involvement of Nax in the [Na+]-specific sensing mechanism in the brain as the [Na+] sensor.20)
Na+-sensitive cells in the brain showed double immunopositivity for glia-specific glutamate transporter (GLAST) and glial fibrillary acidic protein (GFAP), suggesting that cells expressing Nax are glial cells (Fig. 1A).21) Previous studies demonstrated that some GABAergic neurons in the SFO were surrounded by Nax-positive processes (Fig. 1B), whereas GABAergic neurons resided outside of the OVLT and clearly segregated from Nax signals of the OVLT (Fig. 1B).15),16) The double-immunostaining and immunoelectron microscopic analyses of the SFO and OVLT showed the exclusive localization of Nax to the perineuronal lamellate processes that extended from ependymal cells and astrocytes (Fig. 1C).21) The finding that glial cells express Nax indicates that the signal of an increase in [Na+] sensed by Nax needs to be definitely transferred to specific neurons for the control of animal behavior (Fig. 1D).
[Na+] sensitivity of Nax is modulated by endothelin-3.
Our previous in vitro studies of Nax-positive cells identified ∼150 mM as the activation threshold of Nax for [Na+] and ∼157 mM as the half-maximal response concentration (C1/2) (see Fig. 2C).17),18) An increase, but not a decrease, in [Na+]o from physiological levels induced a response by Na-sensitive cells. These cells did not respond to an increase in osmolality or the extracellular concentration of chloride ions.17) Tetrodotoxin at 1 µM did not antagonize these responses, which was in contrast to typical voltage-gated Na channels. Furthermore, inward currents that remained active in ‘high [Na+]’ conditions were detected using whole-cell current recordings.17) Notably, [Na+] in body fluids is usually maintained at 135–145 mM in mammals.8) In order to maintain this physiological level, brain sensor(s) need to be able to detect an increase in [Na+] within this range. This suggests the possibility that the threshold value of Nax for [Na+]o is somewhat shifted to a lower level in vivo compared with that in vitro, and covers this physiological range by some mechanism (see below).

Previous studies reported that the endothelin B receptor (ETBR) is predominantly expressed in glial cells22) and enriched in the SFO.23) Our electrophysiological study using a Nax-positive subset of SFO cells from WT mice showed that the influx of Na+ through Nax was dose-dependently induced by endothelin-3 (ET-3) at concentrations of 0.1–10 nM in a 145 mM [Na+] solution (Fig. 2A).24) This response was also confirmed by intracellular Na+ imaging.24) The plateau of the current amplitude was found to be dependent on the concentration of ET-3 ([ET-3]) with a peak being observed at ∼10 nM with [Na+]o of 140 mM (Fig. 2B); the [ET-3] that elicited C1/2 was 0.52 nM. The preincubation of these cells with 100 nM of ETBR blocker, BQ788 completely suppressed the ET-3-induced current response.24) Glial cells isolated from the SFO of Nax-KO mice did not show any current by ET-3 or [Na+]o.
The [Na+]o-dependent response of Nax reached a maximum at ∼170 mM without ET-3 and the C1/2 value was 157 mM.17) In the presence of 1 nM ET-3, the response curve of [Na+]o dependency shifted to lower level24); the C1/2 value shifted to 133 mM (Fig. 2C). Of note, the slope of the [Na+]o dependence curve with 1 nM ET-3 was more gradual than that without ET-3, indicating that ET-3 signaling increases the sensitivity of Nax channels to a wider range of [Na+]o. These findings suggest that the active range of Nax in vivo covers the whole physiological level. The expression of ET-3 was consistently induced in the SFO in water-depleted conditions when [Na+]o increased (Fig. 2D), along with the phosphorylation of ERK 1/2 (Fig. 2E).24) Therefore, the local increase of ET-3 in the SFO may help and promote the activation of Nax in slightly dehydrated conditions.
Nax activation by ET-3 is mediated by PKC pathways leading to the downstream activation of ERK1/2.
Signaling from ETBR is reportedly transduced via Gαi- and Gαq-dependent pathways.25) Gαi directly activates Src, which leads to the activation of Ras/Raf. In contrast, the Gαq pathway initially activates protein kinase C (PKC) followed by Src and Ras/Raf.26) Both pathways eventually converge to activate ERK1/2 downstream through mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) kinase (MEK). Pharmacological experiments using various kinase inhibitors indicated that the activation of Nax channels by ET-3 occurred downstream of Gαq, but not Gαi, and that the activation of ERK1/2 downstream of PKC was essential for the influx of Na+ (Fig. 2F).24)
Transfer of the signal of an increase in [Na+] from Nax-positive glial cells to neurons.
Nax channels are specifically expressed in the perineuronal processes of astrocytes and ependymal cells enveloping particular neural populations in the SFO, such as GABAergic neurons (Fig. 1C). Therefore, a signal transfer mechanism from Nax-positive glial cells to neurons needs to exist. We revealed that Nax channels stably interacted with the αl and α2 subunits of Na+/K+-ATPase via its carboxyl terminal region.27) This binding of Nax to Na+/K+-ATPase was a requisite to the stimulation of Na+/K+-ATPase activity following the influx of Na+ by the [Na+]o-dependent activation of Nax.27) Because glial cells use an anaerobic pathway for glucose metabolism to regenerate ATP, the activation of Na+/K+-ATPase is anticipated to stimulate lactate production as the end product (Fig. 3A). As expected, lactate release from the SFO tissue of WT, but not Nax-KO, mice was enhanced by incubation with hypertonic Na+ solution (Fig. 3B).

Our electrophysiological experiments revealed the spontaneous firing of GABAergic neurons in slices of the SFO from WT mice, the frequency of which increased gradually with the application of hypertonic Na+ (Fig. 3C).27) In contrast, the activity of GABAergic neurons in the SFO of Nax-KO mice was not potentiated by hypertonic Na+ (Fig. 3C). The firing frequencies of GABAergic neurons of both genotypes increased when lactate was added at 1 mM to the perfusate (Fig. 3D). α-Cyano-4-hydroxycinnamic acid (α-CHCA), an inhibitor of monocarboxylate transporters (MCTs), which transport lactate together with H+ across membranes, inhibited the Na+-dependent potentiation of GABAergic firing.27) Therefore, lactate released from glial cells serves as an energy substrate to generate ATP and increases the firing activity of GABAergic neurons through in activation of KATP channels.27) Consistent with this finding, neuronal activation state estimated by Fos expression was higher in the SFO of Nax-KO mice than that of WT mice after water deprivation when [Na+] in body fluids increased. This is probably because the neural activation of inhibitory GABAergic neurons is deficient in Nax-KO mice (see below).15)
In summary, the existence of a dynamic mechanism, a change in the threshold of [Na+] sensors for body fluids caused by the local expression of the vasorelaxation hormone, ET-3 in the SFO was demonstrated. This mechanism appears to work for [Na+] sensing in body fluids in vivo (Fig. 3E). ETBR signaling subserves the activation of Nax channels and enhances glucose metabolism leading to lactate release in Nax-bearing glial cells. This supply of lactate from glial cells eventually yields the activation of GABAergic neurons in the SFO. The activation of inhibitory neurons in the SFO is thus responsible for the negative control of salt appetite. In dehydrated conditions, Nax signals in the SFO suppress salt intake and those in the OVLT induce water intake, respectively (see below).
2.1.2. SLC9A4.
Transmembrane proteins encoded by the family of slc9 genes [also referred to as Na/hydrogen exchanger (NHE4)] are responsible for the antiport of Na+ and H+ in cells. A previous study reported the crucial roles of these proteins in the regulation of cell volumes, the transepithelial absorption of Na+, and intracellular pH homeostasis.28) The ubiquitous expression of members of the slc9 gene family has been demonstrated including the brain.29) A RNA sequencing analysis of the anteroventral wall of the 3rd ventricle (AV3V), including the OVLT, revealed significantly high expression levels of solute carrier family 9 member A4 (SLC9A4).30) Other transcripts of SLC9 members were also detected in the OVLT, but not at significant levels. In vitro experiments showed that SLC9A4 activity levels were elevated at increased [Na+]o.31)
Our Na imaging experiments on Neuro-2a cells expressing SLC9A4 by transfection showed the activation of SLC9A4 with increases in [Na+]o, but not osmolality.30) [Na+]i in Neuro-2a cells transfected with slc9a4 gradually became elevated as [Na+]o increased from 145 to 170 mM (Fig. 4A). However, a mannitol (50 mM)-induced increase in extracellular osmolality from 330 to 380 mOsm/kg at [Na+]o of 145 mM did not alter [Na+]i. Consistent with our results, SLC9A4 in colonic crypts remained inactive in the presence of increased osmolality in vitro.32) SLC9A4 was activated at [Na+]o higher than ∼150 mM, and its [Na+]o-dependent response plateaued at ∼170 mM with an estimated C1/2 of ∼160 mM (Fig. 4B). Similarities were thus observed in the in vitro functional properties of SLC9A4 and Nax as [Na+] sensors (see Fig. 2C). However, it should be noted that discrepancies have been reported in the effects of increased osmolality on the activation of SLC9A4.31),33),34)
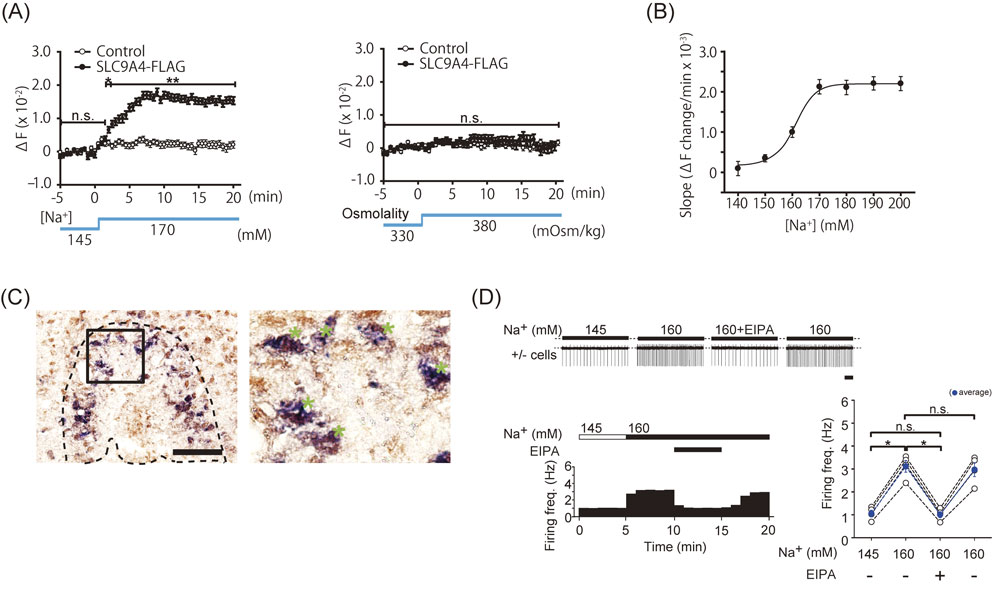
We performed an immunohistochemical analysis of SLC9A4-positive neurons in the OVLT and showed an overlap with Fos-positive cells in water-deprived conditions, and, at the cellular level, the expression of SLC9A4 co-localized with Ang II receptor 1a (AT1a) signals (Fig. 4C).30) The spontaneous firing of SLC9A4-positive neurons in the OVLT at approximately 1 Hz was detected at a physiological level of [Na+] (145 mM). Firing frequency increased to ∼3 Hz at [Na+] of 160 mM, (Fig. 4D) and then decreased to baseline when [Na+] in the perfusate was returned to 145 mM.30) This increase in the firing frequency was shown to be reversibly inhibited by the SLC9-specific antagonist, ethylisopropylamiloride (EIPA).28) Collectively, these findings suggested the facilitation of SLC9A4-positive neuronal activation by [Na+]-sensitive SLC9A4 (Fig. 4D).30) Our functional analyses of SLC9A4 revealed that SLC9A4 works in the OVLT as a [Na+] sensor to regulate water intake behavior (see below).
2.1.3. ENaC.
Epithelial sodium channels (ENaCs) are amiloride-sensitive Na channels highly selective for Na+. These Na channels are non-voltage-dependent, and conduct Na+ ions across the apical membrane of cells in salt-reabsorbing epithelia, such as in the distal nephron,35) in which they play a master role in regulating the volume of extracellular fluid. ENaCs are also expressed in astrocytes, endothelial cells, and the choroid plexus,36) which may contribute to the maintenance of physiological [Na+] in CSF. Besides central neurons,36),37) ENaCs function as Na detectors of the salt taste receptors in the tongue.38),39) Loewy’s group reported that ENaC-positive neurons are highly concentrated in sCVOs.35) These neurons are activated after various manipulations of peripheral [Na+], including systemic injections with hypertonic saline, Na repletion after prolonged Na deprivation, or dietary Na deprivation. Further study on animals with the site-directed deletion of ENaC is required to conclude that ENaC also functions as the brain [Na+] sensor for the control of water/salt intake.
2.2. Osmosensors.
Cell membranes are more permeable to water than to ions because of the presence of aquaporins; therefore, the shrinkage or swelling of cells occurs with an increase or decrease in extracellular osmolality, respectively.12),40) Aquaporins, which are plasma membrane water-transporting proteins, have been shown to play a direct role in cellular shrinkage and swelling.41) The mammalian brain has been suggested to detect systemic hypertonicity through a process involving cellular dehydration. Cellular shrinkage or swelling negatively affects the morphology of cells and lipid bilayer tension. These changes in extracellular osmolality are regarded as a form of mechanical stimulation, which indicates that mechanosensors also function as osmosensors. In the last two decades, a number of transient receptor potential (TRP) cation channels have been proposed to act as mechanosensitive ion channels,42),43) such as TRPC6.44) Previous findings indicated that TRP vanilloid 1 (TRPV1) and TRPV4 have the ability to detect changes in cell volumes caused by increases
or decreases in extracellular osmolality,45)–48) and both are expressed in sCVOs.49)–52)
2.2.1. TRPV1.
TRPV1 is a heat-, ligand- (e.g., capsaicin), and pH-sensitive, voltage-dependent cation channel that is involved in thermal nociception.53) Increases in intracellular calcium (Ca2+) concentration ([Ca2+]i) were not observed in cells expressing full-length TRPV1 in response to hypotonic solutions,47),48) and hypertonic solutions did not affect current at room temperature.54) We previously reported the heterologous expression of TRPV1 in mammalian cells as well as its hypertonicity-induced activation at approximately body temperature46); however, Ca2+ influx induced by increases or decreases in extracellular osmolality was apparently insufficient to produce adequate signals. Another study showed that water intake was significantly lower in TRPV1-KO mice than in WT mice treated with an intraperitoneal (i.p.) injection of a hypertonic solution.49) An N-terminal variant of TRPV1 (ΔN-TRPV1) has been proposed to play a role in osmosensing.55),56) However, normal thirst responses as well as the central induction of Fos were observed in TRPV1-KO mice in systemic hypertonic conditions, and were similar to those induced by a subcutaneous injection of hypertonic saline, water deprivation, or sole access to a salt solution.57),58) We independently investigated voluntary water intake induced immediately after the i.c.v. administration of a hypertonic NaCl solution and found that it was normal in TRPV1-KO mice.59)
2.2.2. TRPV4.
The original role proposed for TRPV4 was the detection of extracellular hypotonicity.47) Liedtke et al. reported the down-regulated expression of Fos in OVLT neurons and decreases in water intake and vasopressin (VP) production in TRPV4-KO mice in response to systemic hypertonicity.51) And yet not the same time, cells expressing TRPV4 in the SFO were previously reported to contribute to reductions in drinking behavior in hypoxic conditions.60) Ciura et al. demonstrated that osmosensory neurons in the OVLT were inhibited by TRPV4-positive glial cells via taurine, a glycine receptor agonist.61)
In contrast, a previous study reported normal water intake behavior, but the excessive production of VP in TRPV4-KO mice subjected to water deprivation and the systemic administration of propylene glycol.62) Another group confirmed normal water intake behavior and the central expression of Fos in TRPV4-KO mice following a subcutaneous injection of hypertonic saline.57) These different results may be attributed, at least partly, to distinct systemic dehydration methods, which affect multiple peripheral organs. This means that direct stimulation of the sensing system in the brain is important for obtaining more precise information. We investigated the functional involvement of brain sensors in water intake behavior using the i.c.v. infusion of hypertonic solutions into the KO mice of these specific sensor genes.59) Our findings suggested that hypertonicity in CSF was not directly sensed by TRPV1 or TRPV4 in sCVOs (see below).
2.2.3. G protein-coupled receptors.
Gudermann’s group reported that activated Gq/11-coupled receptors transmit signals in G protein- and phospholipase C-dependent manners to TRPC channels.63) The cell-attached patch-clamp configuration revealed that only the co-expression of G protein-coupled receptors (GPCRs) with TRPC6 indirectly activated mechanosensitive channels. Zou et al. reported that mechanical stress activated AT1 receptors independently of Ang II, and this activation may be inhibited by an inverse agonist of AT1 receptors. Mechanical stretch induced the association of AT1 receptors with Janus kinase 2 as well as the translocation of G proteins into the cytosol.64)
It is well known that AT1 receptors are abundantly expressed in neurons in the SFO and OVLT. Some groups reported that mechanical stretch induced β-arrestin-biased signaling downstream of AT1 receptors in the absence of ligands or G protein activation65)–67): β-Arrestins, which were originally characterized as terminators of heterotrimeric GPCR signaling, also function as important signal transducers.68) In HEK293 cells transfected with siRNA directed against β-arrestins 1 and 2, hypo-osmotic stretch attenuated ERK phosphorylation significantly more than in cells transfected with control siRNA. Mechanical stretch triggered an AT1 receptor-mediated conformational change in β-arrestins, similar to that induced by β-arrestin-biased ligands, such as TRV120023, to selectively stimulate receptor signaling in the absence of detectable G protein activation.65) AT1 receptors are thus postulated to be mechanical sensors; however, it currently remains unclear whether this system contributes to osmosensing in the brain. In summary, the molecular entity of the brain osmosensor is still enigmatic.
3. Water intake control
Homeostatic control mechanisms ensure that the physiological water balance in body fluids is maintained through the regulation of water intake and excretion.69) Thirst sensation was an essential step in the evolution and adaptation of vertebrates to terrestrial life. A number of different factors, such as lifestyle, eating behavior, body temperature, and body-fluid conditions, have been shown to affect water intake behavior in humans.70) Hyponatremia (hypotonicity) is caused by an excessive water intake that exceeds water excretion by the kidneys, whereas dehydration results in hypernatremia (hypertonicity) in body fluids.71),72) Thirst is generally aroused by a deficit in systemic water70),73),74) and orients the actions of animals towards finding and consuming water, thereby restoring the proper physiological level of the Na-water balance.74),75)
Thirst-driving systems are present in the forebrain sCVOs,4),70),73),75)–78) such as the SFO79) and OVLT,4),80) which contain sensors for many thirst-driving signals in body fluids, including [Na+], hypertonicity, and circulating Ang II.81)–83) A recent study showed that thirst modalities caused by osmotic or hypovolemic stress were evoked by distinct genetic types of excitatory neurons in the SFO and OVLT.84) Previous studies demonstrated that water drinking was immediately elicited following local injections of Ang II into the SFO and OVLT, which express AT1a.83),85)–87)
3.1. Activation mechanisms of thirst.
3.1.1. Increases in [Na+] and osmolality in body fluids.
Are TRPV1 and TRPV4 brain osmosensors?
We previously investigated whether water intake differed among WT, TRPV1-KO, TRPV4-KO, and Nax-KO mice in hydrated (normal) and dehydrated conditions59); systemic dehydration was achieved by water deprivation for 24 or 48 h. Following the dehydration, similar increases were observed in water intake without genotypic differences. This finding suggested that systemic dehydration is also sensed by peripheral sensing systems other than the brain. Therefore, we used an i.c.v. injection to test whether these molecules are brain [Na+]/osmosensors that monitor body-fluid conditions. Rapid water intake responses were observed in all genotypes of mice following the i.c.v. administration of 4 µL of hypertonic sorbitol artificial CSF (aCSF) (aCSF containing 0.145 M NaCl and 1.71 M sorbitol; ∼2.0 osmol/kg H2O), and these responses ended within 20 min.59) Notably, the i.c.v. administration of hypertonic sorbitol aCSF induced similar water intakes, but lower levels in all genotypes, indicating that the sensing of increases in CSF osmolality was normal in all genotypes. This finding indicated that TRPV1, TRPV4, and Nax are not osmosensors for CSF in the brain.
[Na+] sensing by Nax leads to activation of TRPV4-positive neurons for water intake induction.
Following the administration of hypertonic NaCl aCSF (aCSF containing 1 M NaCl; ∼2.0 osmol/kg H2O), genotypic differences were observed in water intake, suggesting roles for Nax and TRPV4, but not TRPV1, as sensors of increases in CSF [Na+].59) Water intakes by Nax-KO, TRPV4-KO, and Nax/TRPV4-double KO (DKO) mice were decreased similarly to ∼50% as compared to the level of WT mice, suggesting the functional involvement of Nax and TRPV4 in a single signaling pathway. A treatment with HC-067047, a TRPV4-specific antagonist, before the i.c.v. administration of hypertonic NaCl aCSF did not alter water intake behavior by TRPV4-KO or Nax-KO mice, whereas that by WT mice was decreased by ∼50% to similar levels as those by TRPV4-KO mice and Nax-KO mice. Therefore, Nax and TRPV4 appear to play roles in the same signaling pathway to stimulate water intake behavior.
Water intake behavior in all genotypes was more robust after i.c.v. infusion of hypertonic NaCl aCSF than hypertonic sorbitol aCSF with the same osmolality and, thus, the increase in [Na+] appeared to be a more effective stimulus for water intake than osmolality.59) Conversely, an i.c.v. injection of hypotonic aCSF did not lead to the intake of 0.3 M NaCl solution in 30 min in WT or Nax-KO mice, suggesting that rapid [Na+] decreases in CSF are not salt appetite-inducing stimuli (our unpublished observation).
OVLT is the main sensing site of [Na+] increases for water intake induction.
To specify the brain locus detecting [Na+] increases in CSF for water intake induction, electrolytic lesion experiments on the SFO or OVLT were performed on WT mice.88) Sham-operated and SFO-lesioned mice showed no significant differences in water intake after the i.c.v. administration of hypertonic aCSF with NaCl or sorbitol, suggesting that thirst driving neurons in the SFO are not activated by increases in [Na+] or osmolality in CSF. This is consistent with previous findings.89)–92) However, OVLT-lesioned mice drank markedly less water than sham-operated mice after the i.c.v. administration of hypertonic aCSF. The reduced water intake by Nax-KO mice was restored to WT levels by the site-specific introduction of the Nax gene into the OVLT in Nax-KO mice using an adenoviral expression vector.88) Therefore, the Nax signal in the OVLT appears to play a role in controlling water intake behavior.
TRPV4 is activated by EETs to induce water intake.
Arachidonic acid (AA) activates TRPV4 in HEK-293 cells via the formation of epoxyeicosatrienoic acids (EETs),93) and 5,6-EET has been identified as the most potent EET for the activation of TRPV4. The preliminary administration of a CYP450 epoxygenase inhibitor, miconazole, which has been shown to inhibit EET synthesis from AA,94) to WT mice decreased hypertonic NaCl aCSF-induced water intake to the same level as that by TRPV4-KO mice.59) However, since water intake remained unchanged in Nax-KO mice upon treatment with miconazole, the Nax signal for water intake behavior appears to require the activation of TRPV4.
In i.c.v. injection experiments using hypertonic NaCl aCSF containing various amounts of 5,6-EET, dose-dependent increases were observed in water intake plateauing at 0.64 ng.88) The i.c.v. injection of 4 µL of hypertonic NaCl aCSF containing 0.64 ng of 5,6-EET restored water intake by Nax-KO mice to the level of WT mice, whereas that by TRPV4-KO mice was not affected (Fig. 5A). These findings provide strong supportive evidence for the activation of TRPV4 by EETs downstream of Nax. Because hypertonic NaCl aCSF-induced water intake behavior remained unchanged in WT mice when injected with these metabolites, the endogenous production of EETs induced in the OVLT of treated WT mice was sufficient for the activation of TRPV4.59) The amount of 5.6-EET in the OVLT reached was measured as 4.16 ± 0.36 pg by the i.c.v. injection of 0.64 ng 5.6-EET,30) which was similar to that in the OVLT in water-deprived WT mice (3.53 ± 0.27 pg) (Fig. 5B). In situ hybridization and immunohistochemical analyses revealed the overlap of TRPV4 mRNA signals with the neural marker, NeuN, but not with the Nax signal.59) Collectively, these findings strongly supported the view that Nax-positive glial cells produce and release EETs in response to [Na+] increases in body fluids, and EETs activate neighboring neurons with TRPV4 in the OVLT to induce water intake.59)

Following the i.c.v. administration of a hypertonic NaCl solution, water intake by Nax-KO mice was significantly less than that by WT mice.59) Nevertheless, water intake was still induced, suggesting the existence of an unknown [Na+]-dependent pathway other than the Nax/TRPV4 pathway for the stimulation of water intake behavior. SLC9A4 (also called NHE4) was identified by our group in an RNA sequencing analysis as one of the molecules enriched in the OVLT.30)
A double immunohistochemical analysis of the OVLT of AT1alacZ/+ mice using anti-SLC9A4 and anti-β-galactosidase antibodies revealed the coexpression of SLC9A4 and AT1a.30) Consistent with the coexpression of SLC9A4 and AT1a at the cellular level, increases in the firing frequency of SLC9A4-positive neurons were detected by [Na+]o of 160 mM as well as the addition of 0.1 µM Ang II to [Na+] of 145 mM.30) The firing activities of SLC9A4-positive neurons in the presence of [Na+]o of 160 mM (∼2.6 Hz) were increased further by the application of Ang II (∼4.1 Hz) (Fig. 5C). Therefore, SLC9A4-positive neurons appeared to be activated by increases in both [Na+]o and the concentration of Ang II ([AngII]). A recent study detected similar neurons, namely, those that respond to elevated [Na+] and [Ang II], in the OVLT of rats.95) Water intake did not significantly differ between WT and AT1a-KO mice administered hypertonic NaCl or sorbitol aCSF, which indicated that AT1a is not involved in the water intake responses stimulated by rapid increases in [Na+] or osmolality in CSF. In systemic dehydration, [Na+] and Ang II gradually reach high levels in blood.96),97) Therefore, SLC9A4 and AT1a double-positive neurons appeared to respond to [Na+] and [Ang II], respectively, both of which are elevated in continuous water-deprived conditions (Fig. 5D).
Knockdown of Slc9a4 in the OVLT impairs water intake.
SLC9A4 expression in the OVLT was knocked down by the expression of Slc9a4 miRNA using an adeno-associated virus (AAV). Water intake induced by an i.c.v. injection of hypertonic NaCl aCSF was significantly lower in Slc9a4-knockdown mice (Slc9a4-KD mice) than in control mice.30) In contrast, water intake was only slightly induced by the i.c.v. administration of equiosmolar hypertonic sorbitol aCSF, but was similar to that by WT mice, indicating that osmosensitivity in the OVLT in Slc9a4-KD mice was unaffected.
The pre-administration of EIPA, an SLC9 family inhibitor, dose-dependently inhibited water intake by WT mice until a plateau at 0.4 mM.30) Similar water intake volumes were observed in EIPA-treated WT mice and Slc9a4-KD mice. In EIPA-treated Nax-KO mice and Nax-KO mice with Slc9a4-KD in the OVLT (Slc9a4-KD/Nax-KO), water intake induced by the i.c.v. administration of hypertonic NaCl aCSF was further reduced to the same level as that stimulated by the i.c.v. administration of hypertonic sorbitol aCSF.30) Collectively, these findings indicated that an unidentified osmosensing system other than the [Na+] sensing systems by Nax and SLC9A4 also plays a role in the control of water intake (Fig. 5D).
Independence of the SLC9A4 pathway from the Nax/TRPV4 pathway.
The i.c.v. administration of 5,6-EET with hypertonic NaCl aCSF increased water intake by Nax-KO mice to the level of WT mice.30) Water intake by Slc9a4-KD/Nax-KO mice also increased to the level of Slc9a4-KD mice following the i.c.v. administration of 5,6-EET, whereas that by Slc9a4-KD mice remained unchanged.30) These findings supported the SLC9A4 signal exerting its effects on water intake control independently of the Nax/TRPV4 pathway.
SLC9A4 is a major proton (H+) extruding system driven by an inward Na+ gradient. SLC9A4 is not electrogenic but plays an important role in signal transduction. The activation of SLC9A4 by elevated [Na+]o reduces extracellular pH (and increases intracellular pH) in OVLT neurons. Psalmotoxin-1 (Pctx1), a specific inhibitor of acid-sensing ion channel 1a (ASIC1a), was found to significantly decrease water intake stimulated by the i.c.v. administration of hypertonic NaCl aCSF.30) Therefore, SLC9A4-positive thirst-driving neurons appear to be activated in a H+-dependent manner via ASIC1a. SLC9A4 thus functioned as an independent [Na+] sensor in the OVLT for the regulation of water intake behavior (Fig. 5D). Our recent study revealed the presence of such neurons activated by increases in extracellular [H+] via ASIC1a in the OVLT.98)
[Na+] sensors and osmosensors for water intake control.
In summary, the stimulation of water intake by elevated [Na+] in body fluids is mainly attributed to the Nax/TRPV4 and SLC9A4/ASIC1a pathways in the OVLT. It is important to note that Ang II activated thirst-driving neurons with SLC9A4/ASIC1a in the OVLT as well as AT1a-positive neurons in the SFO to induce water intake in persistent water-deprived conditions (see below). Physiologically, Na+ is the main factor influencing osmolality in body fluids, and, thus, these two [Na+]-dependent pathways are the main system inducing water intake (Fig. 5D). Nevertheless, the residual amount of water intake controlled by osmosensors have been suggested to play a key role in specific conditions (Fig. 5D). In some situations when extracellular osmolytes besides Na+, for instance glucose in diabetes, may affect the osmolality of body fluids.
3.1.2. Angiotensin II.
The peptide hormone Ang II in the brain induces thirst, salt appetite, and BP elevations.70),99) Angiotensinogen, the precursor of Ang II, is mainly released from the liver and cleaved in the circulation by renin, which is secreted from the juxtaglomerular apparatus of the kidneys to form Ang I.100),101) Circulating Ang I is then activated to the octapeptide Ang II by angiotensin-converting enzyme (ACE), a membrane-bound metalloproteinase that is predominantly expressed in high concentrations on the surface of endothelial cells in the pulmonary circulation.101),102) Ang II in body fluids is physiologically increased by hypovolemia, dehydration, and Na deficiency.70),96),103),104) Because AT1a is highly expressed in sCVOs in the brain, these structures may be the main sites for the transduction of signals by circulating Ang II to neural signals.4),105) On the other hand, the activation of the renin-Ang II system in the SFO was also shown to induce marked increases in water and salt intakes, urine volume, and sympathetic nerve activity (SNA).106)
The systemic or i.c.v. administration of Ang II has been shown to induce water and salt intake.107)–109) Ang II-induced drinking behavior is evolutionarily conserved in many animal species, including humans.70) The stimulatory effects of Ang II on salt ingestion was initially suggested based on brief infusions of large doses of Ang II into the cerebral ventricles or surrounding tissue of the anterior forebrain.110) More localized infusions of Ang II into the AV3V in the immediate vicinity of the OVLT elicited both water and salt ingestion,111) and lesions in the SFO attenuated some models of salt appetite.112),113) We functionally characterized AT1a-expressing neurons in the sCVOs in an attempt to elucidate the neural mechanisms underlying the control of thirst and salt appetite.96)
Roles of the AT1a signal in thirst generation.
A previous study that immunostained β-galactosidase in AT1alacZ/+ mice to investigate the distribution of AT1a-positive neurons revealed their presence in a number of brain nuclei, including three sCVOs.96) In water-depleted conditions in mice achieved by dehydration for 48 h, [Ang II] increased ∼3.0-fold in blood with an elevation in [Na+] of ∼15 mM. The two-bottle test was conducted using WT mice and AT1a-KO mice114); both groups were offered water and 0.3 M NaCl solution in the absence of food.96) The results showed that water intake by WT mice was higher in water-depleted conditions than in control conditions; however, the intake of salt solution was markedly less than that of water, which reflected salt avoidance because of high [Na+] in body fluids due to dehydration. Here, water intake by AT1a-KO mice increased to the same level as that by WT mice in the water-depleted conditions, whereas salt intake was negligible, indicating the presence of an AT1a-independent mechanism, such as a Na+ signal, to induce water intake in water-depleted conditions.96) The number of Fos-positive neurons in the SFO and OVLT of AT1alacZ/+ mice increased with water depletion: fractions of β-gal-positive neurons in Fos-positive neurons were 49.7 ± 4.0% in the SFO and 32.7 ± 2.8% in the OVLT.96) The number of β-gal-positive fractions in Fos-positive neurons decreased in AT1a-KO mice (27.4 ± 3.3% in the SFO and 19.6 ± 1.0% in the OVLT). This finding in the OVLT was consistent with that AT1a-KO mice drink water after dehydration.
To achieve the site-specific deletion of Agtr1a in the SFO or OVLT, the respective brain nuclei of loxP-flanked Agtr1a (AT1aloxP/loxP) mice were injected with an AAV carrying the gene encoding Cre-recombinase (AAV-Cre). The individual deletion of Agtr1a in the SFO or OVLT significantly decreased water intake induced by furosemide treatment, respectively. Recent studies, including ours described below, showed that AT1a-positive OVLT neurons induced thirst and BP elevations by negative reinforcement.30),98),115)
AT1a-positive neurons in the SFO.
The mouse line Vglut2-Cre;AT1alacZ/+ was generated from AT1alacZ/+ mice crossed with vesicular glutamate transporter 2 (Vglut2)-Cre mice.116) In Vglut2-Cre;AT1alacZ/+ mice injected with AAV carrying double-loxP-flanked inverted orientation (DIO)-enhanced green fluorescent protein (EGFP) into the SFO, β-gal (namely AT1a) signals largely overlapped with EGFP in glutamatergic neurons, indicating that AT1a neurons are excitatory neurons.96) The majority of AT1a-positive neurons expressed neural nitric oxide synthase (nNOS), also indicating that AT1a-positive neurons in the SFO are excitatory neurons.117) These glutamatergic SFO neurons innervated the OVLT, median preoptic nucleus (MnPO), the ventral part of the bed nucleus of stria terminalis (vBNST), paraventricular nucleus (PVN), and supraoptic nucleus (SON)96); OVLT was extensively innervated by AT1a-positive glutamatergic SFO neurons. On the other hand, a small number of fibers from GABAergic SFO neurons were detected in the OVLT and MnPO, whereas none projected to the vBNST, PVN, or SON.96)
The SFO→OVLT pathway controls water intake.
In the two-bottle test, water intake, but not salt intake, was rapidly stimulated in mice in normal water-satiated conditions by the optical excitation of the SFO→OVLT pathway (glutamatergic neurons) using the standard optogenetic technique with channelrhodopsin 2 (ChR2) and blue light exposure.96) The intake of ∼1.0 mL of water within 20 min is striking because it represents approximately one-third of the daily drinking volume.96) Water intake by the majority of mice immediately stopped and they left the spout following the termination of the light stimulation shortly after the induction of drinking, which suggested a direct relationship between the neural stimulation and water intake behavior (or the recognition of thirst). However, salt intake was not affected by the optical excitation of the SFO→OVLT pathway.96)
Water intake by mice was typically increased in water-depleted conditions without the optical stimulation, but was significantly decreased by the optical silencing of SFO(→OVLT) neurons using archaerhodopsin 3.0 (ArchT), a yellow light-driven outward H+ pump.96) In contrast, the optical silencing of the same pathway did not alter salt intake in Na-depleted conditions.96) Therefore, SFO(→OVLT) neurons (water neurons) mediate Ang II-dependent thirst control, but not salt appetite control (Fig. 6A).

Consistent with our results, similar studies demonstrated that the stimulation of excitatory neurons in the SFO and OVLT together with the MnPO induced robust water ingestion (Fig. 6A).115),118) Allen et al. revealed that thirst regulates motivated behavior through modulation of brain wide neural population dynamics.119) The optical activation of glutamatergic neurons in the SFO reproduced neural activities throughout the brain along with water intake, which were detected in thirsty mice,119) indicating that the SFO and OVLT are the origins of thirst-encoding information in the brain.
3.1.3. Dipsogenic hormones.
Secretin.
Since its discovery by Bayliss and Starling in 1902,120) the hormone secretin (SCT) has been shown to regulate water homeostasis throughout the body and influence the environment of the duodenum by regulating secretions in the stomach, pancreas, and liver.121) Recent studies reported that SCT and its receptor function in the circulation as well as the central nervous system.122),123) Water intake behavior was induced in WT mice by the i.c.v. administration of Ang II; however, the volume of water consumed by SCT receptor (SCTR)- and SCT-KO mice was much reduced124); therefore, an intact SCT/SCTR system in mice appears to be needed for Ang II to induce its dipsogenic effects. The up-regulated expression of SCT and SCTR was observed in magnocellular cells in the PVN after the i.c.v. administration of Ang II and the chronic feeding of saline.124)
Fluorescence resonance energy transfer (FRET) assays on CHO cells showed that transfected SCTR and AT1a formed homomers by themselves and heteromers by interacting with each other.125) SCRT and AT1a are expressed in the PVN, and they appeared to form heteromers in order to facilitate the response to Ang II induced by hyperosmolality in vivo. High SCT and SCTR transcript and protein levels have been detected in the PVN and SON. In rats exposed to hyperosmolality, mRNA levels for SCT and SCTR were increased in the hypothalamus and pituitary gland.122) Following the i.c.v. administration of SCT, significant increases were observed in VP and oxytocin (OT)-encoding gene transcript levels in vasopressinergic and oxytocinergic neurons, suggesting that SCT stimulates magnocellular cells. An i.c.v. injection of SCT also consistently triggered the release of AVP in the circulation.124)
Relaxin-3.
Relaxin, a 59-amino acid peptide hormone in the insulin superfamily, was initially discovered in 1926.126),127) It is secreted by the corpus luteum of the ovary during pregnancy.128) An i.c.v. injection of relaxin-3 significantly increased water intake and induced Fos expression in the SFO, OVLT, PVN, and SON.129),130) In addition, relaxin-3 receptor (RXFP3)-positive neurons have been detected in several brain regions, such as the SFO, PVN, and SON.131),132) RXFP3-positive neurons in the PVN were previously reported to be involved in motivation and feeding behavior.133) In the PVN and SON, relaxin-3-mediated Fos expression was induced in magnocellular neurons producing VP and OT, which are directly involved in fluid regulation.134),135) Furthermore, an i.c.v. injection of a RXFP3 antagonist suppressed salt appetite in Na-depleted mice, but not RXFP3-KO mice.136) These findings suggested that relaxin-3 is involved in water drinking behavior during pregnancy and/or other conditions.
3.2. Inhibitory mechanisms of thirst.
3.2.1. [Na+] decrease in body fluids.
Cholecystokinin-dependent modulation of water intake.
Cholecystokinin (CCK) has been reported to inhibit water intake when injected into the brain.137) Water intake decreased from 1.33 ± 0.06 mL to 0.87 ± 0.09 mL in 30 min after the direct injection of a sulfated octapeptide of CCK into the SFO in water-depleted conditions.138)
CCK neurons in the SFO.
To clarify the location of CCK-producing cells, we measured CCK concentration ([CCK]) in blood and SFO tissues in water- and Na-depleted conditions.138) Plasma [CCK] remained unchanged between the different conditions, whereas [CCK] in the SFO was markedly higher in Na-depleted conditions than in control conditions (∼9.0-fold). When AAV-DIO-mCherry was stereotaxically injected into the SFO of CCK-Cre mice, mCherry-positive cells in the SFO (CCK-positive cells) overlapped with nNOS-immunoreactive cells, indicating that they belong to excitatory neurons in the SFO.138) The findings of qPCR also revealed significantly higher CCK mRNA levels (∼2.1-fold) in the SFO in Na-depleted conditions than in control conditions, suggesting the up-regulation of CCK in CCK-positive neurons in the SFO.138)
GABAergic neurons are innervated by CCK-positive neurons in the SFO.
CCK-positive neurons and GABAergic neurons in the SFO of CCK-Cre;GAD67-GFP mice were simultaneously visualized using a direct injection of AAV-DIO-mCherry (Fig. 7A, left). The close apposition of mCherry-positive fibers and their varicosities of CCK-positive neurons to the dendrites and cell bodies of GABAergic neurons was observed (Fig. 7A, right). A subset of GABAergic neurons innervated by CCK-positive neurons exhibited excitatory postsynaptic currents (EPSC) that were completely suppressed by CNQX, a AMPA glutamate receptor antagonist (Fig. 7B).

Two types of CCK receptors (CCK-AR and CCK-BR) are present in the brain, with CCK-BR being predominantly expressed.139) GABAergic neurons in the SFO also expressed CCK-B receptors.138) CCK stimulated, whereas a CCK-B receptor antagonist, L-365,260, suppressed the firing frequencies of these GABAergic neurons (Fig. 7C). Consistent with this, water intake was not inhibited by the injection of a specific agonist for CCK-A receptors (A-71623) into the SFO.138) Collectively, these findings indicate that water intake is inhibited by GABAergic neurons with CCK-B receptors, which transmit inhibitory signals from CCK-positive neurons to water neurons in the SFO.
Ang II-dependent water intake is controlled by CCK through GABAergic neurons.
WT mice do not consume water in Na-depleted conditions, whereas plasma [Ang II] is high. To elucidate the inhibitory mechanisms suppressing the activity of SFO(→OVLT) neurons, the firing activities of a set of SFO(→OVLT) neurons and their synaptically connected GABAergic neurons were examined in brain slices from GAD67-GFP mice injected with a highly efficient retrograde gene transfer lentivirus carrying the mCherry gene (HiRet-mCherry) into the OVLT (Fig. 7D, left). The firing activity of SFO(→OVLT) neurons was not detected in WT mice with physiological [Na+] of 145 mM; however, it was induced in ∼50% of these neurons after the addition of Ang II to the perfusate (Fig. 7D). After the application of CCK, GABAergic activities increased and the Ang II-induced firing activity of SFO(→OVLT) neurons decreased (Fig. 7D). Therefore, CCK inhibited the Ang II-dependent activities of water neurons.
Water intake was significantly inhibited by the optical stimulation of CCK-positive neurons in the SFO in water-depleted conditions, in which thirst responses were strongly induced.138) The significant suppression of water intake from 1.40 ± 0.14 mL to 0.84 ± 0.12 mL in water-depleted conditions was also observed in 30 min by the optical stimulation of GABAergic neurons in the SFO. This decrease was similar to that induced in mice in which CCK was injected into the SFO.138) These findings are consistent with the CCK-dependent inhibition of water neurons in the SFO via GABAergic neurons (Fig. 8, right).
Distinct CCK-positive neuron subpopulations are present in the SFO.
Calcium images of CCK-positive neurons in the SFO were obtained at a single-cell resolution using a fluorescent calcium indicator (Fig. 9A). We found the stronger activation of CCK-positive neurons (group 1) in Na-depleted conditions than in control conditions (Fig. 9B and C). Following the initiation of salt ingestion (0.3 M NaCl), the strong activity of group 1 CCK-positive neurons in Na-depleted conditions was markedly reduced, reaching a plateau in 20 min (Fig. 9C). Notably, the neuronal activity did not immediately or transiently decrease after the ingestion of the salt solution (no effects within 40 s).138) However, the activity of CCK-positive neurons was not reduced by the ingestion of sucrose solution (0.3 M) even with similar intake volumes to those of the salt solution (Fig. 9C).

The rapid and transient activation of a number of CCK-positive neurons (group 2) was noted after the onset of water intake under hypertonic Na loading condition (Fig. 9B), but the activity ceased within 20 s. More importantly, this subpopulation was not activated in Na-depleted conditions, which suggests that their activity directly responds to water intake itself. The third subpopulation of CCK-positive neurons (group 3) was not strongly activated in either condition (Fig. 9B). Collectively, these findings indicated the presence of at least three distinct subpopulations of CCK-positive neurons in the SFO.
Group 1 CCK-positive neurons are involved in the continuous suppression of water intake in Na-depleted conditions.
The number of Fos-positive CCK neurons markedly increased in Na-depleted conditions.138) Next, optical silencing of CCK-positive neurons in the SFO by the stimulation of a light-activated chloride pump (eNpHR3.0) in Na-depleted conditions, in which group 1 CCK-positive neurons are selectively activated, significantly induced water intake.138) Water intake was again stimulated in Na-depleted conditions by the chemogenetic silencing of CCK-positive neurons in the SFO.138) Therefore, group 1 CCK-positive SFO neurons appeared to be crucially involved in the inhibition of water intake in Na-depleted (or hydrated) conditions.
3.2.2. Feedback by water intake.
Recent studies showed that water infusion into the gut inhibited the activities of water (thirst) neurons in the SFO.140)–142) During water drinking, GLP1R-positive GABAergic neurons in the SFO and MnPO respond to oral and gastric stimuli, respectively.141),143) A very recent study reported that GLP1 agonists reduced fluid intake and thirst perception in primary polydipsia patients.144) These findings indicated that water drinking signals sensed in the oral cavity and/or gastrointestinal tract were transmitted to central inhibitory systems through divergent neural routes (Fig. 6B).
Group 2 CCK-positive neurons tentatively calm thirst in response to water drinking.
Our recent study revealed that group 2 CCK-positive SFO neurons respond to water intake. The activities of individual CCK-positive SFO neurons were observed during water intake stimulated by the subcutaneous administration of a hypertonic NaCl solution (3.0 M NaCl), (Fig. 9D). These neurons were not activated when the spout of an empty bottle was licked; however, activity was observed within 10 s of the initiation of water drinking followed by a gradual reduction after 20 s (Fig. 9D). Fos proteins were not expressed by CCK-positive neurons in the SFO in water-depleted conditions; however, their expression was up-regulated with the initiation of water drinking.138) Collectively, these findings indicate that CCK-positive neurons activated by the feedback signals of water intake rapidly inhibit water neurons.
The optical silencing of these neurons was conducted in water-depleted conditions when group 1 CCK-positive neurons were inactive.138) Total water intake by dehydrated mice markedly increased in the first 5 min of optical silencing, which supported the inhibitory role of group 2 CCK-positive neurons in the regulation of water intake behavior. To establish the role of group 2 neurons in natural water intake in water- and Na-replete conditions, in which group 1 neurons are inactive,138) the activities of all CCK-positive neurons in the SFO were continuously suppressed by chemogenetic silencing. During the experimental period (6 h), hourly water intake volumes were significantly higher after the subcutaneous administration of CNO than after that of the vehicle. This finding indicated that the transient activation of group 2 CCK-positive SFO neurons in response to water drinking also contributes to the restriction of overall water intake through transient inhibition of thirst.138)
In summary, distinct CCK-positive SFO neurons (groups 1 and 2) are involved in the persistent or transient suppression of water intake through the CCK-B receptor-mediated activation of GABAergic interneurons (Fig. 10). Blood-borne persistent signals (such as [Na+], Ang II, and aldosterone) as well as oropharyngeal/gastrointestinal transient signals induced by water drinking may contribute to the regulation of water neurons in the SFO. Regarding the central mechanisms underlying thirst regulation, water intake was inhibited by the activation of these CCK-positive neurons and induced by their suppression, even in water-replete conditions (Fig. 10).
Feedback regulation through the LPBN.
A previous study reported that acid-sensing taste receptor cells in the tongue responded to water and transmitted the hedonic value to the brain.145) The lateral parabrachial nucleus (LPBN) neurons were also shown to be involved in the inhibitory system of fluid intake through the sensation of water drinking.146),147) LPBN neurons expressing the prodynorphin (PDYN) gene monitor the mechanosensory signals induced by the intake of both fluids and solids from the upper digestive tract and contribute to the negative feedback control of intake behaviors in order to avoid overconsumption (Fig. 6B).148) Furthermore, the activation of OT receptor-positive neurons in the LPBN, which are innervated by projections from OT-positive neurons in the PVN, suppressed fluid intake and responded to drinking stimuli (Fig. 6B).149) OT receptor-positive neurons are a distinct population from PDYN-positive neurons in the LPBN.148) CGRP-positive neurons in the LPBN are also reportedly involved in meal termination.150),151) Excitatory neurons in the pre-locus coeruleus (pre-LC), which is located in the proximity of the LPBN, responded to food or water consumption, and the inhibition of these neurons prolonged the duration of ingestion.152)
Water-predicting cues.
Dopamine neurons in the ventral tegmental area (VTA) were reported to be responsive to a water-predicting cue and were controlled by excitatory neurons in the SFO.153) The activation of thirst-inducible SFO neurons resulted in a negative valence signal that appeared to be consistent with the negative feelings associated with thirst arising from homeostatic deficits.154) Consistent with this finding, the optical activation of thirst-promoting neurons in the SFO and MnPO was associated with aversive behavior in the place-preference test.115),155) These findings suggested that thirst-inducible neurons in the forebrain produce a negative hedonic value via the VTA.
On the other hand, the activities of thirst-promoting SFO neurons were found to be suppressed by predicting changes in osmolality due to drinking and eating.156) Furthermore, water-predicting cues altered hypothalamic neuronal activity in thirsty mice.119) Clock-driven VP-positive neurons in the suprachiasmatic nucleus (SCN) projecting to the OVLT were shown to be involved in anticipatory water intake via VP receptor 1a (Fig. 6A).157) Additionally, VP-secreting neurons in the SON projecting to the posterior pituitary gland predictively responded to water availability in order to protect the body from osmotic stress.158) These findings suggested that predictive signals for water ingestion, such as visual and/or olfactory signals, contribute to the suppression of water intake. A recent study reported that a high dietary salt intake also increased the osmoresponsiveness of VP-secreting magnocellular neurons in the SON.159)
3.2.3. Anti-dipsogenic hormones.
Estrogen.
Estrogen is a steroid hormone involved in reproductive behavior in females and has also been implicated in the control of body fluid balance in female rodents. In ovariectomized female rats, estrogen exerted suppressive effects on Ang II-mediated water intake mediated by neural pathways from the SFO.160)–162) In the SFO of the rat, estrogen receptors co-localized with AT1a.163) Furthermore, estrogen inhibited Ang II-mediated effects through the attenuation of intracellular nitric oxide (NO), reactive oxygen species (ROS), and Ca2+ production.164)
Ghrelin.
Ghrelin is a well-known peptide that is primarily produced in the gastrointestinal tract to increase food intake and exerts its effects in the brains of mammals, including humans.165)–167) Ghrelin-responsive cells have been detected in sCVOs in addition to the arcuate nucleus as a feeding center.168) In the SFO, ghrelin-responsive neurons were identified in an electrophysiological study and were shown to play a role in the suppression of water intake induced by Ang II.169),170) This hormone may regulate the balance between feeding and drinking behaviors.
Atrial natriuretic peptide.
Previous studies demonstrated that an injection of atrial natriuretic peptide (ANP) into the AV3V region induced the dose-dependent inhibition of water intake after dehydration in rats and sheep.171)–177) Pretreatment with various doses of ANP microinjected into the SFO of rats reduced drinking induced by a subsequent Ang II injection into the same site, and also blocked Ang II-induced neuronal excitation in the SFO.178)–180) Furthermore, a small dose of ANP markedly reduced the intake of 1.5% NaCl by salt-depleted rats.181) Therefore, ANP suppresses salt as well as water intake induced by dehydration and Ang II.
An increase in extracellular [Na+] in the OVLT or SFO by a microinjection of hypertonic NaCl into the AV3V region induced a rapid elevation in plasma ANP followed by natriuresis. Lesions at the AV3V caused decreases in both basal and blood volume expansion (BVE)-induced plasma ANP concentrations, indicating that brain ANP plays an important role in mediating the release of ANP after BVE.182) The release of ANP in response to volume expansion involves an afferent baroreceptor input to the AV3V region, which mediates the increased release of ANP by activating the hypothalamic ANP neuronal system.183) An afferent pathway to the AV3V region via serotonergic neurons with cell bodies in the raphe nuclei has been identified,184) suggesting that serotonin (5-HT) contributes to the control of ANP neurons in the AV3V region.185)–187) The raphe nuclei may be stimulated by an afferent input from baroreceptors via the nucleus of the solitary tract (NTS) and contribute to the stimulation of ANP release following BVE.187)
4. Salt intake control
What causes salt appetite?
Salt appetite (also known as Na appetite) characteristically increases after a prolonged period of Na deficiency; however, the underlying regulatory mechanisms have not yet been elucidated in detail. Na deprivation and/or hypovolemia increases the circulating levels of hormones, such as Ang II and aldosterone.70) [Ang II] rapidly increases in response to hypovolemia. Ang II plays an important role in the stimulation of Na appetite, but it does not selectively stimulate the ingestion of salt relative to water, an appropriate response to hypovolemia in a teleological sense. In contrast to thirst, Na appetite does not increase until well after the onset of hypovolemia, up to many hours later depending on the experimental conditions. In addition, most stimuli for Na appetite, including dietary Na deprivation, do not reduce plasma Na concentrations in a short period.188)–191) This distinction was the reason for initial searches for a reservoir organ in the body.192) Long-term studies reported the osmotically inactive storage of Na+ in humans that consume a high-salt (HS) diet,193),194) and this has been experimentally demonstrated in animals.195) Glycosaminoglycans in the cartilage and skin were recently suggested as a reservoir regulating the storage and release of Na+.196),197)
Studies on the regulation of Na appetite were pioneered by Curt Richter’s classic studies showing that the drive to consume Na is hormonally regulated.198) Internal Na depletion increases the combined hormonal secretion of Ang II and aldosterone.199) The homeostatic salt balance is tightly controlled by the renin-angiotensin-aldosterone system (RAAS).70) Salt depletion is initially detected by juxtaglomerular cells in the kidneys, which respond by secreting renin. Renin is an enzyme that converts angiotensinogen (secreted by the liver) into Ang I, which is then converted to Ang II within the lungs by ACE. Ang II stimulates the adrenal secretion of aldosterone and acts on smooth muscle to constrict blood vessels and increase BP. Peripherally administered subthreshold doses of aldosterone and centrally administered Ang II synergized to stimulate more saline intake than either hormone alone.200) Simultaneous elevations in peripheral aldosterone and Ang II produced by the brain’s own RAAS201) may exert synergistic effects on Na appetite. In support of this hypothesis, accumulated evidence indicates that original RAAS components are expressed in the brain, including sCVOs.202),203) Therefore, salt appetite is not likely to be directly induced by sensing decreases in [Na+] in the brain.
The regulation of Na appetite was previously suggested to be dependent on two forebrain sCVOs, the OVLT and SFO, as well as brainstem sensory pathways.4),204),205) The SFO and OVLT detect elevations in plasma Ang II via AT1a receptors, and transmit this information to a range of downstream brain regions that mediate central responses.204) Previous studies demonstrated that the ventrolateral bed nucleus of the stria terminalis (vlBNST) received direct inputs from Ang II-sensitive neurons in the SFO and OVLT206) as well as neurons in the caudal nucleus of the solitary tract (cNTS).207),208) The second major pathway is via neurons residing in the NTS that express mineralocorticoid receptors (MRs) and 11β-hydroxysteroid dehydrogenase type 2 (HSD11β2). The expression of HSD11β2 allows these neurons to be sensitive to circulating aldosterone. Aldosterone-sensitive HSD11β2 neurons in the NTS were previously shown to be activated by chronic dietary Na deprivation and were then inactivated when salt was ingested.209)–211)
4.1. Activation mechanisms of salt appetite.
4.1.1. Angiotensin II.
Roles of the AT1a signal in the generation of salt appetite.
We experimentally established Na-depleted conditions in mice using the combination of a furosemide treatment with water satiation for 24 h. In Na-depleted conditions, [Na+] in blood slightly decreased, whereas [Ang II] increased by ∼3.0-fold, a similar level to that in water-depleted conditions. The number of Fos-positive neurons significantly increased in the SFO and OVLT, but not the AP or PVN, and were largely AT1a-positive.96) Fos expression was not observed in the SFO or OVLT in AT1a-KO mice in these conditions. The loss of SFO and OVLT neuronal activation and the complete suppression of salt intake were detected following continuous i.c.v. administration of the AT1 blocker losartan in Na-depleted conditions, which indicated the essential role of AT1a in salt intake in these conditions.96) Salt, but not water, intake was stimulated in WT mice in Na-depleted conditions, whereas neither was observed in AT1a-KO mice.
AT1a-positive neurons in the SFO.
As described above, the deletion of Agtr1a from the SFO in AT1a-floxed mice with AAV-Cre markedly decreased the consumption of 0.3 M NaCl in the two-bottle test in Na-depleted conditions. Decreases in the intake of 0.3 M NaCl showed a linear correlation with Cre-positive cell numbers in the SFO, with salt intake being completely suppressed in the most effective case.96) However, the deletion of Agtr1a from the OVLT did not affect the intake of 0.3 M NaCl. Therefore, AT1a signals in the SFO appear to contribute to the intake of both water and salt, whereas those in the OVLT only induce water intake.96) Regarding water intake, the remainder of water intake may be partly mediated by AT2 receptors because water intake was reduced in AT2-KO mice following a central injection of Ang II.212)
The SFO→vBNST pathway controls salt appetite.
A subset of glutamatergic SFO neurons specifically project to the vBNST. These SFO(→vBNST) neurons were positive for AT1a and became positive for Fos in Na-depleted conditions.96) Furthermore, the expression of Fos in the vBNST increased ∼2.0-fold as a result of the depletion of Na. The intake of 0.3 M NaCl by WT mice in Na-depleted conditions markedly decreased following the bilateral electrolytic lesions in the vBNST. The genetic ablation of AT1a from SFO(→vBNST) neurons decreased the intake of 0.3 M NaCl to less than that by the control group.96) These findings suggest that SFO(→vBNST) neurons (salt neurons) are Ang II-dependent driving neurons of salt appetite (Fig. 8). However, the intake of 0.3 M NaCl was not affected by the individual deletion of AT1a in other SFO neurons projecting to the OVLT, PVN, or SON.96)
In water-depleted conditions, the intake of 0.3 M NaCl solution was completely avoided by mice, but the intake of 0.15 M NaCl solution was preferred. Following the optical excitation of SFO(→vBNST) neurons using ChR2, avoidance of the 0.3 M NaCl solution was suppressed and the intake of 0.15 M NaCl solution was increased. On the other hand, optical silencing of the SFO→vBNST pathway using ArchT in Na-depleted conditions significantly reduced salt intake without any effect on water intake, again indicating that the activity of the SFO→vBNST pathway is specifically involved in the control of salt intake (Fig. 11).96)
Previous studies demonstrated that aversive behaviors were induced and feeding was inhibited in food-deprived mice following the optical excitation of glutamatergic neurons in the vBNST, whereas the activation of GABAergic neurons in the vBNST induced reward and feeding behaviors.213),214) These neurons mainly projecting to the ventral tegmental area (VTA) appear to be involved in the reward system.213) A recent study reported that the optical excitation of dopamine neurons in the VTA reduced salt appetite, but did not alter salt concentration preferences.215) These findings suggest that the SFO→vBNST pathway regulates the reward system in the VTA in order to induce salt appetite.
Intracellular signaling by AT1 receptors is mediated by the G-protein-mediated activation of phospholipase C, inositol trisphosphate (IP3), and PKC, and by the β-arrestin-induced activation of MAPK.216) A PKC inhibitor attenuated water intake, but not saline intake, induced by Ang II, while an inhibitor of MAPK markedly reduced saline intake, but not water intake,217) suggesting that distinct Ang II-mediated intracellular signaling pathways through PKC or MAPK control water and salt neurons, respectively.
4.1.2. Aldosterone.
The AP is another important CVO for the normal regulation of salt intake. Many Ang II receptor-expressing cells have been detected in the AP, SFO, and OVLT.218),219) However, in contrast to the SFO and OVLT, the AP does not induce the water and saline intake behaviors in response to Ang II.220) This structure appears to be involved in the generation of inhibitory signals.221)–223)
The AP is located at the dorsal midline of the caudal medulla in the vicinity of the NTS. Loewy’s group analyzed brainstem cell groups involved in Na homeostasis. A specific group of neurons in the NTS involved in salt appetite control were found to express the MRs and the glucocorticoid-inactivating enzyme HSD11β2, making them selective for the detection of aldosterone.224) These ‘HSD11β2 neurons’ become positive for Fos with Na depletion.224) Salt appetite was found to be enhanced in brain-specific HSD11β2-KO mice due to the activation of HSD11β2-positive neurons in response to cortisol.225) Therefore, the chemogenetic activation of HSD11β2-positive neurons in the NTS drives salt appetite, and these neurons project not only to the vBNST, but also to salt neurons in the SFO.226),227) Therefore, the integration of Ang II signals from salt neurons in the SFO and aldosterone signals from HSD11β2-positive neurons in the NTS appears to occur in the vBNST to control salt appetite (Fig. 11A).

AP neurons project to the aldosterone-sensitive HSD11β2 neurons of the NTS as well as to other NTS neuronal phenotypes, including neurotensin-immunoreactive neurons.228) AP neurons also project directly to FoxP2-positive neurons in the pre-LC and the inner division of the external lateral parabrachial subnucleus (PBel-inner).229) HSD11β2 neurons in the NTS and FoxP2-positive neurons in the pre-LC and PBel-inner were shown to express Fos for 1 week after Na deprivation.224),230) FoxP2-positive pre-LC neurons project to many brain regions, including the BNST, PVN, dorsomedial nuclei of the hypothalamus (DMH), and VTA. FoxP2-positive PBel-inner neurons also project to the BNST, PVN, and DMH, with weaker connections to the LHA and VTA. The pre-LC and PBel-inner both project to central sites implicated in Na appetite and related responses, including seeking behavior, hedonic responses to salt intake, Na balance, and cardiovascular regulation.231)
Fos is highly expressed in neurons in the pre-LC in Na-depleted conditions. Lee et al. reported that most neurons activated by Na depletion in the pre-LC expressed FoxP2 and PDYN.232) The optogenetic activation of pre-LCPDYN neurons induced a marked increase in the intake of Na from a 0.5 M NaCl solution, a concentration that is generally avoided in satiated conditions. The photoinhibition of pre-LCPDYN neurons specifically suppressed Na ingestion in Na-depleted conditions. These findings showed the essential involvement of pre-LCPDYN neurons in Na appetite and intake. The majority of pre-LCPDYN neurons were innervated by monosynaptic excitatory inputs from NTSHSD11β2 neurons, and, thus, the optogenetic stimulation of NTSHSD11β2 neurons in turn activated pre-LCPDYN neurons in satiated animals.232) Hence, pre-LCPDYN neuronal activation is dependent on excitatory NTSHSD11β2→pre-LCPDYN connections in Na-depleted conditions (Fig. 11A).
4.1.3. Opioids.
Endogenous opioid signaling has been established as a potent controller of salt consumption.233) An acute morphine injection increased the consumption of highly concentrated (usually aversive) salt solutions by rats. In contrast, non-selective opioid receptor antagonists significantly reduced salt consumption.234) Previous studies identified several specific neuronal pathways through which opioid signaling modulated salt consumption, including the mesolimbic dopaminergic system235)–237) and a number of interconnected regions, such as the parabrachial nucleus (PBN)238),239) and central amygdala (CeA).240) Endogenous opioid signaling may also tightly control the sweet taste reward value241) and the processing of other gustatory information controlling feeding, and, thus, is generally more relevant to the reward system.242),243)
Roles of the CeA.
Facilitatory mechanisms for Na intake were suggested in the CeA by electrolytic lesions or muscimol, the GABAA agonist, injections reducing Na intake in different studies.244)–247) Injections of DAMGO (a µ-opioid agonist) into the CeA increased salt intake, while the preliminary administration of CTAP (a µ-opioid antagonist) in the same area eliminated these effects.248) µ-Opioid receptor agonists generate neuronal hyperpolarization in general through by potassium channel opening, thereby inhibiting CeA neurons.249),250) However, DAMGO in the CeA markedly increased salt intake, suggesting that it stimulates its facilitatory mechanisms. In order to produce this effect, DAMGO may remove the effects of some inhibitory mechanisms, such as the GABAergic control in the CeA. Similar mechanisms are proposed for nociception in other central areas.251)
The CeA receives enkephalinergic projections from different areas, such as the ventromedial nucleus of the hypothalamus (VMH), PBN, and mainly from the BNST, which play roles in the control of the fluid and electrolyte balance.252) Signals from the LPBN may affect the release of opioids in the CeA by direct or indirect connections with the CeA or other areas, such as amygdaloid body. The activity of the CeA thus appears to be essential for the ingestion of water and salt caused by the inactivation of LPBN mechanisms, and Na intake induced by opioid activation in the CeA is parallel to the blockade of LPBN mechanisms.
4.2. Inhibitory mechanisms of salt appetite.
4.2.1. [Na+] increase in body fluids.
[Na+] sensing by Nax in the brain to suppress salt intake.
Nax is specifically expressed in the SFO and OVLT at which [Na+] sensing occurs in the brain. As described above, salt avoidance by Nax-KO mice was not observed in dehydrated conditions when [Na+] was elevated in body fluids: Nax-KO mice consumed 0.3 M NaCl and water equally even when dehydrated. To specify the brain locus involved in salt intake control, the Nax gene was locally introduced into the brain of Nax-KO mice. Abnormal salt intake behavior by Nax-KO mice in dehydrated conditions was completely recovered by the site-directed transfer of the Nax gene with an adenoviral vector into the SFO, indicating that the SFO is the primary locus of [Na+] sensing for the suppression of salt intake behavior.20) In this experiment, adenoviral vector which preferentially infects glial cells was used because Nax is expressed in glial cells in vivo.
Control of the SFO→vBNST pathway: Relationship between Nax signals and AT1a-dependent salt appetite.
The strong inhibition of salt intake by WT mice was observed in water-depleted conditions when blood [Ang II] was elevated, which was consistent with salt avoidance due to increased [Na+] in body fluids. The intakes of 0.3 M NaCl by AT1a-KO and Nax/AT1a-DKO mice were very low (Fig. 12A), indicating the dependence of salt appetite on AT1a, and, thus, Ang II. The Nax signal also appeared to be upstream of the AT1a-dependent control of salt intake. The proportion of Fos-positive cells among SFO(→vBNST) neurons (salt neurons) in water-depleted conditions was significantly higher in Nax-KO mice than in WT mice, which suggested the negative regulation of SFO(→vBNST) neurons by the Nax signal (Fig. 8, left).
Nax-dependent modulation of SFO(→vBNST) neuronal activity.
To clarify whether GABAergic neurons are involved in the regulation of the SFO→vBNST pathway, an electrophysiological examination was performed on brain slices from GAD67-GFP and GAD67-GFP;Nax-KO mice, in which the vBNST was infused with the cholera toxin subunit b conjugated with Alexa Fluor 555 dye (CTb-555) (Fig. 12B, left). GABAergic neurons and SFO(→vBNST) neurons were identified by green (GFP) and red (CTb-555) fluorescence, respectively. When GABAergic neurons were stimulated, inhibitory postsynaptic currents (IPSC) from neighboring CTb-555-labeled SFO(→vBNST) neurons connected synaptically to GABAergic neurons were recorded together as a pair (Fig. 12B, center). In physiological [Na+] of 145 mM, SFO(→vBNST) neuronal activity was not detected in GAD67-GFP mice or GAD67-GFP;Nax-KO mice; however, firing was induced by the application of Ang II (Fig. 12B, right). In contrast, the firing activity of GABAergic neurons remained unchanged following the addition of Ang II (Fig. 12B, right).
When the perfusate was replaced with a hypertonic Na solution (160 mM Na+), an increase was observed in the firing frequency of GABAergic neurons (Figs. 12B and C). Similar responses were not detected in slices from Nax-KO mice (Fig. 12C, right), suggesting the regulation of GABAergic activity by Nax signals. Consistent with the increased firing frequency of GABAergic neurons in the hypertonic Na solution, decreases were observed in the Ang II-induced firing activity of the SFO(→vBNST) neurons of GAD67-GFP mice (Fig. 12C). The return of [Na+] to physiological levels abolished this effect by GABAergic neurons on SFO(→vBNST) neurons (Fig. 12C). The [Na+]-dependent regulation of SFO(→vBNST) neurons was not detected in Nax-KO mice (Fig. 12C). Collectively, these findings provide supportive evidence for the regulation of the SFO→vBNST pathway by GABAergic neurons, which are controlled by [Na+] signals sensed by Nax. CCK did not alter the activity of GABAergic neurons innervating SFO(→vBNST) neurons.96) Therefore, specific subpopulations of GABAergic neurons in the SFO are involved in the selective regulation of SFO(→vBNST) neurons (salt neurons) or SFO(→OVLT) neurons (water neurons) (see Fig. 8).
The regulation of the GABAergic neurons by Nax signals is mediated by lactate from Nax-positive glial cells as described above (Fig. 3C), we investigated the impact of inhibition of MCTs involved in the intercellular transport of lactate. The [Na+]-dependent modulations of GABAergic neurons and SFO(→vBNST) neurons were both abolished by 5 mM α-CHCA.96) In addition, the optical excitation of GABAergic neurons in the SFO significantly reduced salt intake in Na-depleted conditions (Fig. 12D). The optical stimulation of GABAergic neurons also significantly reduced water intake in water-depleted conditions,138) suggesting that at least two different populations of GABAergic neurons control SFO(→vBNST) neurons and SFO(→OVLT) neurons, respectively.
In summary, our findings showed that water and salt appetites are specifically encoded by distinct AT1a neurons in the SFO projecting to the OVLT and vBNST, respectively. In the SFO, increases in CCK in Na-depleted (water-satiated) conditions inhibit water neurons via GABAergic neurons. Another population of GABAergic neurons inhibit the activity of salt neurons in water-depleted conditions: In this system, Nax signals activate the GABAergic neurons via lactate. Figure 8 summarizes the neural mechanisms by which two distinct GABAergic neuron populations separately regulate water and salt intake behaviors based on body fluid conditions.
4.2.2. Feedback by salt intake.
Taste receptor cells located in the tongue sense Na through ENaC.38) Taste stimuli are relayed to neurons in the NTS.253) Neurons in the LPBN are activated after the ingestion of salt,254)–256) suggesting that LPBN cells are activated by signals from visceral or taste organs via the NTS. The LPBN is reciprocally connected to forebrain areas, such as the PVN and MnPO, and to medullary regions, including the AP and medial portion of the NTS (mNTS).257)–264) Therefore, the LPBN may convey signals from the AP/mNTS to the forebrain areas involved in the control of body fluid balance. Multiple neurotransmitters modulate LPBN inhibitory mechanisms. Some neurotransmitters, such as 5-HT, CCK, corticotrophin-releasing factor, and glutamate, have been shown to enhance the inhibitory effects of the LPBN and reduce salt intake, whereas other ones, including opioids, GABA, ATP, and noradrenaline, suppress LPBN inhibitory mechanisms and increase salt intake.147),265)–275) A microdialysis study also demonstrated reductions in the release of 5-HT and its metabolite 5-hydroxyindoleacetic acid in the LPBN in rats following the combined treatment with furosemide and captopril, while the enhancement by the intake of salt.276)
A recent study using fiber photometry recordings showed the rapid and persistent inhibition of pre-LCPDYN neuronal activity following the intake of Na.232) The oral consumption of NaCl markedly decreased pre-LCPDYN neuronal activity, whereas the intragastric infusion of NaCl did not.232) Gastric preloading with NaCl did not alter subsequent Na ingestion by Na-depleted mice, whereas oral NaCl rapidly suppressed salt appetite.232) These findings indicated that the oral detection of Na decreased salt appetite via the sustained inhibition of pre-LCPDYN neuronal activity. Notably, the blockade of Na taste receptors with amiloride abolished the NaCl-induced suppression of pre-LCPDYN neurons.38),277) Furthermore, high salt concentrations activated aversive pathways encoding sour and bitter tastes in order to prevent detrimental effects on health.278) Therefore, oral chemosensory signals, potentially via the taste system, appear to play a role in the acute modulation of Na appetite neurons (Fig. 11B).
4.2.3. Anti-salt appetite hormones.
Oxytocin.
Oxytocin (OT) is a posterior pituitary hormone that exerts a wide range of physiological effects. OT is involved in reproduction, but also plays a role in fluid intake.279) Previous studies demonstrated that the central administration of OT inhibited salt appetite in rats.280) In addition, OT-KO mice showed a significant preference for salt solution in normal conditions in the two-bottle test.281) However, the systemic administration of OT did not reduce salt intake, while the blockade of peripheral OT receptors also did not potentiate it.282) OT-positive neurons have been detected in the parvocellular division of the PVN, SON, and ME.283) A recent study revealed that OT-positive neurons in the PVN innervated OT receptor-positive neurons in the PBN, which decreased salt intake (Fig. 11B).149)
Serotonin.
Serotonin (5-HT) is a widespread neurotransmitter in the brain.284) Salt appetite in Na-depleted rats was suppressed following the stimulation of specific 5-HT receptors, including 5-HT2B/2C and 5-HT3.285) Furthermore, 5-HT depletion in the brain by the administration of p-chlorophenylalanine, a competitor of tryptophan and an irreversible inhibitor of tryptophan hydroxylase, enhanced salt intake in Na-depleted conditions,286) suggesting that brain 5-HT is involved in the suppression of salt appetite. Bilateral injections of methysergide, a non-selective 5-HT receptor antagonist, in the LPBN consistently increased salt intake in Na-depleted conditions.287) 5-HT2C-expressing neurons in the LPBN was recently shown to inhibit sodium appetite in Na-depleted conditions, and the optical activation of these neurons projecting to the CeA suppressed salt intake in Na-depleted conditions.288) Serotonergic neurons in the AP and dorsal raphe nucleus were previously reported to project to FoxP2-positive neurons in the PBN and pre-LC.289) PBN neurons are responsive to salt tastants,290) suggesting that feedback signals from the oral cavity are relayed to central systems that control salt appetite through 5-HT transmitters.
5. [Na+] and blood pressure
Salt (NaCl) intake has long been suggested to positively correlate with blood pressure (BP).291),292) A high-salt (HS) diet has been shown to increase plasma and CSF [Na+] in numerous experimental and clinical studies using time-controlled sampling.293)–295) Increases in intravascular fluid volume296) and the secretion of VP from the pituitary gland into blood297) have been attributed to elevated plasma [Na+], with both ultimately resulting in elevated BP. VP has been shown to activate V1a receptors in vascular smooth muscle cells, which causes vasoconstriction.297)
In addition to these mechanisms, elevations in [Na+] in plasma and CSF have been suggested to enhance synpathetic nerve activity (SNA), which in turn increases BP.295),298) Animal studies demonstrated that the administration of hypertonic NaCl via intracarotid or i.c.v. routes altered SNA and elevated BP.299),300) Moreover, increases in BP caused by a HS diet or elevated [Na+] in body fluids were markedly decreased by complete inhibition of SNA.300),301)
Nax mediates between high [Na+] in body fluids and increases in BP via SNA.
Consistent with previous findings on rats and mice,301)–303) the replacement of drinking water with 2% NaCl (HS ingestion) was shown to increase BP.98) After the intake of a HS diet (food and drink) for 1 week, blood and CSF [Na+] increased from the physiological level of ∼145 mM to ∼160 mM in both WT and Nax-KO mice (Fig. 13A). Mean arterial BP (MBP) significantly increased in HS diet-fed WT mice, while the diurnal cycle remained present; however, Nax-KO mice did not show HS-induced increases in MBP (Fig. 13B, left). Average MBP over 24 h was significantly higher (by ∼7 mmHg) in WT mice than in Nax-KO mice even though both were fed the same HS diet (Fig. 13B, right). In mice not fed the HS diet, the i.p. administration of chlorisondamine (CSD), a blocker of autonomic ganglia, reduced MBP by ∼28 mmHg in both WT and Nax-KO mice (Fig. 13C). A marked CSD-induced decrease in MBP (∼41 mmHg) was detected in HS diet-fed WT mice, whereas the reduction observed in their counterpart was similar (∼28 mmHg) to that in control conditions (Fig. 13C).

The effects of the i.p. administration of hypertonic saline (3 M NaCl, 10 µL/20 g body weight/min for 10 min) were also investigated, and the obtained findings revealed simultaneous increases in blood and CSF [Na+] in both genotypes that reached a plateau after 15–30 min (165 and 160 mM, respectively).98) A parallel increase in MBP that reached ∼15 mmHg after 30 min was detected in WT mice, but not Nax-KO mice. The CSD-induced blockade of SNA eliminated the MBP response observed in WT mice, but it had no effect on that in Nax-KO mice.98)
The direct stimulation of brain Nax in the cerebral ventricles by the i.c.v. administration of aCSF containing 450 mM [Na+] (0.4 µL/min for 10 min) also induced increases in CSF [Na+] that reached ∼160 mM after 10 min. Upon a pretreatment with the VP receptor 1a blocker, Manning compound (MC), MBP gradually increased in WT mice from 5 min after its i.c.v. administration, with MBP plateauing at ∼8 mmHg (Fig. 13D, left). In contrast, the same pretreatment completely abolished increases in MBP in Nax-KO mice, which indicated that MBP was elevated in a VP-dependent manner in Nax-KO mice. Furthermore, the addition of CSD abolished the MBP response in MC-pretreated WT mice, but no effect in Nax-KO mice (Fig. 13D, right). Therefore, the Nax signal appears to contribute only to sympathetically mediated increases in BP. Raw lumbar SNA was also increased by the i.c.v. administration of a hypertonic Na solution in anesthetized WT mice, but not Nax-KO mice (Fig. 13E, left). The time course of these increases in lumbar SNA (Fig. 13E, right) was similar to that of the CSD-sensitive component in MBP elevation (Fig. 13D). Together, these findings indicate that the Nax signal elicits [Na+]-dependent increases in BP by enhancing SNA.
OVLT(→PVN) neurons mediate salt-induced increases in BP.
The SFO and OVLT innervate the PVN of the hypothalamus, which contains presympathetic neurons that regulate SNA.298) Consistent with previous findings,304) focal lesion experiments revealed the involvement of the OVLT, but not the SFO, in sensing CSF [Na+] by Nax for BP control (Fig. 13F). The optogenetic stimulation of Vglut2-positive OVLT(→PVN) neurons using ChR2 by a pulsed blue laser set at 5 and 20 Hz induced rapid increases in MBP in a manner that was dependent on the pulse rate (∼15 and ∼30 mmHg, respectively) (Fig. 13G). Furthermore, an immediate increase was observed in MBP following the optical excitation of the OVLT, which persisted throughout the stimulation; however, MBP returned to baseline within 1 min of the stimulation being removed.98) A pretreatment with CSD completely suppressed this elevation in MBP98); therefore, the activation of OVLT(→PVN) neurons appears to induce increases in BP via SNA.
[Na+]-dependently released H+ from Nax-positive glial cells activates OVLT(→PVN) neurons.
Sympathetically mediated increases in MBP induced by the i.c.v. administration of a hypertonic Na solution were completely suppressed by the continuous infusion of α-CHCA, an inhibitor of MCTs, into the OVLT (Fig. 13H). As described above, α-CHCA significantly reduced lactate release from Nax-positive cells in the OVLT when Nax-positive cells were exposed to a hypertonic Na solution.138) MCTs are lactate/H+ symporters and release H+ together with lactate. As expected, pH in the intercellular space actually decreased in OVLT slices from WT mice, but not Nax-KO mice (Fig. 13I), and the release of H+ in WT slices was inhibited by the application of α-CHCA (Fig. 13J).
To clarify the mechanisms by which Nax signals activate OVLT(→PVN) neurons, their firing properties were investigated in acute brain slices (Fig. 14A). The findings obtained showed that firing activity was continuous at ∼1 Hz in the OVLT(→PVN) neurons of both genotypes perfused with isotonic Na solution ([Na+] = 145 mM) (Fig. 14A). An increase in the firing rate of OVLT(→PVN) neurons to ∼3 Hz was observed in WT brain slices, but not Nax-KO slices, when the perfusate was changed to a hypertonic Na solution ([Na+] = 160 mM) (Fig. 14A). The activation of OVLT(→PVN) neurons was detected by the recording solution at pH 6.8 without lactate (Fig. 14B), indicating that H+ released from Nax-positive glial cells functions as a gliotransmitter to activate these neurons. At basal pH (pH 7.4), OVLT(→PVN) neurons were not activated by lactate alone; however, the firing activity of these neurons, which was already enhanced by mild acidification (pH 7.0), was further increased by lactate.98) These findings indicate that increases in [Na+] induced the release of H+ from Nax-positive glial cells and led to the activation of OVLT(→PVN) neurons. We performed optical silencing of the OVLT(→PVN) neurons by using eNpHR3.0, a light-activated chloride pump, and successfully decreased the MBP.98) We did not employ ArchT for this experiment because ArchT, a light-driven proton pump, induces extracellular acidification by itself.

Pretreatment with PcTx1, a specific inhibitor of ASIC1a,305) completely suppressed the increases in MBP induced by elevated CSF [Na+] in the OVLT of WT mice; however, no effects were observed following the application of APETx2, a specific inhibitor of ASIC3306) (Fig. 14C). In support of this notion, continuous infusion into the OVLT of the ASIC1 activator, MitTx,307) at 0.1 and 1.0 µM induced dose-dependent increases in MBP in both genotypes.98) Electrophysiological experiments on acute brain slices showed that OVLT(→PVN) neurons were activated by an increase in extracellular [Na+], and this response was completely inhibited by PcTx1,98) further supporting the conclusion.
The Nax signal elevates BP through the PVN-RVLM pathway.
SNA increases induced by the activation of OVLT(→PVN) neurons were previously suggested to be mediated through the PVN-RVLM pathway.298) A previous study demonstrated the control of spinal sympathetic pre-ganglionic neurons by tyrosine hydroxylase (TH)-positive RVLM neurons.308) Following the intake of a HS diet, we found that PVN neurons innervating the RVLM (PVN[→RVLM] neurons) were activated in WT mice, but not Nax-KO mice.98) Moreover, TH-expressing RVLM neurons were activated in WT mice, but not Nax-KO mice under the same condition.98)
The neural mechanisms underlying NaCl-induced BP increases may be summarized as follows (Fig. 14D). Increases in extracellular [Na+] activate Nax in the OVLT, which consequently leads to the release of H+ and lactate from Nax-positive glial cells through MCTs.27),98) H+ then activates Vglut2-positive OVLT(→PVN) neurons via ASIC1a. OVLT(→PVN) neurons activate PVN(→RVLM) neurons, followed by the activation of TH-positive neurons to enhance SNA, ultimately increasing BP. These molecular and cellular events are the initial steps in the neurogenic mechanisms underlying increases in BP due to elevated blood and CSF [Na+].
6. Summary
Thirst and Na appetite sensations may have evolved for vertebrate terrestrial adaptation. A battery of studies over the past three decades have significantly contributed to a more detailed understanding of the central systems maintaining body fluid homeostasis (Na homeostasis). In this review, we mainly focused on the central sensing mechanisms of [Na+] and signaling mechanisms that control water and salt intakes based on recent discoveries, including our own findings. Na+ is the main electrolyte and major factor influencing osmolality in body fluids.
Water/salt intake behaviors are basically controlled by the balance between [Na+] and [Ang II] in body fluids. Ang II plays a primary role in regaining water and Na contents in the body in conditions such as hypovolemia. Increases in [Ang II] in CSF and blood are sensed by AT1a-expressing neurons in sCVOs (Figs. 5D and 8). However, Ang II signals in the SFO are controlled by the Nax or CCK signal, which are dependent on [Na+] in body fluids, to selectively suppress the neural activity regulating salt appetite or thirst, respectively (Fig. 8). Besides the Ang II signal in the circulation, Ang II produced in the brain may be involved in the control of BP via SNA. Transgenic mice artificially expressing renin and angiotensinogen in the brain exhibited the phenotypes of polydipsia and hypertension, which were attenuated by the SFO-specific deletion of angiotensinogen,202) implicating the production of central Ang II in the SFO in the control of fluid intake and BP.
Increases in [Na+] in CSF and blood are sensed by Nax, a brain [Na+] sensor, in specialized glial cells in the sCVOs of the brain (Fig. 3E). We reported a case in which autoantibodies targeting Nax caused adipsic hypernatremia without hypothalamic structural lesions demonstrable by magnetic resonance imaging.309) Subsequently, we reported newly identified cases with similar symptoms caused by autoantibodies targeting sCVOs.310),311) The common features of these cases were extensive hypernatremia without any sensation of thirst along with a normal VP response to serum hypertonicity. Therefore, the autoimmune destruction of the SFO may be the cause of adipsic hypernatremia. Several genetic populations involved in thirst motivation in the SFO and OVLT have recently been identified (Fig. 6).84)
In the SFO, the Nax signal is transmitted to GABAergic neurons using lactate and leads to the suppression of salt neurons with AT1a, which drives salt appetite (Figs. 3E and 8). Salt neurons are AT1a-positive excitatory neurons that project to the vBNST. Increases in circulating Ang II stimulate the secretion of aldosterone from the adrenal cortex in Na-depleted conditions, and aldosterone activates HSD11β2 neurons in the NTS to promote salt intake (Fig. 11). On the other hand, decreases in [Na+] and stimuli of water drinking are converted to CCK signals in the SFO in order to activate specific GABAergic neurons that suppress water neurons, which drive water intake (Fig. 10). Water neurons, which are AT1a-positive excitatory neurons, innervate the OVLT and MnPO. However, the mechanisms underlying the activation of CCK neurons have not yet been clarified.
In the OVLT, the Nax signal is converted to 5,6-EET, which stimulates TRPV4-positive neurons to induce water intake behavior (Fig. 5D). SLC9A4 also functions as a [Na+] sensor in the OVLT and is activated by elevated [Na+] in body fluids. SLC9A4-positive neurons have ASIC1a, which is activated by H+ and induces water intake via an autocrine mechanism. SLC9A4-positive neurons are also activated by circulating Ang II. These mechanisms coordinate the control of Na appetite and thirst to maintain the Na/water balance in body fluids. The Nax signal in the OVLT is also converted to a H+ signal and activates ASIC1a-positive neurons innervating the PVN and RVLM, thereby activating SNA and, consequently, increasing BP (Fig. 14D).
In humans, several factors have been shown to affect water intake behavior, such as lifestyle, eating behavior, body temperature, and body-fluid conditions. Besides the [Na+]- and Ang II-mediated systems, many central regulatory systems contribute to water/salt intake control. Brain-wide neuronal activities in thirst conditions were recently demonstrated in mice and humans.119),312) Furthermore, the medial prefrontal cortex, which is critically involved in higher cognitive function, also participates in the decision-making of water ingestion.313) Further studies on complex neural systems involving various neurons and neural pathways that integrate multiple sources of information for appropriate behavior are necessary to obtain a more complete picture of body fluid homeostasis.
Acknowledgements
We thank Tomomi Tanaka for her secretarial assistance. M. Noda is funded by MEXT/JSPS KAKENHI (Grant Number; 19H05659), Core Research for Evolutional Science and Technology of the Japan Science and Technology Agency (CREST, JST, grant number JPMJCR1754). T. Matsuda is funded by MEXT/JSPS KAKENHI (Grant Number; 20K15928 and 21H05615) and the Takeda Science Foundation.
Notes
Edited by Shigetada NAKANISHI, M.J.A.
Correspondence should be addressed to: Homeostatic Mechanism Research Unit, Institute of Innovative Research, Tokyo Institute of Technology, Nagatsuta-cho 4259, Midori-ku, Yokohama, Kanagawa 226-8503, Japan (e-mail: noda.m.ae@m.titech.ac.jp).
References
- 1) Cox, P.S., Denton, D.A., Mouw, D.R. and Tarjan, E. (1987) Natriuresis induced by localized perfusion within the third cerebral ventricle of sheep. Am. J. Physiol. 252, R1–R6.
- 2) Park, R., Denton, D.A., McKinley, M.J., Pennington, G. and Weisinger, R.S. (1989) Intracerebroventricular saccharide infusions inhibit thirst induced by systemic hypertonicity. Brain Res. 493, 123–128.
- 3) Denton, D.A., McKinley, M.J. and Weisinger, R.S. (1996) Hypothalamic integration of body fluid regulation. Proc. Natl. Acad. Sci. U.S.A. 93, 7397–7404.
- 4) McKinley, M.J., McAllen, R.M., Davern, P., Giles, M.E., Penschow, J., Sunn, N. et al. (2003) The sensory circumventricular organs of the mammalian brain. Adv. Anat. Embryol. Cell Biol. 172, III–XII, 1–122, back cover.
- 5) Johnson, A.K. and Gross, P.M. (1993) Sensory circumventricular organs and brain homeostatic pathways. FASEB J. 7, 678–686.
- 6) Miyata, S. (2015) New aspects in fenestrated capillary and tissue dynamics in the sensory circumventricular organs of adult brains. Front. Neurosci. 9, 390.
- 7) Kiecker, C. (2018) The origins of the circumventricular organs. J. Anat. 232, 540–553.
- 8) Peruzzo, M., Milani, G.P., Garzoni, L., Longoni, L., Simonetti, G.D., Bettinelli, A. et al. (2010) Body fluids and salt metabolism - part II. Ital. J. Pediatr. 36, 78.
- 9) Andersson, B. (1977) Regulation of body fluids. Annu. Rev. Physiol. 39, 185–200.
- 10) Andersson, B. (1971) Thirst—and brain control of water balance. Am. Sci. 59, 408–415.
- 11) Fitzsimons, J.T. (1989) Bengt Andersson’s pioneering demonstration of the hypothalamic “drinking area” and the subsequent osmoreceptor/sodium receptor controversy. Acta Physiol. Scand. Suppl. 583, 15–25.
- 12) McKinley, M.J., Denton, D.A. and Weisinger, R.S. (1978) Sensors for antidiuresis and thirst—osmoreceptors or CSF sodium detectors? Brain Res. 141, 89–103.
- 13) Weisinger, R.S., Considine, P., Denton, D.A., McKinley, M.J. and Mouw, D. (1979) Rapid effect of change in cerebrospinal fluid sodium concentration on salt appetite. Nature 280, 490–491.
- 14) Goldin, A.L., Barchi, R.L., Caldwell, J.H., Hofmann, F., Howe, J.R., Hunter, J.C. et al. (2000) Nomenclature of voltage-gated sodium channels. Neuron 28, 365–368.
- 15) Watanabe, E., Fujikawa, A., Matsunaga, H., Yasoshima, Y., Sako, N., Yamamoto, T. et al. (2000) Nav2/NaG channel is involved in control of salt-intake behavior in the CNS. J. Neurosci. 20, 7743–7751.
- 16) Watanabe, E., Hiyama, T.Y., Kodama, R. and Noda, M. (2002) Nax sodium channel is expressed in non-myelinating Schwann cells and alveolar type II cells in mice. Neurosci. Lett. 330, 109–113.
- 17) Hiyama, T.Y., Watanabe, E., Ono, K., Inenaga, K., Tamkun, M.M., Yoshida, S. et al. (2002) Nax channel involved in CNS sodium-level sensing. Nat. Neurosci. 5, 511–512.
- 18) Noda, M. and Hiyama, T.Y. (2005) Sodium-level-sensitive Sodium Channel and Salt-Intake Behavior. Chem. Senses 30, i44–i45.
- 19) Matsumoto, M., Hiyama, T.Y., Kuboyama, K., Suzuki, R., Fujikawa, A. and Noda, M. (2015) Channel properties of Nax expressed in neurons. PLoS One 10, e0126109.
- 20) Hiyama, T.Y., Watanabe, E., Okado, H. and Noda, M. (2004) The subfornical organ is the primary locus of sodium-level sensing by Nax sodium channels for the control of salt-intake behavior. J. Neurosci. 24, 9276.
- 21) Watanabe, E., Hiyama, T.Y., Shimizu, H., Kodama, R., Hayashi, N., Miyata, S. et al. (2006) Sodium-level-sensitive sodium channel Nax is expressed in glial laminate processes in the sensory circumventricular organs. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R568–R576.
- 22) Hori, S., Komatsu, Y., Shigemoto, R., Mizuno, N. and Nakanishi, S. (1992) Distinct tissue distribution and cellular localization of two messenger ribonucleic acids encoding different subtypes of rat endothelin receptors. Endocrinology 130, 1885–1895.
- 23) Hindmarch, C., Fry, M., Yao, S.T., Smith, P.M., Murphy, D. and Ferguson, A.V. (2008) Microarray analysis of the transcriptome of the subfornical organ in the rat: regulation by fluid and food deprivation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R1914–R1920.
- 24) Hiyama, T.Y., Yoshida, M., Matsumoto, M., Suzuki, R., Matsuda, T., Watanabe, E. et al. (2013) Endothelin-3 expression in the subfornical organ enhances the sensitivity of Nax, the brain sodium-level sensor, to suppress salt intake. Cell Metab. 17, 507–519.
- 25) Rauh, A., Windischhofer, W., Kovacevic, A., DeVaney, T., Huber, E., Semlitsch, M. et al. (2008) Endothelin (ET)-1 and ET-3 promote expression of c-fos and c-jun in human choriocarcinoma via ET(B) receptor-mediated G(i)- and G(q)-pathways and MAP kinase activation. Br. J. Pharmacol. 154, 13–24.
- 26) Robin, P., Boulven, I., Desmyter, C., Harbon, S. and Leiber, D. (2002) ET-1 stimulates ERK signaling pathway through sequential activation of PKC and Src in rat myometrial cells. Am. J. Physiol. Cell Physiol. 283, C251–C260.
- 27) Shimizu, H., Watanabe, E., Hiyama, T.Y., Nagakura, A., Fujikawa, A., Okado, H. et al. (2007) Glial Nax channels control lactate signaling to neurons for brain [Na+] sensing. Neuron 54, 59–72.
- 28) Orlowski, J. and Grinstein, S. (2011) Na+/H+ exchangers. Compr. Physiol. 2011, 2083–2100.
- 29) Verma, V., Bali, A., Singh, N. and Jaggi, A.S. (2015) Implications of sodium hydrogen exchangers in various brain diseases. J. Basic Clin. Physiol. Pharmacol. 26, 417–426.
- 30) Sakuta, H., Lin, C.H., Hiyama, T.Y., Matsuda, T., Yamaguchi, K., Shigenobu, S. et al. (2020) SLC9A4 in the organum vasculosum of the lamina terminalis is a [Na+] sensor for the control of water intake. Pflugers Arch. 472, 609–624.
- 31) Bookstein, C., Musch, M.W., DePaoli, A., Xie, Y., Rabenau, K., Villereal, M. et al. (1996) Characterization of the rat Na+/H+ exchanger isoform NHE4 and localization in rat hippocampus. Am. J. Physiol. 271, C1629–C1638.
- 32) Arena, E.A., Longo, W.E., Roberts, K.E., Geibel, P., Nateqi, J., Brandstetter, M. et al. (2012) Functional role of NHE4 as a pH regulator in rat and human colonic crypts. Am. J. Physiol. Cell Physiol. 302, C412–C418.
- 33) Bookstein, C., Musch, M.W., DePaoli, A., Xie, Y., Villereal, M., Rao, M.C. et al. (1994) A unique sodium-hydrogen exchange isoform (NHE-4) of the inner medulla of the rat kidney is induced by hyperosmolarity. J. Biol. Chem. 269, 29704–29709.
- 34) Chambrey, R., Achard, J.M. and Warnock, D.G. (1997) Heterologous expression of rat NHE4: a highly amiloride-resistant Na+/H+ exchanger isoform. Am. J. Physiol. 272, C90–C98.
- 35) Miller, R.L., Wang, M.H., Gray, P.A., Salkoff, L.B. and Loewy, A.D. (2013) ENaC-expressing neurons in the sensory circumventricular organs become c-Fos activated following systemic sodium changes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R1141–R1152.
- 36) Amin, M.S., Wang, H.W., Reza, E., Whitman, S.C., Tuana, B.S. and Leenen, F.H. (2005) Distribution of epithelial sodium channels and mineralocorticoid receptors in cardiovascular regulatory centers in rat brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R1787–R1797.
- 37) Teruyama, R., Sakuraba, M., Wilson, L.L., Wandrey, N.E. and Armstrong, W.E. (2012) Epithelial Na+ sodium channels in magnocellular cells of the rat supraoptic and paraventricular nuclei. Am. J. Physiol. Endocrinol. Metab. 302, E273–E285.
- 38) Chandrashekar, J., Kuhn, C., Oka, Y., Yarmolinsky, D.A., Hummler, E., Ryba, N.J.P. et al. (2010) The cells and peripheral representation of sodium taste in mice. Nature 464, 297–301.
- 39) Lin, W., Finger, T.E., Rossier, B.C. and Kinnamon, S.C. (1999) Epithelial Na+ channel subunits in rat taste cells: localization and regulation by aldosterone. J. Comp. Neurol. 405, 406–420.
- 40) Ramsay, D.J., Thrasher, T.N. and Keil, L.C. (1983) The organum vasculosum laminae terminalis: a critical area for osmoreception. Prog. Brain Res. 60, 91–98.
- 41) Borgnia, M., Nielsen, S., Engel, A. and Agre, P. (1999) Cellular and molecular biology of the aquaporin water channels. Annu. Rev. Biochem. 68, 425–458.
- 42) Montell, C., Birnbaumer, L. and Flockerzi, V. (2002) The TRP channels, a remarkably functional family. Cell 108, 595–598.
- 43) Clapham, D.E. (2003) TRP channels as cellular sensors. Nature 426, 517–524.
- 44) Spassova, M.A., Hewavitharana, T., Xu, W., Soboloff, J. and Gill, D.L. (2006) A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc. Natl. Acad. Sci. U.S.A. 103, 16586–16591.
- 45) Bourque, C.W. (2008) Central mechanisms of osmosensation and systemic osmoregulation. Nat. Rev. Neurosci. 9, 519–531.
- 46) Nishihara, E., Hiyama, T.Y. and Noda, M. (2011) Osmosensitivity of transient receptor potential vanilloid 1 is synergistically enhanced by distinct activating stimuli such as temperature and protons. PLoS One 6, e22246.
- 47) Liedtke, W., Choe, Y., Martí-Renom, M.A., Bell, A.M., Denis, C.S., Sali, A. et al. (2000) Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103, 525–535.
- 48) Strotmann, R., Harteneck, C., Nunnenmacher, K., Schultz, G. and Plant, T.D. (2000) OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat. Cell Biol. 2, 695–702.
- 49) Ciura, S. and Bourque, C.W. (2006) Transient receptor potential vanilloid 1 is required for intrinsic osmoreception in organum vasculosum lamina terminalis neurons and for normal thirst responses to systemic hyperosmolality. J. Neurosci. 26, 9069–9075.
- 50) Hollis, J.H., McKinley, M.J., D’Souza, M., Kampe, J. and Oldfield, B.J. (2008) The trajectory of sensory pathways from the lamina terminalis to the insular and cingulate cortex: a neuroanatomical framework for the generation of thirst. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1390–R1401.
- 51) Liedtke, W. and Friedman, J.M. (2003) Abnormal osmotic regulation in trpv4−/− mice. Proc. Natl. Acad. Sci. U.S.A. 100, 13698–13703.
- 52) Mannari, T., Morita, S., Furube, E., Tominaga, M. and Miyata, S. (2013) Astrocytic TRPV1 ion channels detect blood-borne signals in the sensory circumventricular organs of adult mouse brains. Glia 61, 957–971.
- 53) Zheng, J. (2013) Molecular mechanism of TRP channels. Compr. Physiol. 3, 221–242.
- 54) Ahern, G.P., Brooks, I.M., Miyares, R.L. and Wang, X.B. (2005) Extracellular cations sensitize and gate capsaicin receptor TRPV1 modulating pain signaling. J. Neurosci. 25, 5109–5116.
- 55) Sharif Naeini, R., Witty, M.F., Séguéla, P. and Bourque, C.W. (2006) An N-terminal variant of Trpv1 channel is required for osmosensory transduction. Nat. Neurosci. 9, 93–98.
- 56) Zaelzer, C., Hua, P., Prager-Khoutorsky, M., Ciura, S., Voisin, D.L., Liedtke, W. et al. (2015) ΔN-TRPV1: A molecular co-detector of body temperature and osmotic stress. Cell Rep. 13, 23–30.
- 57) Kinsman, B., Cowles, J., Lay, J., Simmonds, S.S., Browning, K.N. and Stocker, S.D. (2014) Osmoregulatory thirst in mice lacking the transient receptor potential vanilloid type 1 (TRPV1) and/or type 4 (TRPV4) receptor. Am. J. Physiol. Regul. Integr. Comp. Physiol. 307, R1092–R1100.
- 58) Taylor, A.C., McCarthy, J.J. and Stocker, S.D. (2008) Mice lacking the transient receptor vanilloid potential 1 channel display normal thirst responses and central Fos activation to hypernatremia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1285–R1293.
- 59) Sakuta, H., Nishihara, E., Hiyama, T.Y., Lin, C.H. and Noda, M. (2016) Nax signaling evoked by an increase in [Na+] in CSF induces water intake via EET-mediated TRPV4 activation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 311, R299–R306.
- 60) Yang, F., Zhou, L., Wang, D., Yang, L.L., Yuan, G.R. and Huang, Q.Y. (2016) Suppression of TRPV4 channels ameliorates anti-dipsogenic effects under hypoxia in the subfornical organ of rats. Sci. Rep. 6, 30168.
- 61) Ciura, S., Prager-Khoutorsky, M., Thirouin, Z.S., Wyrosdic, J.C., Olson, J.E., Liedtke, W. et al. (2018) Trpv4 mediates hypotonic inhibition of central osmosensory neurons via taurine gliotransmission. Cell Rep. 23, 2245–2253.
- 62) Mizuno, A., Matsumoto, N., Imai, M. and Suzuki, M. (2003) Impaired osmotic sensation in mice lacking TRPV4. Am. J. Physiol. Cell Physiol. 285, C96–C101.
- 63) Mederos y Schnitzler, M., Storch, U., Meibers, S., Nurwakagari, P., Breit, A., Essin, K. et al. (2008) Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 27, 3092–3103.
- 64) Zou, Y., Akazawa, H., Qin, Y., Sano, M., Takano, H., Minamino, T. et al. (2004) Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat. Cell Biol. 6, 499–506.
- 65) Rakesh, K., Yoo, B., Kim, I.-M., Salazar, N., Kim, K.-S. and Rockman, H.A. (2010) β-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci. Signal. 3, ra46.
- 66) Tang, W., Strachan, R.T., Lefkowitz, R.J. and Rockman, H.A. (2014) Allosteric modulation of β-arrestin-biased angiotensin II type 1 receptor signaling by membrane stretch. J. Biol. Chem. 289, 28271–28283.
- 67) Wang, J., Hanada, K., Gareri, C. and Rockman, H.A. (2018) Mechanoactivation of the angiotensin II type 1 receptor induces β-arrestin-biased signaling through Gα(i) coupling. J. Cell. Biochem. 119, 3586–3597.
- 68) Lefkowitz, R.J. (1998) G protein-coupled receptors: III. New roles for receptor kinases and β-arrestins in receptor signaling and desensitization. J. Biol. Chem. 273, 18677–18680.
- 69) Saltmarsh, M. (2001) Thirst: or, why do people drink? Nutr. Bull. 26, 53–58.
- 70) Fitzsimons, J.T. (1998) Angiotensin, thirst, and sodium appetite. Physiol. Rev. 78, 583–686.
- 71) Adrogué, H.J. and Madias, N.E. (2000) Hypernatremia. N. Engl. J. Med. 342, 1493–1499.
- 72) Adrogué, H.J. and Madias, N.E. (2000) Hyponatremia. N. Engl. J. Med. 342, 1581–1589.
- 73) Leib, D.E., Zimmerman, C.A. and Knight, Z.A. (2016) Thirst. Curr. Biol. 26, R1260–R1265.
- 74) Ramsay, D.J. and Thrasher, T.N. (1991) Regulation of fluid intake in dogs following water deprivation. Brain Res. Bull. 27, 495–499.
- 75) McKinley, M.J. and Johnson, A.K. (2004) The physiological regulation of thirst and fluid intake. News Physiol. Sci. 19, 1–6.
- 76) Zimmerman, C.A., Leib, D.E. and Knight, Z.A. (2017) Neural circuits underlying thirst and fluid homeostasis. Nat. Rev. Neurosci. 18, 459–469.
- 77) Ichiki, T., Augustine, V. and Oka, Y. (2019) Neural populations for maintaining body fluid balance. Curr. Opin. Neurobiol. 57, 134–140.
- 78) Noda, M. and Sakuta, H. (2013) Central regulation of body-fluid homeostasis. Trends Neurosci. 36, 661–673.
- 79) Mangiapane, M.L. and Simpson, J.B. (1980) Subfornical organ: forebrain site of pressor and dipsogenic action of angiotensin II. Am. J. Physiol. 239, R382–R389.
- 80) Thrasher, T.N. and Keil, L.C. (1987) Regulation of drinking and vasopressin secretion: role of organum vasculosum laminae terminalis. Am. J. Physiol. 253, R108–R120.
- 81) Anderson, J.W., Washburn, D.L.S. and Ferguson, A.V. (2000) Intrinsic osmosensitivity of subfornical organ neurons. Neuroscience 100, 539–547.
- 82) Noda, M. and Hiyama, T.Y. (2015) Sodium sensing in the brain. Pflugers Arch. 467, 465–474.
- 83) Simpson, J.B. and Routtenberg, A. (1973) Subfornical organ: site of drinking elicitation by angiotensin II. Science 181, 1172.
- 84) Pool, A.H., Wang, T., Stafford, D.A., Chance, R.K., Lee, S., Ngai, J. et al. (2020) The cellular basis of distinct thirst modalities. Nature 588, 112–117.
- 85) el Ghissassi, M., Thornton, S.N. and Nicolaïdis, S. (1995) Angiotensin II-induced thirst, but not sodium appetite, via AT1 receptors in organum cavum prelamina terminalis. Am. J. Physiol. 268, R1401–R1405.
- 86) Lenkei, Z., Corvol, P. and Llorens-Cortes, C. (1995) The angiotensin receptor subtype AT1A predominates in rat forebrain areas involved in blood pressure, body fluid homeostasis and neuroendocrine control. Brain Res. Mol. Brain Res. 30, 53–60.
- 87) Barth, S.W. and Gerstberger, R. (1999) Differential regulation of angiotensinogen and AT1A receptor mRNA within the rat subfornical organ during dehydration. Brain Res. Mol. Brain Res. 64, 151–164.
- 88) Sakuta, H., Lin, C.-H., Yamada, M., Kita, Y., Tokuoka, S.M., Shimizu, T. et al. (2020) Nax-positive glial cells in the organum vasculosum laminae terminalis produce epoxyeicosatrienoic acids to induce water intake in response to increases in [Na+] in body fluids. Neurosci. Res. 154, 45–51.
- 89) Buggy, J. and Johnson, A.K. (1977) Preoptic-hypothalamic periventricular lesions: thirst deficits and hypernatremia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 233, R44–R52.
- 90) Hosutt, J.A., Rowland, N. and Stricker, E.M. (1981) Impaired drinking responses of rats with lesions on the subfornical organ. J. Comp. Physiol. Psychol. 95, 104–113.
- 91) Johnson, R.F., Beltz, T.G., Thunhorst, R.L. and Johnson, A.K. (2003) Investigations on the physiological controls of water and saline intake in C57BL/6 mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 285, R394–R403.
- 92) Lind, R.W., Thunhorst, R.L. and Johnson, A.K. (1984) The subfornical organ and the integration of multiple factors in thirst. Physiol. Behav. 32, 69–74.
- 93) Watanabe, H., Vriens, J., Prenen, J., Droogmans, G., Voets, T. and Nilius, B. (2003) Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 424, 434–438.
- 94) Roman, R.J. (2002) P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol. Rev. 82, 131–185.
- 95) Kinsman, B.J., Simmonds, S.S., Browning, K.N., Wenner, M.M., Farquhar, W.B. and Stocker, S.D. (2020) Integration of hypernatremia and angiotensin II by the organum vasculosum of the lamina terminalis regulates thirst. J. Neurosci. 40, 2069.
- 96) Matsuda, T., Hiyama, T.Y., Niimura, F., Matsusaka, T., Fukamizu, A., Kobayashi, K. et al. (2017) Distinct neural mechanisms for the control of thirst and salt appetite in the subfornical organ. Nat. Neurosci. 20, 230–241.
- 97) Mann, J.F., Johnson, A.K. and Ganten, D. (1980) Plasma angiotensin II: dipsogenic levels and angiotensin-generating capacity of renin. Am. J. Physiol. 238, R372–R377.
- 98) Nomura, K., Hiyama, T.Y., Sakuta, H., Matsuda, T., Lin, C.H., Kobayashi, K. et al. (2019) [Na+] increases in body fluids sensed by central Nax induce sympathetically mediated blood pressure elevations via H+-dependent activation of ASIC1a. Neuron 101, 60–75.e66.
- 99) Bellin, S.I., Bhatnagar, R.K. and Johnson, A.K. (1987) Periventricular noradrenergic systems are critical for angiotensin-induced drinking and blood pressure responses. Brain Res. 403, 105–112.
- 100) Deschepper, C.F. (1994) Angiotensinogen: hormonal regulation and relative importance in the generation of angiotensin II. Kidney Int. 46, 1561–1563.
- 101) Hall, J.E. (2003) Historical perspective of the renin-angiotensin system. Mol. Biotechnol. 24, 27–39.
- 102) Paul, M., Poyan Mehr, A. and Kreutz, R. (2006) Physiology of local renin-angiotensin systems. Physiol. Rev. 86, 747–803.
- 103) Abdelaal, A.E., Mercer, P.F. and Mogenson, G.J. (1976) Plasma angiotensin II levels and water intake following β-adrenergic stimulation, hypovolemia, cellular dehydration and water deprivation. Pharmacol. Biochem. Behav. 4, 317–321.
- 104) Weisinger, R.S., Blair-West, J.R., Burns, P., Denton, D.A., McKinley, M.J. and Tarjan, E. (1996) The role of angiotensin II in ingestive behaviour: a brief review of angiotensin II, thirst and Na appetite. Regul. Pept. 66, 73–81.
- 105) Premer, C., Lamondin, C., Mitzey, A., Speth, R.C. and Brownfield, M.S. (2013) Immunohistochemical localization of AT1a, AT1b, and AT2 angiotensin II receptor subtypes in the rat adrenal, pituitary, and brain with a perspective commentary. Int. J. Hypertens. 2013, 175428.
- 106) Grobe, J.L., Grobe, C.L., Beltz, T.G., Westphal, S.G., Morgan, D.A., Xu, D. et al. (2010) The brain Renin-angiotensin system controls divergent efferent mechanisms to regulate fluid and energy balance. Cell Metab. 12, 431–442.
- 107) Johnson, A.K. and Schwob, J.E. (1975) Cephalic angiotensin receptors mediating drinking to systemic angiotensin II. Pharmacol. Biochem. Behav. 3, 1077–1084.
- 108) Fitzsimons, J.T. and Simons, B.J. (1969) The effect on drinking in the rat of intravenous infusion of angiotensin, given alone or in combination with other stimuli of thirst. J. Physiol. 203, 45–57.
- 109) Epstein, A.N., Fitzsimons, J.T. and Rolls, B.J. (1970) Drinking induced by injection of angiotensin into the rain of the rat. J. Physiol. 210, 457–474.
- 110) Buggy, J. and Fisher, A.E. (1974) Evidence for a dual central role for angiotensin in water and sodium intake. Nature 250, 733–735.
- 111) Fitts, D.A. and Masson, D.B. (1990) Preoptic angiotensin and salt appetite. Behav. Neurosci. 104, 643–650.
- 112) Thunhorst, R.L., Ehrlich, K.J. and Simpson, J.B. (1990) Subfornical organ participates in salt appetite. Behav. Neurosci. 104, 637–642.
- 113) Weisinger, R.S., Denton, D.A., Di Nicolantonio, R., Hards, D.K., McKinley, M.J., Oldfield, B. et al. (1990) Subfornical organ lesion decreases sodium appetite in the sodium-depleted rat. Brain Res. 526, 23–30.
- 114) Matsusaka, T., Asano, T., Niimura, F., Kinomura, M., Shimizu, A., Shintani, A. et al. (2010) Angiotensin receptor blocker protection against podocyte-induced sclerosis is podocyte angiotensin II type 1 receptor-independent. Hypertension 55, 967–973.
- 115) Leib, D.E., Zimmerman, C.A., Poormoghaddam, A., Huey, E.L., Ahn, J.S., Lin, Y.C. et al. (2017) The forebrain thirst circuit drives drinking through negative reinforcement. Neuron 96, 1272–1281.e1274.
- 116) Vong, L., Ye, C., Yang, Z., Choi, B., Chua, S. Jr. and Lowell, B.B. (2011) Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71, 142–154.
- 117) Oka, Y., Ye, M. and Zuker, C.S. (2015) Thirst driving and suppressing signals encoded by distinct neural populations in the brain. Nature 520, 349–352.
- 118) Abbott, S.B.G. and Saper, C.B. (2017) Median preoptic glutamatergic neurons promote thermoregulatory heat loss and water consumption in mice. J. Physiol. 595, 6569–6583.
- 119) Allen, W.E., Chen, M.Z., Pichamoorthy, N., Tien, R.H., Pachitariu, M., Luo, L. et al. (2019) Thirst regulates motivated behavior through modulation of brainwide neural population dynamics. Science 364, 253.
- 120) Bayliss, W.M. and Starling, E.H. (1902) The mechanism of pancreatic secretion. J. Physiol. 28, 325–353.
- 121) Bai, J.J., Tan, C.D. and Chow, B.K.C. (2016) Secretin, at the hub of water-salt homeostasis. Am. J. Physiol. Renal Physiol. 312, F852–F860.
- 122) Chu, J.Y.S., Lee, L.T.O., Lai, C.H., Vaudry, H., Chan, Y.S., Yung, W.H. et al. (2009) Secretin as a neurohypophysial factor regulating body water homeostasis. Proc. Natl. Acad. Sci. U.S.A. 106, 15961–15966.
- 123) Ng, S.S., Yung, W.H. and Chow, B.K. (2002) Secretin as a neuropeptide. Mol. Neurobiol. 26, 97–107.
- 124) Lee, V.H., Lee, L.T., Chu, J.Y., Lam, I.P., Siu, F.K., Vaudry, H. et al. (2010) An indispensable role of secretin in mediating the osmoregulatory functions of angiotensin II. FASEB J. 24, 5024–5032.
- 125) Lee, L.T., Ng, S.Y., Chu, J.Y., Sekar, R., Harikumar, K.G., Miller, L.J. et al. (2014) Transmembrane peptides as unique tools to demonstrate the in vivo action of a cross-class GPCR heterocomplex. FASEB J. 28, 2632–2644.
- 126) Hisaw, F.L. (1926) Experimental relaxation of the pubic ligament of the guinea pig. Proc. Soc. Exp. Biol. Med. 23, 661–663.
- 127) Hudson, P., Haley, J., John, M., Cronk, M., Crawford, R., Haralambidis, J. et al. (1983) Structure of a genomic clone encoding biologically active human relaxin. Nature 301, 628–631.
- 128) Sherwood, O.D., Crnekovic, V.E., Gordon, W.L. and Rutherford, J.E. (1980) Radioimmunoassay of relaxin throughout pregnancy and during parturition in the rat. Endocrinology 107, 691–698.
- 129) Otsubo, H., Onaka, T., Suzuki, H., Katoh, A., Ohbuchi, T., Todoroki, M. et al. (2010) Centrally administered relaxin-3 induces Fos expression in the osmosensitive areas in rat brain and facilitates water intake. Peptides 31, 1124–1130.
- 130) de Avila, C., Chometton, S., Lenglos, C., Calvez, J., Gundlach, A.L. and Timofeeva, E. (2018) Differential effects of relaxin-3 and a selective relaxin-3 receptor agonist on food and water intake and hypothalamic neuronal activity in rats. Behav. Brain Res. 336, 135–144.
- 131) Smith, C.M., Shen, P.J., Banerjee, A., Bonaventure, P., Ma, S., Bathgate, R.A. et al. (2010) Distribution of relaxin-3 and RXFP3 within arousal, stress, affective, and cognitive circuits of mouse brain. J. Comp. Neurol. 518, 4016–4045.
- 132) Albert-Gasco, H., Ma, S., Ros-Bernal, F., Sanchez-Perez, A.M., Gundlach, A.L. and Olucha-Bordonau, F.E. (2017) GABAergic neurons in the rat medial septal complex express relaxin-3 receptor (RXFP3) mRNA. Front. Neuroanat. 11, 133.
- 133) Ma, S., Smith, C.M., Blasiak, A. and Gundlach, A.L. (2017) Distribution, physiology and pharmacology of relaxin-3/RXFP3 systems in brain. Br. J. Pharmacol. 174, 1034–1048.
- 134) McKinley, M.J., Mathai, M.L., McAllen, R.M., McClear, R.C., Miselis, R.R., Pennington, G.L. et al. (2004) Vasopressin secretion: osmotic and hormonal regulation by the lamina terminalis. J. Neuroendocrinol. 16, 340–347.
- 135) Olszewski, P.K., Klockars, A., Schioth, H.B. and Levine, A.S. (2010) Oxytocin as feeding inhibitor: maintaining homeostasis in consummatory behavior. Pharmacol. Biochem. Behav. 97, 47–54.
- 136) Smith, C.M., Walker, L.L., Chua, B.E., McKinley, M.J., Gundlach, A.L., Denton, D.A. et al. (2015) Involvement of central relaxin-3 signalling in sodium (salt) appetite. Exp. Physiol. 100, 1064–1072.
- 137) Willis, G.L., Hansky, J. and Smith, G.C. (1984) Ventricular, paraventricular and circumventricular structures involved in peptide-induced satiety. Regul. Pept. 9, 87–99.
- 138) Matsuda, T., Hiyama, T.Y., Kobayashi, K., Kobayashi, K. and Noda, M. (2020) Distinct CCK-positive SFO neurons are involved in persistent or transient suppression of water intake. Nat. Commun. 11, 5692.
- 139) Honda, T., Wada, E., Battey, J.F. and Wank, S.A. (1993) Differential gene expression of CCK(A) and CCK(B) receptors in the rat brain. Mol. Cell. Neurosci. 4, 143–154.
- 140) Zimmerman, C.A., Huey, E.L., Ahn, J.S., Beutler, L.R., Tan, C.L., Kosar, S. et al. (2019) A gut-to-brain signal of fluid osmolarity controls thirst satiation. Nature 568, 98–102.
- 141) Augustine, V., Ebisu, H., Zhao, Y., Lee, S., Ho, B., Mizuno, G.O. et al. (2019) Temporally and spatially distinct thirst satiation signals. Neuron 103, 242–249.e244.
- 142) Augustine, V., Lee, S. and Oka, Y. (2020) Neural control and modulation of thirst, sodium appetite, and hunger. Cell 180, 25–32.
- 143) Augustine, V., Gokce, S.K., Lee, S., Wang, B., Davidson, T.J., Reimann, F. et al. (2018) Hierarchical neural architecture underlying thirst regulation. Nature 555, 204–209.
- 144) Winzeler, B.F., Sailer, C.O., Coynel, D., Deborah, V., Davide, Z., Urwyler, S. et al. (2021) GLP1 receptor agonists reduce fluid intake in primary polydipsia. J. Endocr. Soc. 5, A514–A515.
- 145) Zocchi, D., Wennemuth, G. and Oka, Y. (2017) The cellular mechanism for water detection in the mammalian taste system. Nat. Neurosci. 20, 927–933.
- 146) Davern, P.J. (2014) A role for the lateral parabrachial nucleus in cardiovascular function and fluid homeostasis. Front. Physiol. 5, 436.
- 147) Menani, J.V., De Luca, L.A. Jr. and Johnson, A.K. (2014) Role of the lateral parabrachial nucleus in the control of sodium appetite. Am. J. Physiol. Regul. Integr. Comp. Physiol. 306, R201–R210.
- 148) Kim, D.Y., Heo, G., Kim, M., Kim, H., Jin, J.A., Kim, H.K. et al. (2020) A neural circuit mechanism for mechanosensory feedback control of ingestion. Nature 580, 376–380.
- 149) Ryan, P.J., Ross, S.I., Campos, C.A., Derkach, V.A. and Palmiter, R.D. (2017) Oxytocin-receptor-expressing neurons in the parabrachial nucleus regulate fluid intake. Nat. Neurosci. 20, 1722–1733.
- 150) Carter, M.E., Soden, M.E., Zweifel, L.S. and Palmiter, R.D. (2013) Genetic identification of a neural circuit that suppresses appetite. Nature 503, 111–114.
- 151) Campos, C.A., Bowen, A.J., Schwartz, M.W. and Palmiter, R.D. (2016) Parabrachial CGRP neurons control meal termination. Cell Metab. 23, 811–820.
- 152) Gong, R., Xu, S., Hermundstad, A., Yu, Y. and Sternson, S.M. (2020) Hindbrain double-negative feedback mediates palatability-guided food and water consumption. Cell 182, 1589–1605.e1522.
- 153) Hsu, T.M., Bazzino, P., Hurh, S.J., Konanur, V.R., Roitman, J.D. and Roitman, M.F. (2020) Thirst recruits phasic dopamine signaling through subfornical organ neurons. Proc. Natl. Acad. Sci. U.S.A. 117, 30744–30754.
- 154) Betley, J.N., Xu, S., Cao, Z.F.H., Gong, R., Magnus, C.J., Yu, Y. et al. (2015) Neurons for hunger and thirst transmit a negative-valence teaching signal. Nature 521, 180–185.
- 155) Allen, W.E., DeNardo, L.A., Chen, M.Z., Liu, C.D., Loh, K.M., Fenno, L.E. et al. (2017) Thirst-associated preoptic neurons encode an aversive motivational drive. Science 357, 1149–1155.
- 156) Zimmerman, C.A., Lin, Y.C., Leib, D.E., Guo, L., Huey, E.L., Daly, G.E. et al. (2016) Thirst neurons anticipate the homeostatic consequences of eating and drinking. Nature 537, 680–684.
- 157) Gizowski, C., Zaelzer, C. and Bourque, C.W. (2016) Clock-driven vasopressin neurotransmission mediates anticipatory thirst prior to sleep. Nature 537, 685–688.
- 158) Mandelblat-Cerf, Y., Kim, A., Burgess, C.R., Subramanian, S., Tannous, B.A., Lowell, B.B. et al. (2017) Bidirectional anticipation of future osmotic challenges by vasopressin neurons. Neuron 93, 57–65.
- 159) Levi, D.I., Wyrosdic, J.C., Hicks, A.I., Andrade, M.A., Toney, G.M., Prager-Khoutorsky, M. et al. (2021) High dietary salt amplifies osmoresponsiveness in vasopressin-releasing neurons. Cell Rep. 34, 108866.
- 160) Tanaka, J., Kariya, K., Miyakubo, H., Sakamaki, K. and Nomura, M. (2002) Attenuated drinking response induced by angiotensinergic activation of subfornical organ projections to the paraventricular nucleus in estrogen-treated rats. Neurosci. Lett. 324, 242–246.
- 161) Krause, E.G., Curtis, K.S., Davis, L.M., Stowe, J.R. and Contreras, R.J. (2003) Estrogen influences stimulated water intake by ovariectomized female rats. Physiol. Behav. 79, 267–274.
- 162) Tanaka, J., Miyakubo, H., Fujisawa, S. and Nomura, M. (2003) Reduced dipsogenic response induced by angiotensin II activation of subfornical organ projections to the median preoptic nucleus in estrogen-treated rats. Exp. Neurol. 179, 83–89.
- 163) Rosas-Arellano, M.P., Solano-Flores, L.P. and Ciriello, J. (1999) Co-localization of estrogen and angiotensin receptors within subfornical organ neurons. Brain Res. 837, 254–262.
- 164) Xue, B., Johnson, A.K. and Hay, M. (2013) Sex differences in angiotensin II- and aldosterone-induced hypertension: the central protective effects of estrogen. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R459–R463.
- 165) Kojima, M., Hosoda, H., Date, Y., Nakazato, M., Matsuo, H. and Kangawa, K. (1999) Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402, 656–660.
- 166) Tschop, M., Smiley, D.L. and Heiman, M.L. (2000) Ghrelin induces adiposity in rodents. Nature 407, 908–913.
- 167) Wren, A.M., Seal, L.J., Cohen, M.A., Brynes, A.E., Frost, G.S., Murphy, K.G. et al. (2001) Ghrelin enhances appetite and increases food intake in humans. J. Clin. Endocrinol. Metab. 86, 5992–5995.
- 168) Cabral, A., Fernandez, G. and Perello, M. (2013) Analysis of brain nuclei accessible to ghrelin present in the cerebrospinal fluid. Neuroscience 253, 406–415.
- 169) Pulman, K.J., Fry, W.M., Cottrell, G.T. and Ferguson, A.V. (2006) The subfornical organ: a central target for circulating feeding signals. J. Neurosci. 26, 2022–2030.
- 170) Mietlicki, E.G., Nowak, E.L. and Daniels, D. (2009) The effect of ghrelin on water intake during dipsogenic conditions. Physiol. Behav. 96, 37–43.
- 171) Antunes-Rodrigues, J., McCann, S.M., Rogers, L.C. and Samson, W.K. (1985) Atrial natriuretic factor inhibits dehydration- and angiotensin II-induced water intake in the conscious, unrestrained rat. Proc. Natl. Acad. Sci. U.S.A. 82, 8720.
- 172) Masotto, C. and Negro-Vilar, A. (1985) Inhibition of spontaneous or angiotensin II-stimulated water intake by atrial natriuretic factor. Brain Res. Bull. 15, 523–526.
- 173) Tarjan, E., Denton, D.A. and Weisinger, R.S. (1988) Atrial natriuretic peptide inhibits water and sodium intake in rabbits. Regul. Pept. 23, 63–75.
- 174) Itoh, H., Nakao, K., Yamada, T., Shirakami, G., Kangawa, K., Minamimo, N. et al. (1988) Antidipsogenic action of a novel peptide, ‘brain natriuretic peptide’, in rats. Eur. J. Pharmacol. 150, 193–196.
- 175) Nakamura, M., Katsuura, G., Nakao, K. and Imura, H. (1985) Antidipsogenic action of α-human atrial natriuretic polypeptide administered intracerebroventricularly in rats. Neurosci. Lett. 58, 1–6.
- 176) Zhu, B. and Herbert, J. (1996) Central antagonism of atrial natriuretic peptides on behavioral and hormonal responses to angiotensin II: mapping with c-fos. Brain Res. 734, 55–60.
- 177) Weisinger, R.S., Blair-West, J.R., Denton, D.A. and Tarjan, E. (1992) Central administration of atrial natriuretic peptide suppresses sodium and water intake of sheep. Brain Res. 579, 113–118.
- 178) Buranarugsa, P. and Hubbard, J.I. (1988) Excitatory effects of atrial natriuretic peptide on rat subfornical organ neurons in vitro. Brain Res. Bull. 20, 627–631.
- 179) Ehrlich, K.J. and Fitts, D.A. (1990) Atrial natriuretic peptide in the subfornical organ reduces drinking induced by angiotensin or in response to water deprivation. Behav. Neurosci. 104, 365–372.
- 180) Hattori, Y., Kasai, M., Uesugi, S., Kawata, M. and Yamashita, H. (1988) Atrial natriuretic polypeptide depresses angiotensin II induced excitation of neurons in the rat subfornical organ in vitro. Brain Res. 443, 355–359.
- 181) Antunes-Rodrigues, J., McCann, S.M. and Samson, W.K. (1986) Central administration of atrial natriuretic factor inhibits saline preference in the rat. Endocrinology 118, 1726–1728.
- 182) Antunes-Rodrigues, J., Ramalho, M.J., Reis, L.C., Menani, J.V., Turrin, M.Q., Gutkowska, J. et al. (1991) Lesions of the hypothalamus and pituitary inhibit volume-expansion-induced release of atrial natriuretic peptide. Proc. Natl. Acad. Sci. U.S.A. 88, 2956–2960.
- 183) Antunes-Rodrigues, J., Machado, B.H., Andrade, H.A., Mauad, H., Ramalho, M.J., Reis, L.C. et al. (1992) Carotid-aortic and renal baroreceptors mediate the atrial natriuretic peptide release induced by blood volume expansion. Proc. Natl. Acad. Sci. U.S.A. 89, 6828.
- 184) Bosler, O. and Descarries, L. (1988) Monoamine innervation of the organum vasculosum laminae terminalis (OVLT): a high resolution radioautographic study in the rat. J. Comp. Neurol. 272, 545–561.
- 185) Stein, J.M., Lind, R.W. and Johnson, A.K. (1987) Central serotonergic influences on renal electrolyte and water excretion. Neuropharmacology 26, 1685–1692.
- 186) Reis, L.C., Ramalho, M.J. and Antunes-Rodrigues, J. (1991) Participation of the median raphe nucleus and central serotoninergic pathways in the control of water electrolyte excretion. Braz. J. Med. Biol. Res. 24, 847–854.
- 187) Reis, L.C., Ramalho, M.J., Favaretto, A.L., Gutkowska, J., McCann, S.M. and Antunes-Rodrigues, J. (1994) Participation of the ascending serotonergic system in the stimulation of atrial natriuretic peptide release. Proc. Natl. Acad. Sci. U.S.A. 91, 12022–12026.
- 188) McCance, R. (1936) Experimental human salt deficiency. Lancet 1, 823–830.
- 189) Bojesen, E. (1966) Concentrations of aldosterone and corticosterone in peripheral plasma of rats. The effects of salt depletion, salt repletion, potassium loading and intravenous injections of renin and angiotensin II. Eur. J. Steroids 1, 145–169.
- 190) Contreras, R.J. and Hatton, G.I. (1975) Gustatory adaptation as an explanation for dietary-induced sodium appetite. Physiol. Behav. 15, 569–576.
- 191) Stricker, E.M., Thiels, E. and Verbalis, J.G. (1991) Sodium appetite in rats after prolonged dietary sodium deprivation: a sexually dimorphic phenomenon. Am. J. Physiol. 260, R1082–R1088.
- 192) Wolf, G. and Stricker, E.M. (1967) Sodium appetite elicited by hypovolemia in adrenalectomized rats: Reevaluation of the “reservoir” hypothesis. J. Comp. Physiol. Psychol. 63, 252–257.
- 193) Heer, M., Baisch, F., Kropp, J., Gerzer, R. and Drummer, C. (2000) High dietary sodium chloride consumption may not induce body fluid retention in humans. Am. J. Physiol. Renal Physiol. 278, F585–F595.
- 194) Titze, J., Maillet, A., Lang, R., Gunga, H.C., Johannes, B., Gauquelin-Koch, G. et al. (2002) Long-term sodium balance in humans in a terrestrial space station simulation study. Am. J. Kidney Dis. 40, 508–516.
- 195) Titze, J., Lang, R., Ilies, C., Schwind, K.H., Kirsch, K.A., Dietsch, P. et al. (2003) Osmotically inactive skin Na+ storage in rats. Am. J. Physiol. Renal Physiol. 285, F1108–F1117.
- 196) Titze, J., Shakibaei, M., Schafflhuber, M., Schulze-Tanzil, G., Porst, M., Schwind, K.H. et al. (2004) Glycosaminoglycan polymerization may enable osmotically inactive Na+ storage in the skin. Am. J. Physiol. Heart Circ. Physiol. 287, H203–H208.
- 197) Wiig, H., Luft, F.C. and Titze, J.M. (2018) The interstitium conducts extrarenal storage of sodium and represents a third compartment essential for extracellular volume and blood pressure homeostasis. Acta Physiol. (Oxf.) 222, e13006.
- 198) Richter, C.P. (1936) Increased salt appetite in adrenalectomized rats. Am. J. Physiol. Leg. Cont. 115, 155–161.
- 199) Sakai, R.R., Nicolaidis, S. and Epstein, A.N. (1986) Salt appetite is suppressed by interference with angiotensin II and aldosterone. Am. J. Physiol. Regul. Integr. Comp. Physiol. 251, R762–R768.
- 200) Fluharty, S.J. and Epstein, A.N. (1983) Sodium appetite elicited by intracerebroventricular infusion of angiotensin II in the rat: II. Synergistic interaction with systemic mineralocorticoids. Behav. Neurosci. 97, 746–758.
- 201) Epstein, A.N. (1982) Mineralocorticoids and cerebral angiotensin may act together to produce sodium appetite. Peptides 3, 493–494.
- 202) Coble, J.P., Grobe, J.L., Johnson, A.K. and Sigmund, C.D. (2015) Mechanisms of brain renin angiotensin system-induced drinking and blood pressure: importance of the subfornical organ. Am. J. Physiol. Regul. Integr. Comp. Physiol. 308, R238–R249.
- 203) de Kloet, A.D., Liu, M., Rodríguez, V., Krause, E.G. and Sumners, C. (2015) Role of neurons and glia in the CNS actions of the renin-angiotensin system in cardiovascular control. Am. J. Physiol. Regul. Integr. Comp. Physiol. 309, R444–R458.
- 204) Johnson, A.K. and Thunhorst, R.L. (1997) The neuroendocrinology of thirst and salt appetite: visceral sensory signals and mechanisms of central integration. Front. Neuroendocrinol. 18, 292–353.
- 205) Geerling, J.C. and Loewy, A.D. (2008) Central regulation of sodium appetite. Exp. Physiol. 93, 177–209.
- 206) Sunn, N., McKinley, M.J. and Oldfield, B.J. (2003) Circulating angiotensin II activates neurones in circumventricular organs of the lamina terminalis that project to the bed nucleus of the stria terminalis. J. Neuroendocrinol. 15, 725–731.
- 207) Ricardo, J.A. and Koh, E.T. (1978) Anatomical evidence of direct projections from the nucleus of the solitary tract to the hypothalamus, amygdala, and other forebrain structures in the rat. Brain Res. 153, 1–26.
- 208) Terenzi, M.G. and Ingram, C.D. (1995) A combined immunocytochemical and retrograde tracing study of noradrenergic connections between the caudal medulla and bed nuclei of the stria terminalis. Brain Res. 672, 289–297.
- 209) Geerling, J.C., Engeland, W.C., Kawata, M. and Loewy, A.D. (2006) Aldosterone target neurons in the nucleus tractus solitarius drive sodium appetite. J. Neurosci. 26, 411–417.
- 210) Geerling, J.C. and Loewy, A.D. (2006) Aldosterone-sensitive NTS neurons are inhibited by saline ingestion during chronic mineralocorticoid treatment. Brain Res. 1115, 54–64.
- 211) Geerling, J.C. and Loewy, A.D. (2007) Sodium depletion activates the aldosterone-sensitive neurons in the NTS independently of thirst. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R1338–R1348.
- 212) Li, Z., Iwai, M., Wu, L., Shiuchi, T., Jinno, T., Cui, T.X. et al. (2003) Role of AT2 receptor in the brain in regulation of blood pressure and water intake. Am. J. Physiol. Heart Circ. Physiol. 284, H116–H121.
- 213) Jennings, J.H., Sparta, D.R., Stamatakis, A.M., Ung, R.L., Pleil, K.E., Kash, T.L. et al. (2013) Distinct extended amygdala circuits for divergent motivational states. Nature 496, 224–228.
- 214) Jennings, J.H., Rizzi, G., Stamatakis, A.M., Ung, R.L. and Stuber, G.D. (2013) The inhibitory circuit architecture of the lateral hypothalamus orchestrates feeding. Science 341, 1517–1521.
- 215) Sandhu, E.C., Fernando, A.B.P., Irvine, E.E., Tossell, K., Kokkinou, M., Glegola, J. et al. (2018) Phasic stimulation of midbrain dopamine neuron activity reduces salt consumption. eNeuro 5, ENEURO.0064-18.2018.
- 216) Wei, H., Ahn, S., Shenoy, S.K., Karnik, S.S., Hunyady, L., Luttrell, L.M. et al. (2003) Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. U.S.A. 100, 10782–10787.
- 217) Daniels, D., Mietlicki, E.G., Nowak, E.L. and Fluharty, S.J. (2009) Angiotensin II stimulates water and NaCl intake through separate cell signalling pathways in rats. Exp. Physiol. 94, 130–137.
- 218) Mendelsohn, F.A., Quirion, R., Saavedra, J.M., Aguilera, G. and Catt, K.J. (1984) Autoradiographic localization of angiotensin II receptors in rat brain. Proc. Natl. Acad. Sci. U.S.A. 81, 1575–1579.
- 219) Yamada, H. and Mendelsohn, F.A. (1989) Angiotensin II receptor binding in the rat hypothalamus and circumventricular organs during dietary sodium deprivation. Neuroendocrinology 50, 469–475.
- 220) Fitts, D.A. and Masson, D.B. (1989) Forebrain sites of action for drinking and salt appetite to angiotensin or captopril. Behav. Neurosci. 103, 865–872.
- 221) Contreras, R.J. and Stetson, P.W. (1981) Changes in salt intake after lesions of the area postrema and the nucleus of the solitary tract in rats. Brain Res. 211, 355–366.
- 222) Edwards, G.L. and Ritter, R.C. (1982) Area postrema lesions increase drinking to angiotensin and extracellular dehydration. Physiol. Behav. 29, 943–947.
- 223) Curtis, K.S., Verbalis, J.G. and Stricker, E.M. (1996) Area postrema lesions in rats appear to disrupt rapid feedback inhibition of fluid intake. Brain Res. 726, 31–38.
- 224) Geerling, J.C. and Loewy, A.D. (2006) Aldosterone-sensitive neurons in the nucleus of the solitary tract: efferent projections. J. Comp. Neurol. 497, 223–250.
- 225) Evans, L.C., Ivy, J.R., Wyrwoll, C., McNairn, J.A., Menzies, R.I., Christensen, T.H. et al. (2016) Conditional deletion of Hsd11b2 in the brain causes salt appetite and hypertension. Circulation 133, 1360–1370.
- 226) Jarvie, B.C. and Palmiter, R.D. (2017) HSD2 neurons in the hindbrain drive sodium appetite. Nat. Neurosci. 20, 167–169.
- 227) Resch, J.M., Fenselau, H., Madara, J.C., Wu, C., Campbell, J.N., Lyubetskaya, A. et al. (2017) Aldosterone-sensing neurons in the NTS exhibit state-dependent pacemaker activity and drive sodium appetite via synergy with angiotensin II signaling. Neuron 96, 190–206.e197.
- 228) Sequeira, S.M., Geerling, J.C. and Loewy, A.D. (2006) Local inputs to aldosterone-sensitive neurons of the nucleus tractus solitarius. Neuroscience 141, 1995–2005.
- 229) Stein, M.K. and Loewy, A.D. (2010) Area postrema projects to FoxP2 neurons of the pre-locus coeruleus and parabrachial nuclei: brainstem sites implicated in sodium appetite regulation. Brain Res. 1359, 116–127.
- 230) Geerling, J.C., Stein, M.K., Miller, R.L., Shin, J.W., Gray, P.A. and Loewy, A.D. (2011) FoxP2 expression defines dorsolateral pontine neurons activated by sodium deprivation. Brain Res. 1375, 19–27.
- 231) Shin, J.W., Geerling, J.C., Stein, M.K., Miller, R.L. and Loewy, A.D. (2011) FoxP2 brainstem neurons project to sodium appetite regulatory sites. J. Chem. Neuroanat. 42, 1–23.
- 232) Lee, S., Augustine, V., Zhao, Y., Ebisu, H., Ho, B., Kong, D. et al. (2019) Chemosensory modulation of neural circuits for sodium appetite. Nature 568, 93–97.
- 233) Smith, C.M. and Lawrence, A.J. (2018) Salt Appetite, and the Influence of Opioids. Neurochem. Res. 43, 12–18.
- 234) Hubbell, C.L. and McCutcheon, N.B. (1993) Opioidergic manipulations affect intake of 3% NaCl in sodium-deficient rats. Pharmacol. Biochem. Behav. 46, 473–476.
- 235) Lucas, L.R., Grillo, C.A. and McEwen, B.S. (2003) Involvement of mesolimbic structures in short-term sodium depletion: in situ hybridization and ligand-binding analyses. Neuroendocrinology 77, 406–415.
- 236) Lucas, L.R., Grillo, C.A. and McEwen, B.S. (2007) Salt appetite in sodium-depleted or sodium-replete conditions: possible role of opioid receptors. Neuroendocrinology 85, 139–147.
- 237) Zhang, M. and Kelley, A.E. (2002) Intake of saccharin, salt, and ethanol solutions is increased by infusion of a mu opioid agonist into the nucleus accumbens. Psychopharmacology (Berl) 159, 415–423.
- 238) De Oliveira, L.B., De Luca, L.A. and Menani, J.V. (2008) Opioid activation in the lateral parabrachial nucleus induces hypertonic sodium intake. Neuroscience 155, 350–358.
- 239) Pavan, C.G., Roncari, C.F., Barbosa, S.P., De Paula, P.M., Colombari, D.S., De Luca, L.A. Jr. et al. (2015) Activation of μ opioid receptors in the LPBN facilitates sodium intake in rats. Behav. Brain Res. 288, 20–25.
- 240) Smith, C.M., Walker, L.L., Leeboonngam, T., McKinley, M.J., Denton, D.A. and Lawrence, A.J. (2016) Endogenous central amygdala mu-opioid receptor signaling promotes sodium appetite in mice. Proc. Natl. Acad. Sci. U.S.A. 113, 13893–13898.
- 241) Eikemo, M., Løseth, G.E., Johnstone, T., Gjerstad, J., Willoch, F. and Leknes, S. (2016) Sweet taste pleasantness is modulated by morphine and naltrexone. Psychopharmacology (Berl) 233, 3711–3723.
- 242) Kelley, A.E., Bakshi, V.P., Haber, S.N., Steininger, T.L., Will, M.J. and Zhang, M. (2002) Opioid modulation of taste hedonics within the ventral striatum. Physiol. Behav. 76, 365–377.
- 243) Nogueiras, R., Romero-Picó, A., Vazquez, M.J., Novelle, M.G., López, M. and Diéguez, C. (2012) The opioid system and food intake: homeostatic and hedonic mechanisms. Obes. Facts 5, 196–207.
- 244) Galaverna, O., De Luca, L.A., Schulkin, J., Yao, S.-Z. and Epstein, A.N. (1992) Deficits in NaCl ingestion after damage to the central nucleus of the amygdala in the rat. Brain Res. Bull. 28, 89–98.
- 245) Zardetto-Smith, A.M., Beltz, T.G. and Johnson, A.K. (1994) Role of the central nucleus of the amygdala and bed nucleus of the stria terminalis in experimentally-induced salt appetite. Brain Res. 645, 123–134.
- 246) Covian, M.R., Antunes-Rodrigues, J., Gentil, C.G., Saad, W.A., Camargo, L.A. and Neto, C.R.S. (2019) Central control of salt balance. In Neural Integration of Physiological Mechanisms and Behaviour (eds. Mogenson, G. and Calaresu, F.). University of Toronto Press, Toronto, pp. 267–282.
- 247) Wang, Q., Li, J.R., Yang, X.J., Chen, K., Sun, B. and Yan, J.Q. (2012) Inhibitory effect of activation of GABAA receptor in the central nucleus of amygdala on the sodium intake in the sodium-depleted rat. Neuroscience 223, 277–284.
- 248) Yan, J., Li, J., Yan, J., Sun, H., Wang, Q., Chen, K. et al. (2013) Activation of μ-opioid receptors in the central nucleus of the amygdala induces hypertonic sodium intake. Neuroscience 233, 28–43.
- 249) Zhu, W. and Pan, Z.Z. (2005) μ-opioid-mediated inhibition of glutamate synaptic transmission in rat central amygdala neurons. Neuroscience 133, 97–103.
- 250) Chieng, B.C.H., Christie, M.J. and Osborne, P.B. (2006) Characterization of neurons in the rat central nucleus of the amygdala: Cellular physiology, morphology, and opioid sensitivity. J. Comp. Neurol. 497, 910–927.
- 251) Vaughan, C.W., Ingram, S.L., Connor, M.A. and Christie, M.J. (1997) How opioids inhibit GABA-mediated neurotransmission. Nature 390, 611–614.
- 252) Andrade-Franzé, G.M.F., Andrade, C.A.F., De Luca, L.A., De Paula, P.M., Colombari, D.S.A. and Menani, J.V. (2010) Lesions in the central amygdala impair sodium intake induced by the blockade of the lateral parabrachial nucleus. Brain Res. 1332, 57–64.
- 253) Di Lorenzo, P.M. and Victor, J.D. (2003) Taste response variability and temporal coding in the nucleus of the solitary tract of the rat. J. Neurophysiol. 90, 1418–1431.
- 254) Kobashi, M., Ichikawa, H., Sugimoto, T. and Adachi, A. (1993) Response of neurons in the solitary tract nucleus, area postrema and lateral parabrachial nucleus to gastric load of hypertonic saline. Neurosci. Lett. 158, 47–50.
- 255) Yamamoto, T., Shimura, T., Sako, N., Sakai, N., Tanimizu, T. and Wakisaka, S. (1993) C-Fos expression in the parabrachial nucleus after ingestion of sodium chloride in the rat. Neuroreport 4, 1223–1226.
- 256) Franchini, L.F. and Vivas, L. (1999) Distribution of Fos immunoreactivity in rat brain after sodium consumption induced by peritoneal dialysis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 276, R1180–R1187.
- 257) Norgren, R. (1981) The central organization of the gustatory and visceral afferent systems in the nucleus of the solitary tract. Brain mechanisms of sensation, 143–160.
- 258) Ciriello, J., Lawrence, D. and Pittman, Q.J. (1984) Electrophysiological identification of neurons in the parabrachial nucleus projecting directly to the hypothalamus in the rat. Brain Res. 322, 388–392.
- 259) Fulwiler, C.E. and Saper, C.B. (1984) Subnuclear organization of the efferent connections of the parabrachial nucleus in the rat. Brain Res. Rev. 7, 229–259.
- 260) Lança, A.J. and Van Der Kooy, D. (1985) A serotonin-containing pathway from the area postrema to the parabrachial nucleus in the rat. Neuroscience 14, 1117–1126.
- 261) Herbert, H., Moga, M.M. and Saper, C.B. (1990) Connections of the parabrachial nucleus with the nucleus of the solitary tract and the medullary reticular formation in the rat. J. Comp. Neurol. 293, 540–580.
- 262) Krukoff, T.L., Harris, K.H. and Jhamandas, J.H. (1993) Efferent projections from the parabrachial nucleus demonstrated with the anterograde tracer Phaseolus vulgaris leucoagglutinin. Brain Res. Bull. 30, 163–172.
- 263) Jhamandas, J.H., Petrov, T., Harris, K.H., Vu, T. and Krukoff, T.L. (1996) Parabrachial nucleus projection to the amygdala in the rat: Electrophysiological and anatomical observations. Brain Res. Bull. 39, 115–126.
- 264) Jhamandas, J.H., Harris, K.H., Petrov, T. and Krukoff, T.L. (1992) Characterization of the Parabrachial Nucleus Input to the Hypothalamic Paraventricular Nucleus in the Rat. J. Neuroendocrinol. 4, 461–471.
- 265) Menani, J.V. and Johnson, A.K. (1995) Lateral parabrachial serotonergic mechanisms: Angiotensin-induced pressor and drinking responses. Am. J. Physiol. Regul. Integr. Comp. Physiol. 269, R1044–R1049.
- 266) Menani, J.V., Thunhorst, R.L. and Johnson, A.K. (1996) Lateral parabrachial nucleus and serotonergic mechanisms in the control of salt appetite in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 270, R162–R168.
- 267) Menani, J.V. and Johnson, A.K. (1998) Cholecystokinin actions in the parabrachial nucleus: Effects on thirst and salt appetite. Am. J. Physiol. Regul. Integr. Comp. Physiol. 275, R1431–R1437.
- 268) Menani, J.V., Barbosa, S.P., De Luca, L.A., De Gobbi, J.I.F. and Johnson, A.K. (2002) Serotonergic mechanisms of the lateral parabrachial nucleus and cholinergic-induced sodium appetite. Am. J. Physiol. Regul. Integr. Comp. Physiol. 282, R837–R841.
- 269) De luca, L.A., Barbosa, S.P. and Menani, J.V. (2003) Brain serotonin blockade and paradoxical salt intake in rats. Neuroscience 121, 1055–1061.
- 270) Andrade, C.A.F., Barbosa, S.P., De Luca, L.A. and Menani, J.V. (2004) Activation of α2-adrenergic receptors into the lateral parabrachial nucleus enhances NaCl intake in rats. Neuroscience 129, 25–34.
- 271) Andrade, C.A.F., De Luca, L.A., Colombari, D.S.A. and Menani, J.V. (2006) Alpha2-adrenergic activation in the lateral parabrachial nucleus induces NaCl intake under conditions of systemic hyperosmolarity. Neuroscience 142, 21–28.
- 272) De Gobbi, J.I.F., Beltz, T.G., Johnson, R.F., Menani, J.V., Thunhorst, R.L. and Johnson, A.K. (2009) Non-NMDA receptors in the lateral parabrachial nucleus modulate sodium appetite. Brain Res. 1301, 44–51.
- 273) Gasparini, S., de Luca, L.A., Colombari, D.S.A., de Paula, P.M., Barbosa, S.P. and Menani, J.V. (2009) Adrenergic mechanisms of the Kölliker-Fuse/A7 area on the control of water and sodium intake. Neuroscience 164, 370–379.
- 274) De Oliveira, L.B., Kimura, E.H., Callera, J.C., De Luca, L.A., Colombari, D.S.A. and Menani, J.V. (2011) Baclofen into the lateral parabrachial nucleus induces hypertonic sodium chloride and sucrose intake in rats. Neuroscience 183, 160–170.
- 275) Menezes, M.F., Barbosa, S.P., De Andrade, C.A.F., Menani, J.V. and De Paula, P.M. (2011) Purinergic mechanisms of lateral parabrachial nucleus facilitate sodium depletion-induced NaCl intake. Brain Res. 1372, 49–58.
- 276) Tanaka, J., Hayashi, Y., Yamato, K., Miyakubo, H. and Nomura, M. (2004) Involvement of serotonergic systems in the lateral parabrachial nucleus in sodium and water intake: a microdialysis study in the rat. Neurosci. Lett. 357, 41–44.
- 277) Heck, G.L., Mierson, S. and DeSimone, J.A. (1984) Salt taste transduction occurs through an amiloride-sensitive sodium transport pathway. Science 223, 403–405.
- 278) Oka, Y., Butnaru, M., von Buchholtz, L., Ryba, N.J. and Zuker, C.S. (2013) High salt recruits aversive taste pathways. Nature 494, 472–475.
- 279) Gimpl, G. and Fahrenholz, F. (2001) The oxytocin receptor system: structure, function, and regulation. Physiol. Rev. 81, 629–683.
- 280) Stricker, E.M. and Verbalis, J.G. (1996) Central inhibition of salt appetite by oxytocin in rats. Regul. Pept. 66, 83–85.
- 281) Puryear, R., Rigatto, K.V., Amico, J.A. and Morris, M. (2001) Enhanced salt intake in oxytocin deficient mice. Exp. Neurol. 171, 323–328.
- 282) Stricker, E.M. and Verbalis, J.G. (1987) Central inhibitory control of sodium appetite in rats: correlation with pituitary oxytocin secretion. Behav. Neurosci. 101, 560–567.
- 283) Katoh, A., Fujihara, H., Ohbuchi, T., Onaka, T., Hashimoto, T., Kawata, M. et al. (2011) Highly visible expression of an oxytocin-monomeric red fluorescent protein 1 fusion gene in the hypothalamus and posterior pituitary of transgenic rats. Endocrinology 152, 2768–2774.
- 284) Boadle-Biber, M.C. (1993) Regulation of serotonin synthesis. Prog. Biophys. Mol. Biol. 60, 1–15.
- 285) Castro, L., Athanazio, R., Barbetta, M., Ramos, A.C., Angelo, A.L., Campos, I. et al. (2003) Central 5-HT2B/2C and 5-HT3 receptor stimulation decreases salt intake in sodium-depleted rats. Brain Res. 981, 151–159.
- 286) Lima, H.R., Cavalcante-Lima, H.R., Cedraz-Mercez, P.L., Costa, E.S.R.H., Olivares, E.L., Badaue-Passos, D. Jr. et al. (2004) Brain serotonin depletion enhances the sodium appetite induced by sodium depletion or beta-adrenergic stimulation. An. Acad. Bras. Cienc. 76, 85–92.
- 287) Menani, J.V., Colombari, D.S., Beltz, T.G., Thunhorst, R.L. and Johnson, A.K. (1998) Salt appetite: interaction of forebrain angiotensinergic and hindbrain serotonergic mechanisms. Brain Res. 801, 29–35.
- 288) Park, S., Williams, K.W., Liu, C. and Sohn, J.W. (2020) A neural basis for tonic suppression of sodium appetite. Nat. Neurosci. 23, 423–432.
- 289) Miller, R.L., Stein, M.K. and Loewy, A.D. (2011) Serotonergic inputs to FoxP2 neurons of the pre-locus coeruleus and parabrachial nuclei that project to the ventral tegmental area. Neuroscience 193, 229–240.
- 290) Biondolillo, J.W., Williams, L.A. and King, M.S. (2009) Blocking glutamate receptors in the waist area of the parabrachial nucleus decreases taste reactivity behaviors in conscious rats. Chem. Senses 34, 221–230.
- 291) Group, I.C.R. (1988) Intersalt: an international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. Intersalt Cooperative Research Group. BMJ 297, 319.
- 292) Mente, A., O’Donnell, M.J., Rangarajan, S., McQueen, M.J., Poirier, P., Wielgosz, A. et al. (2014) Association of urinary sodium and potassium excretion with blood pressure. N. Engl. J. Med. 371, 601–611.
- 293) Adrogué, H.J. and Madias, N.E. (2007) Sodium and potassium in the pathogenesis of hypertension. N. Engl. J. Med. 356, 1966–1978.
- 294) de Wardener, H.E., He, F.J. and MacGregor, G.A. (2004) Plasma sodium and hypertension. Kidney Int. 66, 2454–2466.
- 295) Stocker, S.D., Monahan, K.D. and Browning, K.N. (2013) Neurogenic and sympathoexcitatory actions of NaCl in hypertension. Curr. Hypertens. Rep. 15, 538–546.
- 296) Gavras, I. and Gavras, H. (2012) ‘Volume-expanded’ hypertension: the effect of fluid overload and the role of the sympathetic nervous system in salt-dependent hypertension. J. Hypertens. 30, 655–659.
- 297) Henderson, K.K. and Byron, K.L. (2007) Vasopressin-induced vasoconstriction: two concentration-dependent signaling pathways. J. Appl. Physiol. 102, 1402–1409.
- 298) Guyenet, P.G. (2006) The sympathetic control of blood pressure. Nat. Rev. Neurosci. 7, 335–346.
- 299) Frithiof, R., Xing, T., McKinley, M.J., May, C.N. and Ramchandra, R. (2014) Intracarotid hypertonic sodium chloride differentially modulates sympathetic nerve activity to the heart and kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 306, R567–R575.
- 300) Kawano, Y. and Ferrario, C.M. (1984) Neurohormonal characteristics of cardiovascular response due to intraventricular hypertonic NaCl. Am. J. Physiol. Heart Circ. Physiol. 247, H422–H428.
- 301) Ribeiro, N., Panizza Hdo, N., Santos, K.M., Ferreira-Neto, H.C. and Antunes, V.R. (2015) Salt-induced sympathoexcitation involves vasopressin V1a receptor activation in the paraventricular nucleus of the hypothalamus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 309, R1369–R1379.
- 302) Choe, K.Y., Han, S.Y., Gaub, P., Shell, B., Voisin, D.L., Knapp, B.A. et al. (2015) High salt intake increases blood pressure via BDNF-mediated downregulation of KCC2 and impaired baroreflex inhibition of vasopressin neurons. Neuron 85, 549–560.
- 303) Nicol, C.J., Adachi, M., Akiyama, T.E. and Gonzalez, F.J. (2005) PPARγ in endothelial cells influences high fat diet-induced hypertension. Am. J. Hypertens. 18, 549–556.
- 304) Stocker, S.D., Lang, S.M., Simmonds, S.S., Wenner, M.M. and Farquhar, W.B. (2015) Cerebrospinal fluid hypernatremia elevates sympathetic nerve activity and blood pressure via the rostral ventrolateral medulla. Hypertension 66, 1184–1190.
- 305) Escoubas, P., De Weille, J.R., Lecoq, A., Diochot, S., Waldmann, R., Champigny, G. et al. (2000) Isolation of a tarantula toxin specific for a class of proton-gated Na+ channels. J. Biol. Chem. 275, 25116–25121.
- 306) Diochot, S., Baron, A., Rash, L.D., Deval, E., Escoubas, P., Scarzello, S. et al. (2004) A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. EMBO J. 23, 1516–1525.
- 307) Bohlen, C.J., Chesler, A.T., Sharif-Naeini, R., Medzihradszky, K.F., Zhou, S., King, D. et al. (2011) A heteromeric Texas coral snake toxin targets acid-sensing ion channels to produce pain. Nature 479, 410–414.
- 308) Burke, P.G., Abbott, S.B., Coates, M.B., Viar, K.E., Stornetta, R.L. and Guyenet, P.G. (2014) Optogenetic stimulation of adrenergic C1 neurons causes sleep state-dependent cardiorespiratory stimulation and arousal with sighs in rats. Am. J. Respir. Crit. Care Med. 190, 1301–1310.
- 309) Hiyama, T.Y., Matsuda, S., Fujikawa, A., Matsumoto, M., Watanabe, E., Kajiwara, H. et al. (2010) Autoimmunity to the sodium-level sensor in the brain causes essential hypernatremia. Neuron 66, 508–522.
- 310) Hiyama, T.Y., Utsunomiya, A.N., Matsumoto, M., Fujikawa, A., Lin, C.H., Hara, K. et al. (2017) Adipsic hypernatremia without hypothalamic lesions accompanied by autoantibodies to subfornical organ. Brain Pathol. 27, 323–331.
- 311) Shirai, Y., Miura, K., Nakamura-Utsunomiya, A., Ishizuka, K., Hattori, M. and Hattori, M. (2021) Analysis of water and electrolyte imbalance in a patient with adipsic hypernatremia associated with subfornical organ-targeting antibody. CEN Case Reports.
- 312) Saker, P., Carey, S., Grohmann, M., Farrell, M.J., Ryan, P.J., Egan, G.F. et al. (2020) Regional brain responses associated with using imagination to evoke and satiate thirst. Proc. Natl. Acad. Sci. U.S.A. 117, 13750–13756.
- 313) Eiselt, A.K., Chen, S., Chen, J., Arnold, J., Kim, T., Pachitariu, M. et al. (2021) Hunger or thirst state uncertainty is resolved by outcome evaluation in medial prefrontal cortex to guide decision-making. Nat. Neurosci. 24, 907–912.
- 314) Noda, M. (2007) Hydromineral neuroendocrinology: mechanism of sensing sodium levels in the mammalian brain. Exp. Physiol. 92, 513–522.
Profile

Masaharu Noda was born in Hiroshima Prefecture, Japan, in 1953. He graduated from Kyoto University Faculty of Engineering in 1977. After finishing its Masters course in 1979, he went on to the Doctorate course of Kyoto University Graduate School of Medicine and received his PhD degree in 1983. During his research as a postgraduate student, Assistant Professor, and Associate Professor at Kyoto University Faculty of Medicine, he succeeded in molecular cloning of the enkephalin precursor, all of the nicotinic acetylcholine receptor subunits, and three brain voltage-gated Na channels and elucidated the structure–function relationship of the Na channel. He subsequently moved to the Max Planck Institute for Developmental Biology at Tübingen, Germany, as a visiting scientist in 1989–1991 to study the molecular mechanisms of the topographic retinotectal projection. He became Professor at the National Institute for Basic Biology and The Graduate University for Advanced Studies, Japan, in 1991 and promoted the following three projects in parallel until 2019: 1) Brain mechanisms for body fluid homeostasis and blood pressure control, 2) Developmental mechanisms for the regional specification in the retina and topographic retinotectal projection, and 3) Physiological roles of receptor-type protein tyrosine phosphatases. Since 2019 to date, he has been working on blood pressure elevation mechanisms by inflammation in the brain as a Professor at the Institute of Innovative Research in Tokyo Institute of Technology.

Takashi Matsuda graduated with Master's degree through the Osaka University of Pharmaceutical Sciences, Laboratory of Cell Biology in 2012. After graduation, he started his research at Masaharu Noda’s laboratory in The Graduate University for Advanced Studies, SOKENDAI, and received his Ph.D. for studies on the central mechanisms for the control of body fluid homeostasis. He identified the thirst-driving neurons (water neurons) and salt appetite-driving neurons (salt neurons) in the subfornical organ and revealed their regulatory mechanisms by Na+-dependent and cholecystokinin-dependent activation of GABAergic neurons (Matsuda et al. Nature Neuroscience, 2017; Matsuda et al. Nature Communications, 2020). Accompanying a transfer of the laboratory, he moved to Tokyo Institute of Technology in 2019 as a Specially Appointed Assistant Professor. He is now expanding his research targets to the central mechanisms for control of blood pressure.



