Abstract
Molecular clouds (MCs) in space are the birthplace of various molecular species. Chemical reactions occurring on the cryogenic surfaces of cosmic icy dust grains have been considered to play important roles in the formation of these species. Radical reactions are crucial because they often have low barriers and thus proceed even at low temperatures such as ∼10 K. Since the 2000s, laboratory experiments conducted under low-temperature, high-vacuum conditions that mimic MC environments have revealed the elementary physicochemical processes on icy dust grains. In this review, experiments conducted by our group in this context are explored, with a focus on radical reactions on the surface of icy dust analogues, leading to the formation of astronomically abundant molecules such as H2, H2O, H2CO, and CH3OH and deuterium fractionation processes. The development of highly sensitive, non-destructive methods for detecting adsorbates and their utilization for clarifying the behavior of free radicals on ice, which contribute to the formation of complex organic molecules, are also described.
1. Introduction
Extremely cold and dense regions in interstellar media are known as molecular clouds (MCs), which comprise large amounts of gases and dust grains, where the dust-to-gas mass ratio is approximately 10−2.1),2) The temperature of MCs is low (10–100 K) because of the presence of dust grains, which shield the inner region of the MCs from the radiation from external stars. The gas density, which mostly corresponds to the abundant H2 molecules, is 103–105 cm−3, and more than 150 types of molecular species have been identified.3) These molecular species evolve from atoms even in such low-temperature environments through processes referred to as chemical evolution.4) In the gas phase, ion–molecule and radical–radical reactions can occur even at low temperatures because they are typically barrierless. However, gas-phase reactions alone cannot explain the large variety and abundance of the molecular species in MCs. Surface physicochemical processes on low-temperature dust surfaces are suggested to play key roles in chemical evolution.5),6)
A dust particle with a typical diameter of ∼0.1 µm has a silicate or carbonaceous core. In the MC environment, the dust core is typically enveloped by an ice mantle; this dust-particle/ice-mantle system is known as icy dust. The ice mantle predominantly comprises amorphous-phase water ice (amorphous solid water; ASW) and other molecules such as CO, CO2, NH3, CH4, H2CO, and CH3OH.7) These molecules are thought to form on dust surfaces via reactions between atoms and primordial molecules such as CO and are eventually released into the gas phase when the temperature of the dust increases. Therefore, the physicochemical processes important for the chemical evolution on icy dust are (i) adsorption of atoms and molecules, (ii) surface diffusion of these species, (iii) bimolecular reactions, and (iv) desorption of products. Dust surface reactions are advantageous over gas-phase reactions in certain contexts. Because of low temperatures, various species can accumulate on dust and encounter each other many times as compared to the gaseous phase collisions. Furthermore, when reactants stay at neighboring sites on dust, they interact with each other for a long time. In other words, the chances of encounters and subsequent reactions between reactants are greater in dust than in gas-phase reactions, where the collision rate between the highly abundant hydrogen is approximately once a year. Another important role of dust is to function as a third body to dissipate the excess energy of the surface reaction. For example, in the gas phase, the collision of two ground-state H atoms (the recombination reaction) does not yield H2 molecules because the heat of reaction (4.5 eV) cannot be released through the emission of a photon; in contrast, the recombination reaction occurs efficiently on dust surfaces.8) Because of their relevance to icy dust, the chemical processes occurring on ASW have been investigated in several laboratory and theoretical studies. Therefore, representative experiments on the physicochemical processes occurring on ASW are discussed in this short review, with a focus on our research.
Among the surface processes relevant to icy dust in MCs, the reactions of abundant H atoms are considered important; the accumulation rate of H atoms on dust is approximately once a day in MCs. Because of their low mass, physisorbed H atoms move efficiently on the surface even at 10 K; furthermore, tunneling reactions are expected to occur owing to the enhancement of the wave nature of H atoms at low temperatures. Laboratory investigations of H-atom reactions on ASW are introduced in Section 2. An important milestone has been reached because of laboratory experiments that clarified the formaldehyde (H2CO) and methanol (CH3OH) formation routes in an MC environment.9),10) As detailed in Section 2.3, several groups have demonstrated that these molecules are formed by successive hydrogenation of CO on the ASW surface; the addition of two H atoms to CO produces H2CO, and the subsequent addition of two H atoms yields CH3OH. Recently, certain H-atom reactions have been found to cause desorption of products. This process, known as reactive desorption or chemical desorption, is an important mechanism of desorption from dust without an external energy input such as photons and ion bombardments (Section 2.4).
Astronomical observations revealed that deuterated species are abundant in MCs.11) Although the abundance ratio of (partially) deuterated isotopologues to the normal isotopolog depends not only on species but also on MCs, it is a few-orders-of-magnitude higher than the abundance ratio of deuterium atoms to hydrogen atoms (D/H = 10−5) in interstellar media.12) This phenomenon, known as deuterium fractionation or enrichment, basically results from the greater thermodynamic stability of deuterated species than that of the corresponding normal species, and leads to the former being more prominent under lower-temperature conditions such as those in MCs. Deuterium enrichment has also long been recognized in meteorites and comets.13) The D/H ratio in terrestrial seawater is approximately one-order-of-magnitude higher than the interstellar abundance ratio. Because a similar degree of deuterium enrichment has been found for H2O molecules in comets,14) deuterium enrichment in the solar system is thought to originate at least partially from the MC that produced the Sun.15) In other words, deuterium fractionation proceeds significantly quickly in MCs at low temperatures, and it does not equilibrate at higher temperatures even after the formation of planetary systems. The extremely high deuterium fractionation in MCs cannot be explained by gas-phase mechanisms alone. Deuterium fractionation has been extensively investigated by our group, especially for formaldehyde and methanol, whose deuteration levels were found to be as high as 0.1 relative to the normal species in MCs (Section 3). We have demonstrated that efficient H–D substitution reactions (such as H2CO + H → HCO + H2 and HCO + D → HDCO) facilitate deuterium enrichment of molecules on icy dust. Our proposed H–D substitution mechanism has been incorporated into standard chemical models for deuterium enrichment in MCs.16)
The H-atom reactions alone cannot explain the formation of complex organic molecules (COMs) that have been observed in various astronomical objects; molecules comprising more than six atoms (excluding methanol) are often termed as COMs in the astrochemical community.17) Skeletal evolution in molecules via C–C bond formation, for instance, is necessary to form COMs. The reactions of radicals such as OH, HCO, CH3O, CH2OH, NH, and NH2 are believed to yield diverse COMs.18) These radicals must diffuse to meet each other on the surface of icy dust and ensure COMs formation; however, a non-diffusive mechanism, in which two radicals are produced in the vicinity, has also been proposed.19) Because these radicals are considerably heavier than the H atom, their diffusion is activated when the temperature of the dust and gas gradually increases from 10 to ∼200 K during star formation. Therefore, the temperature at which each radical begins to diffuse is an important parameter for understanding the formation of COMs. However, this parameter has not been experimentally determined because of the difficulty in in situ detection of radicals on ASW. To experimentally monitor the behavior of radicals on ASW, we developed a new method by combining photostimulated desorption and resonance-enhanced multiphoton ionization (PSD-REMPI) to sensitively detect trace amounts of surface adsorbates.20),21) The application of this method to OH radicals, which are thought to be abundant on ASW, is described in Section 4.1. The Cs+-ion pickup technique was also developed by our group as another sensitive method to study radical reactions on ASW. This method and its utilization for clarifying COM formation processes are detailed in Section 4.2.
Furthermore, our group has contributed experimentally to enhancing the knowledge on the other important physicochemical processes occurring on dust surfaces. Relevant aspects such as the ortho-to-para conversion of H2 on dust grain surfaces, the ortho-to-para ratio of water molecules desorbed from ASW, and the shape of icy grains are briefly discussed in Section 5. Future perspectives are presented in Section 6.
2. Hydrogen reactions on icy grains
2.1. H-atom diffusion and recombination (H2 formation).
Molecular hydrogen (H2) is the most abundant molecule in the Universe and plays important roles in several astronomical processes, including chemical reactions and gas dynamics during star formation. Thus, the mechanisms of H2 formation in interstellar environments are a crucial topic in astrochemical and astrophysical research communities.5),8),22),23) The gas-phase formation of H2 is inefficient because of the forbidden radiative association reaction between H atoms in the ground state (H + H → H2 + hν), and the three-body reaction (H + H + M → H2 + M) is unfeasible under the ultrahigh-vacuum conditions of MCs; moreover, the electron-based reactions (H + e− → H− + hν, H− + H → H2 + e−) are remarkably slow because of the scarcity of electrons.24) Consequently, other H2 formation processes should be considered to account for the observed high abundance in interstellar media. Generally, H2 is thought to be produced on dust grain surfaces,25) which permit interactions between H atoms and act as a third body “M” to dissipate the heat of reaction (4.5 eV). Because the recombination reaction itself is barrierless, the rate of H2 formation on the grain surfaces is dominated by the surface diffusion rate of H atoms. Thus, to elucidate the H2 formation mechanisms in MCs, investigations of H-atom diffusion on low-temperature surfaces, particularly on ASW, are critical.
Most experimental studies on the H-atom diffusion on ASW have employed temperature-programmed desorption (TPD), in which the H2 molecules desorbing from the H-atom-deposited ASW during a temperature increase are detected using a quadrupole mass spectrometer (QMS). The H-atom diffusion is indirectly deduced from TPD spectra, which are plots of the H2 desorption rate as a function of the surface temperature. However, the TPD method can cause a large ambiguity because the TPD spectral analysis involves several parameters such as the number density, desorption rate, and surface diffusion rate to derive the activation energies for H2 desorption (Edes) and H-atom diffusion (Esd) and desorption. Furthermore, TPD is unsuitable for analyzing irregular amorphous surfaces, where the activation energy cannot be represented by a single value but has a wide distribution, and often provides contradictory results. For example, Perets et al. and Matar et al. reported differing Esd values of H atoms (40–55 and 22 ± 2 meV, respectively)26),27); the former suggests that H(D) atoms hardly diffuse at 10 K, whereas the latter indicates that they can diffuse on ASW within the experimental timescale. If the H atoms and H2 molecules on the surface can be detected without employing TPD, the Esd of H atoms can be determined more unambiguously. To that end, we developed a new experimental method known as PSD-REMPI.20),21)
In the experimental setup used for the PSD-REMPI measurements of H atoms and H2 molecules on ASW (Fig. 1a), an aluminum (Al) substrate is located at the center of an ultrahigh-vacuum chamber; the temperature of the substrate can be controlled between 6 and 300 K using a closed-cycle He refrigerator and a heater. To prepare the ASW sample, H2O vapor is introduced into the chamber through a capillary plate and deposited onto the substrate maintained at 8 K. Subsequently, hydrogen atoms produced by a microwave discharge created in a Pyrex tube are introduced into the main chamber through an Al pipe at 100 K to reduce the translational energy. The scheme governing the PSD-REMPI detection is illustrated in Fig. 1b. Essentially, the hydrogen atoms on the ASW surface are photodesorbed using a PSD laser (532 nm, 10 ns duration, <100 µJ per pulse). Although the mechanism of the PSD is unclear, the H-atom desorption presumably results from the propagation of phonons produced on the Al substrate.28) Then, the desorbed H atoms are ionized using a REMPI laser. The (2 + 1) REMPI scheme involving the 2s ← 1s transition was used to ionize H atoms. A linear time-of-flight (TOF) mass spectrometer was used to detect the ionized H atoms (that is, H+ ions). As shown in Fig. 1c, the intensities of the PSD-REMPI signal for H atoms increases linearly with deposition time. Because the H-atom flux is constant and low enough to assume that the surface number density of H atoms (nH) increases linearly with the deposition time, the PSD-REMPI intensities are considered to be proportional to nH.29)

After the deposition of H atoms on the ASW surface, nH is expected to decrease with time (waiting time in Fig. 1b) via monoatomic desorption and/or recombination reactions to form H2. Thus, the rate equation for the time variation of nH is expressed as
| \begin{equation}
\frac{dn_{\text{H}}}{dt} = - 2k_{\text{H-H}}n_{\text{H}}^{2} - k_{\text{des}}n_{\text{H}},
\end{equation}
| [1] |
where
kH-H and
kdes are the rate constants for the recombination reaction and thermal desorption, respectively. Because thermal desorption is negligible at low temperatures such as 8 K, the decrease in
nH is dominated by the recombination. Under this assumption,
Eq. [1] can be solved as follows:
| \begin{equation}
\frac{I_{\text{H}}}{I_{0}} \propto \frac{n_{\text{H}}}{n_{0}} = \frac{1 - b}{2k_{\text{H-H}}n_{0}t + 1} + b,
\end{equation}
| [2] |
where
IH and
I0 are the PSD-REMPI signal intensities at times
t and 0, respectively, and
nH and
n0 are the corresponding number densities of H atoms. An asymptotic value
b was introduced to account for the intact H atoms remaining on the ASW within the experimental timescale (∼2 h). The attenuation curves for the H atoms on ASW (
Fig. 1d) adequately fit
Eq. [2]. The activation energy for surface diffusion (
Esd) was subsequently determined as follows:
| \begin{equation}
2k_{\text{H-H}}n_{0} = \theta \nu \exp \left(- \frac{E_{\text{sd}}}{k_{\text{B}}T} \right),
\end{equation}
| [3] |
where θ is the surface coverage of the H(D) atom, ν is the hopping frequency,
kB is Boltzmann’s constant, and
T is the temperature. Analysis of the
Fig. 1d data yielded
Esd = 22 (23) meV for H(D) atoms on ASW,
28) which is consistent with the TPD-derived value obtained by Matar
et al.27) The dotted line in
Fig. 1d indicates that the decay of H(D) atoms does not occur when assuming
Esd = 30 meV. Hama
et al. classified the adsorption sites on the ASW surface for H atoms into three groups: very-shallow-, middle-, and deep-potential sites (
Esd = ≤18, 22, and ≥30 meV, respectively).
28) At 8 K, H atoms adsorbed onto the very-shallow-potential sites diffuse rapidly to form H
2 during the H-atom deposition, whereas those on the middle-potential sites diffuse on a timescale of tens of minutes; those on the deep-potential sites do not diffuse.
As described above, the surface diffusion of H(D) atoms on ASW occurs through a thermal hopping mechanism. However, the diffusion of H atoms on polycrystalline ice (PCI) occurs remarkably quickly, hindering the scrutiny of nH by PSD-REMPI with varying waiting times after the H-atom deposition.28) This quick decline of H atoms on PCI suggests the fast diffusion through the tunneling effect. Another interesting issue regarding H-atom diffusion is the viability of tunneling diffusion. Kuwahata et al. employed another methodology to assess the diffusion on PCI for evaluating the diffusion-related kinetic isotope effect.30) They monitored the intensities of H or D atoms on PCI under steady-state conditions during continuous deposition of H or D atoms with identical fluxes on PCI at 10 K; the steady-state condition refers to a balance between the supply and loss of H (D) atoms (ps·F = kH-HnH2, where ps and F are the sticking coefficient, which is assumed to be the same for H and D atoms, and atomic flux, respectively). For identical fluxes of H and D atoms in each experiment, the ratio of the surface number densities for H and D atoms can be expressed simply using the recombination rates kH-H and kD-D:
| \begin{equation}
\frac{n_{\text{D}}}{n_{\text{H}}} = \sqrt{\frac{k_{\text{H-H}}}{k_{\text{D-D}}}}.
\end{equation}
| [4] |
The average surface number density of H(D) atoms can be effectively controlled by changing the flux. A higher flux can provide a shorter distance between atoms, that is, the recombination and the ensuing short-distance diffusion can be monitored in a higher flux condition. When the H(D) atom flux increases, the
kH-H/
kD-D ratio for the PCI surface reaches >70, and the large isotope effect cannot be explained by the thermal hopping at 10 K; this indicates that the H-atom diffusion occurs predominantly through the tunneling mechanism. For the ASW surface,
kH-H/
kD-D approaches a maximum of ∼16 as the flux increases. This result suggests the coexistence of thermal hopping and tunneling diffusion on ASW, with the former being the dominant mechanism for determining the surface diffusion rate of H and D atoms on ASW. In fact, theoretical studies by Smoluchowski proposed that diffusion by quantum tunneling is suppressed on ASW because of the nonperiodic potential,
31)–33) whereas on PCI surfaces H atoms would spread instantaneously to their boundaries through quantum tunneling and become localized there.
33)
Because the surface composition of icy dust grains is considered to partly include solid CO, H-atom diffusion on pure solid CO—which is the second most abundant molecule after H2—was investigated by our group.34) By analyzing the attenuation curve of the PSD-REMPI intensities, Kimura et al. determined the Esd values for H atoms on solid CO to be ∼22, 30, and 37 meV at surface temperatures of 8, 12, and 15 K, respectively. These Esd data are representative values at the corresponding temperatures because the surface of the solid CO sample produced by vapor deposition at 10 K is typically rough and has several types of sites distinguished with adsorption potential depths; at higher temperatures, the diffusion is dominated by the activation energy for deeper adsorption sites.
MCs evolve from diffuse clouds in which the temperature is relatively high (∼80 K) and grains are not covered with ice. H2 is abundantly produced in this environment; however, the related mechanism is unclear. Therefore, we studied the H2 formation processes on diamond-like carbon (DLC), which serves as a model surface for bare grains (that is, ice-free grains).35) An analysis similar to that performed by Kuwahata et al.30) revealed the significance of tunneling diffusion in the diffusion of H atoms on the DLC surface. Moreover, we found that H2 was formed efficiently on the DLC even at 20 K, which is higher than the threshold for H2 formation on ASW. However, the formation at 20 K is feasible only under laboratory conditions of high H-atom fluxes; therefore, other mechanisms that are responsible for H2 formation in diffuse clouds must be clarified.
2.2. H2O formation.
The abundance of H2O in space cannot be explained only by the gas-phase route H3O+ + e− → H2O + H36); the surface reactions on dust in MCs, as originally proposed by Tielens and Hagen,37) should also be considered. In fact, H2O is the dominant component of the icy mantle. Several mechanisms have been proposed for H2O formation on dust surfaces based on laboratory experiments, as detailed in a review article.23) The simplest mechanism involves reactions featuring the addition of H-atoms to O atoms ($\text{O}\overset{\text{H}}{\to}\text{OH}\overset{\text{H}}{\to}\text{H$_{2}$O}$). As both steps are barrierless radical–radical reactions, the rate of H2O formation is determined by the surface diffusion of H atoms. Another important H2O-formation process is the successive addition of H atoms to O2 molecules38):
| \begin{equation}
\text{O$_{2}$}\overset{\text{H}}{\to}\text{HO$_{2}$}\overset{\text{H}}{\to}\text{H$_{2}$O$_{2}$}\overset{\text{H}}{\to}\text{H$_{2}$O} + \text{OH}.
\end{equation}
| [5] |
Despite its last step having a relatively large barrier of ∼170 meV in the gas phase,
39),40) this process has been assumed to actively occur in astrochemical models without experimental verification. In their model, Cuppen and Herbst determined Reaction
[5] to be an important pathway for the formation of H
2O in cold MCs.
38) In 2008, our group confirmed the occurrence of this process on solid oxygen at low temperatures for the first time.
41)
Miyauchi et al. performed experiments using an apparatus similar to that shown in Fig. 1a.41) In their experiments, reflection–absorption infrared (IR) spectrometry was used to trace the evolution of a solid O2 sample subjected to H-atom irradiation. The difference IR spectra collected after H-atom irradiation of a solid O2 sample at 10 K (Fig. 2) helped verify the formation of H2O2 and H2O. Interestingly, the shape of the OH-stretching feature in the IR spectrum, which is expected in the region 3000–3700 cm−1, indicated that the solid H2O formed by this reaction was not crystalline but amorphous, which is consistent with the characteristics of abundant ASW in the icy mantle of dust grains. Similar experiments were performed using D atoms, and the formation of D2O2 and D2O was confirmed. The formation rates of H2O2 and D2O2 were similar, as expected for barrierless reactions. However, the rate of H2O formation was one-order-of-magnitude higher than that of D2O, indicating that the last step in Reaction [5] occurred via quantum mechanical tunneling.

The formation of H2O molecules upon the co-deposition of O2 molecules and H atoms was investigated by Oba et al.42) Continuous formation of H2O and H2O2 was observed even at 40 K. A particularly important finding was that amorphous H2O ice formed was compact (nonporous), as indicated by the absence of dangling OH features in the IR spectra, which is consistent with astronomical observations of MCs.43) However, the exact morphology of ASW in space—compact or otherwise—is unclear, because the dangling OH feature was tentatively identified in a recent observation of a dense region of an MC.44)
Another H2O-formation process involving the reaction between H2 and OH radicals (H2 + OH → H + H2O) was examined by Oba et al.45) This reaction has a barrier of ∼180 meV in the gas phase,46) indicating that the thermal reaction does not occur at low temperatures such as 10 K. Using several combinations of reactants (H2, D2, HD, OH, and OD), the authors experimentally confirmed the occurrence of this reaction. Moreover, the product yield in the H-atom abstraction reaction (HD + OH → D + H2O) was one-order-of-magnitude higher than that in the D-atom abstraction reaction (HD + OH → H + HDO). This difference was explained by considering the fact that the effective mass for quantum mechanical tunneling in the former case was approximately half that in the latter case.
2.3. Successive hydrogenation of CO.
Simple organic molecules—H2CO and CH3OH—are abundantly present not only in the gas phase, but also in icy dust and some comets. These molecules appear to have evolved from CO molecules on dust through successive hydrogenation (that is, H-atom addition), photolysis, and/or cosmic-ray proton bombardment on H2O–CO ice. Because of the ubiquity of these molecules in various astronomical objects and their importance as precursors of COMs, the origin of these molecules has been intensively studied. Successive hydrogenation, expressed as follows:
| \begin{equation}
\text{CO}\overset{\text{H}}{\to}\text{HCO}\overset{\text{H}}{\to}\text{H$_{2}$CO}\overset{\text{H}}{\to}\text{CH$_{3}$O/CH$_{2}$OH}\overset{\text{H}}{\to}\text{CH$_{3}$OH},
\end{equation}
| [6] |
has been proposed as the most plausible process in many theoretical studies.
37),47) Experiments conducted by Hiraoka
et al., in which cold H atoms were sprayed onto solid CO at low temperatures, confirmed the formation of H
2CO. However, no CH
3OH was detected through IR measurements.
48),49) Watanabe and Kouchi realized that quantitative information on aspects such as the relationship between yield and H-atom dose was lacking in the preceding study. Consequently, they developed a high-flux H-atom source capable of providing the H-atom fluence expected in an MC (10
18 atoms cm
−2 in 10
5 years) to study the successive hydrogenation reactions of CO.
9) Their experiments clearly indicated that the successive hydrogenation reactions of CO occurred efficiently on the ASW surface. The formation of H
2CO and CH
3OH was observed via IR measurements of the surface under MC-relevant low-temperature and high H-atom fluence conditions.
Subsequently, several types of experiments targeting Reaction [6] on ASW were performed in the 2000s, as elaborated in a review.5) The total reaction rate is one-order-of-magnitude smaller in the experiments conducted using D atoms, presumably because the first and third steps in Reaction [6] have barriers and proceed via quantum mechanical tunneling at low temperatures.50) The conversion rate of the reaction H2CO → CH3OH at 15 K was found to be approximately half that for CO → H2CO.51) The yield of hydrogenation reactions depends significantly on the surface composition (solid CO, CO on ASW, or H2O–CO ice) and surface temperature, which affect the sticking coefficient of H atoms and their residence time on the surface.52)–54)
Based on the aforementioned experimental findings, successive hydrogenation of CO on ASW is thought to occur only at temperatures below 20 K, where H atoms can remain on the surface. Recently, we found that when CO molecules are buried deep in ASW, the successive hydrogenation reactions occur even at 70 K because certain H atoms that collide with the ASW surface can penetrate the ASW along the walls of cracks and may have a prolonged residence time.55) The penetration depths of H atoms for porous and nonporous ASW were determined to be >50 and ∼10 monolayers, respectively,56) where one monolayer of ASW has a thickness of approximately 0.3 nm. This type of reaction is important for any atom or molecule embedded in ASW.21)
2.4. Chemical desorption.
Similar to the aforementioned discussion on the efficient formation of formaldehyde and methanol on ice, diverse COMs have been experimentally confirmed to be produced on ice surfaces.17),57) Certain gas-phase COMs have been detected using radio telescopes even in cold MCs.58) Desorption should occur when these species are produced on ice and detected in the gas phase. Thermal desorption is negligible at temperatures as low as 10 K, and photodesorption is inefficient because of the extremely low flux of UV radiation in the dense MC region. Therefore, an additional desorption mechanism—reactive desorption or chemical desorption—has been proposed.59) Therein, a reaction product desorbs because of the heat of reaction. Although the heat of reaction is generally much larger than the adsorption energy in the physisorption case, the desorption of products following their formation is not obvious. In fact, during the hydrogenation of CO (Reaction [6]), most of the products of H2CO and CH3OH remain on the surface, implying that a significant fraction of the excess energy dissipates into the bulk surface. Although the exact dynamics of chemical desorption remain unclear, the occurrence of chemical desorption has been demonstrated in several laboratory experiments.60)–64) Nevertheless, the efficiency of chemical desorption should depend on the reactions, and it is often included in astrochemical models through an equation that relates the probability of chemical desorption and the excess energy of reaction.65)–68)
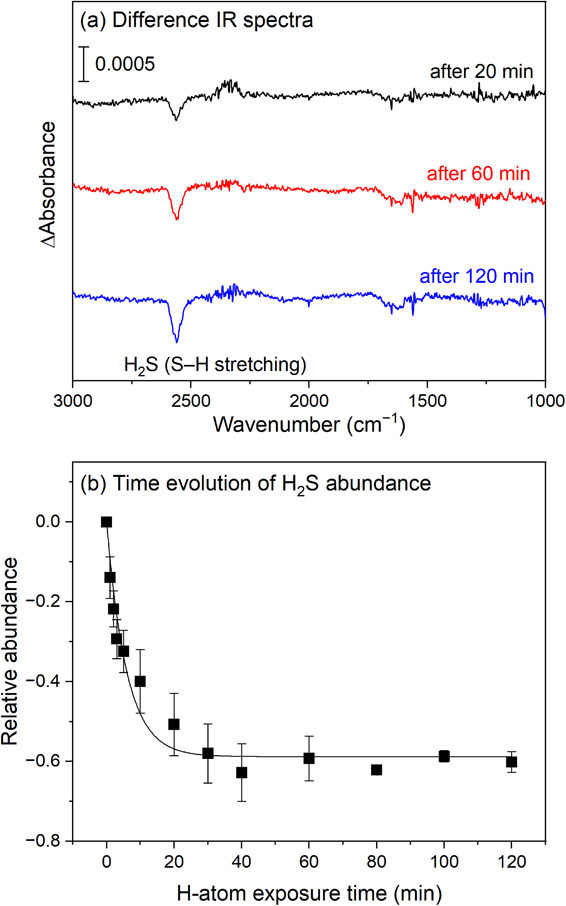
In 2018, Oba et al. analyzed the chemical desorption of H2S through in situ IR measurements.62) They investigated chemical desorption during the reactions H2S + H → HS + H2 and/or HS + H → H2S through IR measurements for the first time. The occurrence of these two reactions was confirmed by monitoring the formation of HDS and D2S upon exposing H2S to D atoms. As shown in Fig. 3, the amount of H2S adsorbed on ASW significantly decreased upon exposure to H atoms at 10 K. Because the parent and final product molecules obtained via the exposure of H2S to H atoms were both H2S as mentioned above, the decrease was presumably due to desorption of products from the surface. The exothermicity values of the reactions H + H2S → H2 + HS and HS + H → H2S are ∼0.6 and 3.8 eV, respectively, and the binding energies calculated for HS and H2S on a water cluster are both ∼0.1 eV. Based on exothermicity, Oba et al. deduced that chemical desorption is more likely to occur upon the formation of H2S. They estimated the effective cross-section for chemical desorption and found that the chemical desorption rate exceeded the expected photodesorption rate in a typical interstellar environment. Furthermore, the effective desorption cross-section is not strongly influenced by the ice structure (porous amorphous, nonporous amorphous, or crystalline) and temperature (10–30 K).63) Recently, Nguyen et al. found that phosphine molecules (PH3) on ice surfaces can also be desorbed into the gas phase via chemical desorption through the reactions H + PH3 → H2 + PH2 and/or PH2 + H → PH3.64)
The aforementioned experimental results indicate that the chemical desorption of H2S and PH3 can potentially affect the chemistry of sulfur and phosphorus in interstellar media. Furuya et al. attempted to constrain the desorption probability of H2S and PH3 per reaction event on ASW by numerically simulating laboratory experiments.69) The desorption probabilities for H2S and PH3, which are often assumed to be 1% in chemical models, were estimated to be (3 ± 1.5)% and (4 ± 2)%, respectively, using the microscopic kinetic Monte Carlo method.
2.5. Other reactions.
To acquire further insight into the chemistry of sulfur and phosphorus in MCs, we extended our investigations to H-atom reactions with relevant species. Carbonyl sulfide (OCS) is an abundant S-bearing molecule in MCs that has been found both in the gas phase and on icy grains.70),71) Thus, the OCS molecule can potentially react with H atoms on icy grains. Three possible pathways exist for hydrogen addition to OCS: cis-HOCS (addition to O), OC(H)S (addition to C), and trans-OCSH (addition to S). The three reactions have relatively large barriers, as evidenced by the values estimated for isolated systems via quantum calculations performed at the CCSD(T)/aug-cc-pVTZ level72) (25.2, 40.4, and 93.4 kJ mol−1, respectively). If the shape of the barrier in these reactions is assumed to be similar, the H-atom tunneling reaction preferentially yields cis-HOCS. Nguyen et al. performed co-deposition experiments of OCS and H atoms.73) They did not observe any mono-hydrogenated OCS, but obtained CO, hydrogen sulfide (H2S), H2CO, and CH3OH as the main products, with a minor yield of thioformic acid (HC(O)SH). Quantum chemical calculations were performed for the successive hydrogenation of OCS on ASW. Similar to the calculations performed on the isolated system, the addition of H to S, yielding OCSH, has the lowest barrier of 0.21 eV (20.3 kJ mol−1) for the first reaction (OCS + H);73) the initial product trans-OCSH readily isomerizes to cis-OSCH owing to the exothermicity of the H-addition reaction.72) A secondary reaction between OSCH and an H atom yields HOCSH, OC(H)SH, and a CO–H2S pair, with the last channel being most favorable energetically. Thus, calculations have suggested that CO and H2S are the main products and OC(H)SH is a minor product, which is consistent with the experimental results, whereas H2CO and CH3OH are thought to be produced via successive hydrogenation of the CO product.
The reactions between PH3 molecules and H(D) atoms on ASW have been studied both experimentally and theoretically.74) The H–D substitution reaction of PH3 and the D–H substitution reaction of PD3 occurred upon D- and H-atom irradiation, respectively, at 10 K on ice surfaces. Although the H- and D-abstraction reactions with a barrier height of ∼0.13 eV likely occurred via quantum mechanical tunneling, the H–D substitution reaction of PH3 was slightly slower than the D–H substitution reaction of PD3 under the employed experimental conditions. These results indicate that the relative rates of the H–D and D–H substitution reactions were dictated not by the kinetic isotope effect in the tunneling, but by other elementary processes such as atomic diffusion on ice. Based on their experimental observations, the authors speculated the presence of a deuterated phosphine isotopolog (PH2D) in PH3-rich interstellar environments. This topic (that is, H-atom reactions with OCS and PH3) has been reviewed from the viewpoint of theoretical calculations.75)
The isomerism of molecules and their mechanisms in the interstellar environment are unresolved questions in the chemistry of organic molecules in space.76) Formic acid (HC(O)OH) is the simplest organic acid exhibiting cis–trans conformational isomerism.77),78) Formic acid is mostly (>99.9%) in its trans conformation under ambient conditions because of its thermodynamic stability, whereas abundant cis-formic acid has been identified in the interstellar photodissociation regions (up to 33%) and cold cores (6%).79),80) Molpeceres et al. attempted to explain the abundance of the cis-form based on the hydrogen abstraction reactions.76) Theoretical calculations revealed a clear dependence of the H-abstraction reactions on the isomer of the reactant, with the rate constants for the cis-isomer at ∼50 K being five-orders-of-magnitude greater than those of the trans-isomer. These results indicated that the H-abstraction reactions reduced the abundance of the cis-form, in contrast to significant presence of cis-form found by astronomical observations. To confirm these theoretical predictions, co-deposition experiments were performed using trans-formic acid and D atoms at 10 K. C–D stretching features were not observed in the IR spectra, indicating that the reaction trans-HC(O)OH + D → cis-HOCO + HD did not occur.76) Although this result is consistent with theoretical predictions, the reaction cis-HC(O)OH + D → trans-HOCO + HD was not investigated because they do not have the means to prepare cis-formic acid. Therefore, additional experiments based on IR-laser-induced trans-to-cis isomerization81) should be conducted to study the isomer-dependent reactivity in the H-abstraction reaction. Nevertheless, other mechanisms such as those leading to the preferential formation of the cis-form over the trans-form should be clarified in the future to explain the astronomically observed abundance.
Hydrogen reactions with aromatic molecules, particularly polycyclic aromatic hydrocarbons (PAHs), are considered important for their relevance to H2 formation.56),82)–84) Compared to the extensive investigations on gas-phase reactions, limited experimental studies have been conducted on the reaction between hydrogen and PAHs on ice. Hydrogenation reactions in solid benzene (C6H6) were studied by Hama et al.,85) who found that the reactivity depended significantly on the crystallinity (amorphous or crystalline) of the sample. The considerably higher reactivity in the amorphous solid was attributed to the difference in geometric constraints between the amorphous and crystalline surfaces, where the reactive benzene, that is “dangling benzene”, exists in considerably larger numbers on the amorphous surface. Although the H-atom addition reactions were found to proceed via quantum mechanical tunneling, a large kinetic isotope effect—characteristic of the tunneling reaction—was not observed.86) The significant reduction in the kinetic isotope effect was due to the control of chemical kinetics by the surface diffusion of the H/D atoms, which proceeds via a thermal hopping mechanism with a small kinetic isotope effect.
3. Deuterium fractionation
3.1. Formaldehyde and methanol.
From astronomical observations, high D/H ratios (abundance ratios of deuterated molecules and their equivalents having only hydrogen) have often been observed in interstellar molecules, particularly in H2O, NH3, H2CO, and CH3OH. In the case of CH3OH, the abundance of its singly deuterated isotopologue—CH2DOH—in the star-forming region IRAS 16293-2422, has been reported to be ∼30% of the total CH3OH content.87) Moreover, even its triply deuterated isotopologue—CD3OH—has been detected in the same region. The formation of multiple deuterated molecules cannot be explained by ion–molecule reactions in the gas phase; therefore, the reactions on icy grains are likely responsible for the significant deuterium enrichment in CH3OH.
As described in Section 2.3, the formaldehyde and methanol in MCs are predominantly formed via successive hydrogenation of CO (Reaction [6]). Nagaoka et al.88) speculated that the reactions of D atoms during the formation of formaldehyde and methanol on dust play a role in the deuterium enrichment; they experimentally observed deuterium-enriched methanol upon exposing solid CO to H and D atoms simultaneously. Analyzing the time variation of the products of formaldehyde, methanol, and their deuterated isotopologues, the authors found that the H–D substitution reactions in methanol and formaldehyde are key routes for deuterium enrichment (Section 3.1.5).88) Figure 4 shows the network of reactions with H and D atoms in the CO–formaldehyde–methanol system. The network includes H (D) addition and H–D (D–H) substitution reactions on the reactant species. We experimentally examined each pathway in the network, as follows, to determine the process most responsible for deuterium enrichment in methanol:

When solid CO was irradiated with D atoms, fully deuterated formaldehyde (D2CO) and methanol (CD3OD) were formed, validating the successive addition of D atoms to CO molecules.50) As expected for tunneling reactions, the effective reaction rate for CO + D → DCO was one-order-of-magnitude smaller than that for CO + H → HCO, indicating that the addition of D to CO was not responsible for deuterium fractionation.
3.1.2. H2CO + D.
In this experiment, H2CO was deposited onto ASW and subsequently irradiated with D atoms.89) Immediately after the D-atom irradiation was initiated, the H2CO content decreased and HDCO was formed, followed by the production of D2CO. This indicates that the H atoms in H2CO were replaced with D atoms, that is, the occurrence of H–D substitution reactions. Two H–D substitution reaction mechanisms are possible in this regard: sequential tunneling H-abstraction (H2CO + D → HCO + HD) and D-addition (HCO + D → HDCO), and direct H–D exchange (H2CO + D → HDCO + H). The sequential and direct mechanisms may compete with each other because of their similar activation energies (1899 and 1799 K, respectively, in the gas phase90)). Longer D-atom irradiation did not yield deuterated methanol isotopologues (CH2DOD, CHD2OD, or CD3OD), but produced D2CO. These results suggest that H–D substitution reactions occur preferentially over D-addition reactions. If the D–H substitution reactions, which reduce the degree of deuterium fractionation, are slow, the observed H–D substitution reaction in H2CO may involve deuterium fractionation of the formaldehyde in MCs.
3.1.3. D2CO + H.
H-atom irradiation experiments were performed for D2CO deposited on ASW.89) After the H-atom irradiation, CHD2OH, HDCO, H2CO, and CH3OH were generated, with CHD2OH being the major product. The formation of HDCO and H2CO occurred via H–D substitution, and the H2CO product was further hydrogenated to yield CH3OH. After prolonged H-atom irradiation, the fluence of which corresponded to that in MCs for 106 years, CHD2OH and CH3OH were obtained in similar yields. A clear difference was observed between the H2CO + D and D2CO + H reactions; only the substitution reaction occurred in the former system, and both substitution and addition reactions occurred in the latter system.
3.1.4. CH3OH + D.
CH2DOH, CHD2OH, and CD3OH were formed upon the D-atom irradiation of a solid CH3OH sample at 10 K.91) The time evolutions of CH2DOH, CHD2OH, and CD3OH indicated that they were produced through a sequence of substitution reactions:
| \begin{equation}
\text{CH$_{3}$OH}\overset{k_{1}}{\to}\text{CH$_{2}$DOH}\overset{k_{2}}{\to}\text{CHD$_{2}$OH}\overset{k_{3}}{\to}\text{CD$_{3}$OH},
\end{equation}
| [7] |
where
k1,
k2, and
k3 are the rate constants of each step. However, methanol isotopologues with deuterated hydroxyl groups (such as CH
3OD and CD
3OD) were not detected. The absence of these species suggests that the hydrogen atoms in the methyl group preferentially react with D atoms at 10 K. The H–D substitution preferentially involves H-abstraction by D atoms (such as CH
3OH + D → CH
2OH + HD) and D addition (CH
2OH + D → CH
2DOH) rather than the direct exchange of H and D atoms (such as CH
3OH + D → CH
2DOH + H) with larger barriers. According to quantum chemical calculations, reactions involving H-abstraction from the methyl group (CH
3OH + D → CH
2OH + HD) and from the hydroxyl group (CH
3OH + D → CH
3O + HD) have activation barriers of 0.33 and 0.53 eV, respectively.
92) Therefore, the former reaction is more efficient than the latter, which is consistent with experimental observations; however, the reaction rate constants should be evaluated based on the potential energy surface calculated for the species on ice, including the tunneling effect.
3.1.5. CH3OH-dn + H(D).
CH3OH-dn solids (n = 1–4), representing deuterated methanol with n deuterium atoms, were irradiated with H.91) However, D–H substitution reactions were not observed in any scenario, indicating that after the CH3OH-dn compounds were produced on ice, they did not transform into CH3OH through surface reactions with H atoms.
Experiments were performed using CH3OH isotopologues (CH3OH, CH2DOH, and CHD2OH) and D atoms to verify the relative rate of Reaction [7]; this parameter was estimated by simply fitting the time-decay curves acquired upon irradiating the isotopologues with D-atoms to a single-exponential function. The relative rates (k1/k2/k3) were determined to be 1/∼0.5/∼0.3. These results indicate that the relative reaction rates are effectively scaled by the number of H atoms in the methyl group, that is, the probability of a reacting D atom locating the H atom is highest in CH3OH and lowest in CHD2OH. Moreover, small differences in the C–H bond strengths of these isotopologues can affect the relative rate.
3.1.6. CO + H(D) reactions and comparison with astronomical observations.
The aforementioned experimental results clearly demonstrate that the deuterium fractionation in formaldehyde and methanol proceeds via H–D substitution reactions, and that the deuterium addition reactions in CO and D–H substitution reactions in CH3OH-dn are inefficient. To estimate the abundance of H2CO-dn and CH3OH-dn on dust grains in MCs, Nagaoka et al.88) performed simulation experiments in which a solid CO sample was irradiated simultaneously with H and D atoms having a flux ratio (D/H) of ∼0.1, which was adopted from theoretical models. Various deuterated products—HDCO, D2CO, CH2DOH, CHD2OH, and CD3OH—were formed during the H- and D-atom irradiation.
The deposition of atoms with fluences corresponding to those expected in MCs for 1–2 × 106 years yielded the following H2CO-dn/H2CO and CH3OH-dn/CH3OH ratios (astronomically observed values corresponding to the low-mass protostar IRAS 16293-242287),93) are shown in parentheses): HDCO/H2CO = 0.1–0.5 (0.13–0.16), D2CO/H2CO = 0.03–0.06 (0.05–0.06), CH2DOH/CH3OH = 0.1–0.3 (0.3), CHD2OH/CH3OH = 0.15–0.3 (0.06), and CD3OH/CH3OH = 0.02–0.1 (0.01). These species are considered to have formed on icy grains sublimated by heat from the protostar. Because this protostar is relatively young (104 years old), the aforementioned species have not been subjected to gas-phase processes. Therefore, the observed H2CO-dn/H2CO and CH3OH-dn/CH3OH ratios are representative of those achieved through reactions on icy grain surfaces.94) A comparison between the laboratory simulation and observations indicates that the deuterium fractionation in formaldehyde and methanol was adequately reproduced in the simulation experiment. In particular, a high abundance of multiple deuterated species was observed experimentally. This level of abundance has not been achieved in chemical models that do not include the H–D substitution reactions. The experimental findings on the H–D substitution reactions have deepened our understanding of deuterium fractionation.
3.2. Other molecules.
3.2.1. Small molecules.
D-atom irradiation experiments have also been performed on solid H2O, NH3, and CH4, which are deuterium-enriched interstellar molecules. Although these experiments were preliminary, we did not observe the H–D substitution reactions at 10 K. These results indicate that deuterated isotopologues of the aforementioned species can be formed only during production, for example, via OH + D → HDO and/or gas-phase reactions. Consequently, the degree of deuterium fractionation in these species is expected to be lower than those of H2CO and CH3OH. According to astronomical observations, the deuterium fractionation degree in H2CO and CH3OH is approximately one-order-of-magnitude larger than that of H2O and CH4.
The absence of H–D substitution reactions in these species can be attributed to reaction thermicity. The H–D substitution reaction is considered a two-step reaction, for example, CH4 + H → CH3 + H2 and CH3 + D → CH3D. According to quantum chemical calculations, the reaction CH4 + H(D) → CH3 + H2 (HD) is endothermic (ΔH ∼ 3 kJ mol−1) and has an activation barrier (Ea) of ∼60 kJ mol−1.92) This reaction cannot occur at low temperatures such as 10 K, even through quantum mechanical tunneling. However, the ΔH and Ea values for the reaction CH3OH + H → CH2OH + H2, whose occurrence at 10 K has been verified (Section 3.1), are reported to be −33 and 36 kJ mol−1, respectively. The H–D substitution reactions in other astrochemically relevant molecules were investigated by our group, and the results are briefly discussed in the following subsections.
3.2.2. Ethanol.
Ethanol (CH3CH2OH) is a highly abundant COM found in star-forming regions. Although deuterated ethanol has not been astronomically detected, its production is believed to occur predominantly through dust-surface reactions. Therefore, Oba et al. performed D(H)-atom irradiation experiments on solid ethanol at 10 K.95) H–D substitution reactions were found to occur on the ethyl (CH3CH2) group but not on the hydroxyl (OH) group. When the deuterated ethanol solid was irradiated with H atoms, the D–H substitution reactions occurred only on the ethyl group. These results are consistent with those of methanol, where only the hydrogen (deuterium) atoms in the methyl group undergo substitution reactions. The rates of H–D and D–H substation reactions were found to be similar to each other whereas the reaction involves quantum mechanical tunneling. They considered that other elementary processes such as atomic diffusion on ice dominate the reaction rates. The deuterium fractionation of dimethyl ether (CH3OCH3) subjected to H–D substitution has also been reported.96) In the case of dimethyl ether, Oba et al. found that the H–D substitution rates for pure solid CH3OCH3 were 20 times greater than that for the CH3OCH3–H2O complex deposited on water ice, indicating a “negative” catalytic effect of water in the H–D substitution reactions. Based on experimentally determined reaction rates, the authors estimated the abundance ratios for CH3CHDOH/CH3CH2OH and CH2DCH2OH/CH3CH2OH after 106 years under MC conditions to be ∼1 × 10−2 and ∼6 × 10−3, respectively, and predicted the existence of deuterated ethanol as well as deuterated dimethyl ether in space.95)
3.2.3. Methylamine.
Methylamine (CH3NH2) is considered one of the simplest building blocks of biomolecules in interstellar media and has been found in the comet Wild 2.97) This species would be formed both in the gas phase and on interstellar grains. Regardless, methylamine can be presumed to exist on the surface of icy grains at low temperatures (∼10 K) and undergo several physicochemical processes, including H(D)-atom reactions. Extensive laboratory simulation experiments have been conducted on this molecule under astrophysical conditions, and the formation of glycine—the simplest amino acid (NH2CH2COOH)—from methylamine has been reported.98),99) However, less attention has been paid to the deuterium fractionation in this molecule. If methylamine serves as a building block for amino acids or other biomolecules, its deuterium enrichment may propagate to its products. Therefore, Oba et al. investigated H–D substitution reactions.100)
After the exposure of solid CH3NH2 to D atoms, H–D substitution reactions were confirmed to occur on both the methyl (CH3) and amino (NH2) groups, which yielded CD3ND2 as the terminal product. In the experiments involving exposure of CD3ND2 to H atoms, CH3NH2 was generated as the product. Experiments on partially deuterated isotopologues (CH3ND2 or CD3NH2) revealed that the H–D (D–H) substitution reactions on the methyl group were faster than those on the amino group. The rates of D–H substitution reactions were found to be slower than H–D substitution reactions, indicating that H(D)-abstraction step via quantum mechanical tunneling is the rate-determining step. According to quantum chemical calculations, the H–D substitution in methylamine occurs either via an abstraction–addition mechanism or through the formation of an intermediate such as [CH3NH2D], followed by a reaction with another H(D) atom. Numerical simulations indicated that the CH2DNH2 isotopologue can be produced on icy grain surfaces within the relevant timescale (106 years); the abundance ratio, CH2DNH2/CH3NH2, can reach 10−1 under the assumption of an atomic D/H ratio of 10−2.
4. Direct detection of radicals on ASW
4.1. Behavior of OH radicals on ice.
4.1.1. Detection of OH radicals on ice by PSD-REMPI.
Hydroxyl (OH) radicals are thought to be abundantly produced on icy grains because they are an intermediate in the formation of H2O—the most abundant component of the ice mantle—and are a UV photolysis product of H2O. Therefore, the OH radical is a critical radical in chemical evolution, that is, the formation of COMs on icy grains.13) The chemical reactions involving OH radicals are activated when the temperature of the dust increases in star-forming regions. To assess the OH chemistry in chemical models, information on the surface diffusion of OH on ice prior to the reaction is desired. That is, in situ detection of the OH radicals on ice is desirable. Because of the high reactivity of OH radicals, achieving a surface density of OH on ice sufficient for detection using conventional experimental methods is difficult. Therefore, the methods typically used for analyzing solids, such as Raman spectroscopy, IR spectroscopy, and electron spin resonance spectroscopy, are inapplicable. Therefore, a new method must be established to sensitively detect OH radicals on ASW. Recently, we successfully utilized the PSD-REMPI method to detect OH radicals on ice. Details concerning the OH radical detection by PSD-REMPI101) are provided in this subsection, and its application to the surface diffusion of OH radicals on ice102) is described thereafter (Section 4.1.2).
The experimental apparatus used for OH radical detection is similar to that shown in Fig. 1a. The OH radicals on ASW are produced by photolyzing H2O using UV photons from a conventional deuterium lamp. The generated OH radicals are desorbed using a PSD laser and subsequently ionized using a REMPI laser at ∼1 mm above the surface. The (2 + 1) REMPI scheme via D 2∑− (v′ = 0) is used (see the rotationally resolved PSD-REMPI spectra in Ref. 101). “Delay time spectra”, which reflect the translational energy distribution of desorbed OH radicals, are acquired by changing the delay between the PSD and REMPI lasers, which corresponds to the TOF of OH from the surface to the focal point of the REMPI laser (Fig. 5a). The observed distribution can be fit appropriately using flux-weighted Maxwell–Boltzmann distributions at ∼3000 K (0.26 eV). This extremely high translational energy cannot be attributed to gentle desorption processes, such as the phonon-induced desorption suggested previously for the PSD of the H atom from ASW.28),30) The PSD-REMPI intensity of OH radicals is independent of the ASW thickness (Fig. 5b) and linearly dependent on the PSD laser power (Fig. 5c), indicating that the desorption is induced by a one-photon process occurring on the surface.

To gain further insight into the mechanism of the PSD process, we performed quantum chemical calculations on the OH radicals adsorbed on ASW.75),101) First, an ice cluster model for ASW comprising 159 H2O molecules was prepared. Second, the vertical excitation energy for the 2Σ+ ← 2Π transition, which corresponds to the A–X transition of isolated OH, was calculated for 18 types of binding sites on OH radicals using time-dependent density functional theory. The calculated excitation energies were generally in the range 3–4 eV (corresponding to 400–300 nm photons), which is similar to that calculated for the H2O–OH complex.103) We observed a largely red-shifted transition at 2.14 eV (580 nm) for a strongly bound OH radical on ice having a binding energy of 0.61 eV. Although this excitation energy does not exactly match the photon energy of the PSD laser (532 nm), certain OH radicals in the strongly bound sites of ASW would absorb the photons at 532 nm to be excited to an electronically excited state. These results indicate that the OH radicals detected by PSD-REMPI originate from those adsorbed on the strongly bound sites. Although the exact mechanism leading to the desorption of OH remains unclear, we proposed a mechanism based on preliminary calculations of the potential energy surface in the excited state. A conical intersection was found near the bottom of the excited state. The transition to another repulsive electronic state via this conical intersection may lead to desorption. Indeed, the measured translational energy of photodesorbed OH radicals (∼0.26 eV) was consistent with the calculated energy gap (0.24 eV) between the conical intersection and the OH + ice dissociation limit. High-level quantum chemical calculations should be performed to elucidate the exact desorption mechanism; however, calculations based on the ASW model are beyond computational feasibility. Therefore, calculations performed using OH–(H2O)n with a sufficiently large n and a sufficiently high level of theory are imperative to obtain further insight into desorption dynamics.21)
The PSD-REMPI technique for OH radicals has been adopted to study the surface diffusion of OH radicals on ice (see next subsection).102) Furthermore, the behavior of OH radicals has been clarified for scenarios with coexisting electrons on the surface, which has led to the discovery of negative current conductivity, that is, proton-hole transfer in ice. Details regarding the latter topic can be found in certain original research papers104),105) and reviews.20),21)
4.1.2. Surface diffusion of OH radicals on ice.
The activation energy for the surface diffusion of OH radicals (Esd) is important for evaluating its reactions with less-mobile species, where the diffusion of OH radicals controls the reaction rate. The experimental determination of Esd is described henceforth. During the continuous production of OH radicals on ASW by UV photolysis, the PSD-REMPI signal for OH radicals on ice reaches a steady state, that is, the signal intensities do not change with time. In this scenario, the production and loss of OH radicals are balanced. The steady-state signal intensities were recorded at temperatures between 54 and 80 K and found to decrease with increasing temperature, from approximately 60 to 80 K (Fig. 6a). In other words, the loss of OH radicals accelerated at higher temperatures. Under steady-state conditions, the number density of surface OH radicals, [OH], can be expressed as
| \begin{equation}
0 = \frac{d[\text{OH}]}{dt} = f\sigma c[\text{H$_{2}$O}] - 2k_{\text{OH-OH}}[\text{OH}]^{2} - k_{\text{des}}[\text{OH}],
\end{equation}
| [8] |
where
kOH-OH is the rate constant for the recombination reaction of two OH radicals, and
kdes is the rate constant for thermal desorption. In
Eq. [8], the reaction between OH radical and H atoms is not taken into account because photolytically produced H atoms readily desorb from the surface at experimental temperatures above 54 K. The production rate of OH radicals is presumed to be temperature-independent and is described as
fσ
c[H
2O], where
f, σ, and
c are the flux of UV photons, absorption coefficient, and quantum yield of OH radicals upon a photon absorption event, respectively. Only the recombination-dominating case is considered henceforth, whereas the desorption-dominating case is discussed in the original article.
102) Because the OH + OH recombination reaction is considered barrierless and its rate is diffusion-limited,
kOH-OH can be expressed as
| \begin{equation}
k_{\text{OH-OH}} = D_{\text{s}} = D_{0}\exp\left(- \frac{E_{\text{sd}}}{k_{\text{B}}T} \right),
\end{equation}
| [9] |
where
Ds is the diffusion coefficient (diffusive area per unit time),
D0 is a prefactor,
Esd is the activation energy for surface diffusion,
kB is Boltzmann’s constant, and
T is the temperature. When the third term on the right side of
Eq. [8] is neglected, the two equations can be combined as follows:
| \begin{equation}
-{\ln}[\text{OH}]^{2} = \ln D_{0} - \ln \frac{f\sigma c[\text{H$_{2}$O}]}{2} - \frac{E_{\text{sd}}}{k_{\text{B}}T}.
\end{equation}
| [10] |
This Arrhenius-type relationship indicates that
Esd can be determined from the slope of a plot between −ln[OH]
2 and
T−1. Because [OH] is proportional to the PSD-REMPI intensity
IOH, the −ln(
IOH2) values can be plotted as a function of
T−1 (
Fig. 6b), yielding an
Esd of 0.14 ± 0.01 eV. This value is larger than or equivalent to those assumed in theoretical models.
18),106) The prefactor
D0 in
Eq. [9] can be expressed as a
2ν, where a and ν are the single-hopping distance and frequency factor, respectively. Using an ‘a’ value of 0.3 nm, which is the average distance between two H
2O molecules on ice, and the typical frequency factor for thermal hopping (ν = 10
12 s
−1), the diffusive area (
Ds) can be obtained as a function of temperature. The time required for diffusing through 100 nm, which is equivalent to the characteristic diameter of dust, can also be estimated. When an
Esd value of 0.14 eV (1650 K) is used, OH radicals can diffuse over 100 nm on icy dust surfaces within 10
5 years at temperatures above 36 K, where the typical lifetime of MCs (10
5 years) is assumed. Thus, we speculate that the diffusive reactions of OH radicals on icy grains begin to be activated at ∼36 K.

Because the experimental determination of Esd is more difficult than that of Edes, which can be derived from TPD experiments, astrochemical models often use a simple scaling relationship Esd = fEdes, where the scaling factor f is set to be a universal value regardless of the adsorbed molecules. Consequently, a wide range of Esd values are used in models, depending on the adopted f and Edes values. Based on the determination of Esd for stable molecules,107)–109) Furuya et al. found f to be molecule-dependent.110) Additionally, through astrochemical model calculations, they confirmed the presence of the following key species whose Esd significantly affected the model predictions: O and N atoms; and HCO, CH2, and NH radicals. Therefore, the determination of Esd for these atoms and radicals is essential for obtaining better predictions using astrochemical models, and the PSD-REMPI method should play an important role in this regard.
4.2. COM formation studied using the ion-pickup method.
4.2.1. Apparatus for Cs+-ion pickup.
As mentioned above, several technical requirements exist for conducting experiments on the behavior of free radicals and trace species on ice. The method should be highly sensitive to low densities, surface selective to distinguish species on the surface and in the bulk, and (ideally) nondestructive to track the time evolution of species. The PSD-REMPI method has been demonstrated to fulfill these requirements and has been successfully applied to H atoms and OH radicals on ice. One disadvantage of the PSD-REMPI method is that the target species must be gently photodesorbed by weak laser radiation; therefore, this technique cannot be feasibly employed for larger species. Moreover, the REMPI scheme cannot be applied to all species (such as methane). Consequently, we developed the “Cs+-ion pickup” method as an alternative to PSD-REMPI.
The Cs+-ion pickup method is a reactive-ion-scattering technique that is utilized in surface science and was developed by Kang et al. at Seoul National University.111) Originally, this method was extensively used to examine the physicochemical processes of molecules on clean, well-defined surfaces. However, its application to species on rough surfaces, such as ASW, has not been successful because of the roughness-induced decrease in detection efficiency. Therefore, we focused on improving the sensitivity of the Cs+-ion pickup method to permit in situ, nondestructive monitoring of the radical reactions on ASW. Because the fundamentals related to this method have been extensively described elsewhere,111) only the operating scheme of this method and the performance of our newly developed apparatus are described herein.
Schematics of the Cs+-ion pickup method are shown in Fig. 7a. Ice samples are produced on a cooled Al substrate located at the center of an ultrahigh-vacuum chamber. A low-energy Cs+ beam (kinetic energy, ∼20 eV; flux, ∼1 nA) is injected into the chamber at an angle of 45° with respect to the normal to the substrate surface. When Cs+ ions (mass number 133) scatter on the substrate, certain Cs+ ions pickup molecules (mass number M) from the outermost surface and scatter as Cs+-ion/molecule complexes (mass number 133 + M). The scattered ions are subjected to mass analysis using a QMS, whose conventional ionization source is removed. Specially designed ion lenses, which are installed in front of the QMS, significantly improve the detection sensitivity, which is up to ∼103 times greater than that of the conventional reactive-ion-scattering setup.

Figure 7b shows the mass spectrum of an ASW sample at 10 K acquired via Cs+-ion pickup. The strongest signal at m/z 133 is attributed to the primary Cs+ ions, whereas the peak at m/z 151 (133 + 18) originated from H2O. As shown in Fig. 7b, isotopologues of water—H217O and H218O—were also detected at their natural abundances of approximately 0.04% and 0.2%, respectively. These results suggest that a detection limit of ∼10−4 monolayer coverage (1 monolayer = 1 × 1015 molecules cm−2) was achieved.
However, this method cannot separate structural isomers. As described in the next subsection, we distinguished isomers by acquiring mass spectra during the sample warm-up to identify the specific sublimation temperature of the target molecule; this method is analogous to the TPD analysis involving ionization and detection of sublimating species.
4.2.2. Formation of methyl formate on ice.
Among the COMs observed in MCs and star-forming regions, the structural isomers of C2H4O2—methyl formate (MF; HCOOCH3), acetic acid (CH3COOH), and glycolaldehyde (HOCH2CHO)—are attracting considerable attention because of their known abundances; thus, understanding their formation pathways will provide insight into the chemical evolution in interstellar media. Among the aforementioned isomers, MF is most abundant in various astronomical objects.112)–114) Therefore, the formation mechanism of MF has been primarily targeted in laboratory experiments. Because solid methanol is thought to exist on the ice mantle and is thus an important precursor of the aforementioned COMs, pure solid methanol has been subjected to radiolysis and photolysis experiments.115)–117) Although MF was formed in these experiments, other products such as ethylene glycol (EG; HOCH2CH2OH) and ethanol (Eth; CH3CH2OH) were present in greater amounts. These product-yield-based relationships are inconsistent with astronomical observations, where MF is considerably more abundant than EG and Eth. The aforementioned compounds can be produced via the following reactions involving CH2OH (Eqs. [11] and [12]) and CH3O radicals (Eq. [13]):
| \begin{equation}
\text{CH$_{2}$OH} + \text{CH$_{2}$OH} \to \text{HOCH$_{2}$CH$_{2}$OH (EG)},
\end{equation}
| [11] |
| \begin{equation}
\text{CH}_{2}\text{OH} + \text{CH}_{3} \to \text{CH}_{3}\text{CH}_{2}\text{OH (Eth)},
\end{equation}
| [12] |
| \begin{equation}
\text{CH$_{3}$O} + \text{HCO} \to \text{HCOOCH$_{3}$ (MF)}.
\end{equation}
| [13] |
Based on these reaction pathways, the formation of CH
2OH dominates CH
3O in the photolysis of solid methanol. We believe that the discrepancy between the experiments and astronomical observations arises from the composition of the samples, that is, the use of pure solid methanol in the experiments. Our recent study suggested that methanol does not exist as a pure solid, but tends to be in close contact with H
2O molecules because CO, the parent molecule of methanol (Section 2.3), forms an island of solid CO and does not cover the ASW surface (Section 5).
108),109) Consequently, methanol formation occurs at the ASW/solid-CO boundary, and the produced methanol is expected to be adjacent to both CO and H
2O molecules. The UV photolysis product of methanol in this environment may differ from that of pure solid methanol. Therefore, UV irradiation experiments should preferentially be conducted on methanol molecules adsorbed onto ASW (H
2O-rich and CH
3OH-poor conditions). However, these experiments have not been realized because of the experimental difficulties in detecting COM products or their precursors using conventional methods such as IR spectroscopy. Using the highly sensitive Cs
+-ion pickup method to detect reactants, radical intermediates, and products, Ishibashi
et al. demonstrated that MF is efficiently produced upon UV irradiation of methanol on water ice at 10 K.
118)
The pickup mass spectrum collected during the UV irradiation of methanol adsorbed onto ASW at 10 K is shown in Fig. 8a. In this spectrum, the peaks induced by UV irradiation (indicated in red) include those of OH (17), HCO (29), and CH3O or CH2OH (31) radicals and the stable products of CO (28), H2CO (30), C2H4O2 (60), and C2H6O2 (62); the mass numbers of species are indicated in parentheses. Notably, only the CO and H2CO products were identified through the IR measurements. Unfortunately, the structural isomers, that is, masses 31, 60, and 62, cannot be distinguished in the pickup mass spectrum. Assignments were made for the stable products (masses 60 and 62) by acquiring their pickup spectra during sample warm-up. After UV photolysis, the sample temperature was gradually increased, and the mass-60 signal was continuously acquired (upper panel of Fig. 8b). The signal intensity dropped rapidly at ∼125 K due to the sublimation of the product with a mass of 60. Reference measurements were performed by depositing MF, acetic acid, and glycolaldehyde vapors on ASW; the temperature at which MF disappeared in the pickup spectra (125 K; lower panel of Fig. 8b) was remarkably consistent with that of the UV-photolysis product. Similarly, the product with a mass of 62 was assigned to methoxymethanol (MM; CH3OCH2OH) from reference measurements by depositing MM and EG vapors on Al substrate.

The formation of MM occurred via the reaction:
| \begin{equation}
\text{CH$_{3}$O} + \text{CH$_{2}$OH} \to \text{CH$_{3}$OCH$_{2}$OH (MM)}.
\end{equation}
| [14] |
A comparison of Reactions
[11] and
[14] suggests that the relative yields of MM and EG depend on the abundance ratio of CH
3O and CH
2OH radicals. EG and Eth were the major products derived from the UV-irradiated solid methanol, indicating that the CH
2OH radicals were predominantly produced by UV photolysis. However, the dominant formation of MM via UV photolysis of methanol on ASW indicates that the content of CH
3O was higher than that of CH
2OH in this scenario. To clarify the difference between the experiments on pure solid methanol and methanol on ASW, an additional pathway of the reaction on ASW should be considered, as described below:
| \begin{equation}
\text{H$_{2}$O} + h\nu \to \text{OH} + \text{H},
\end{equation}
| [15] |
| \begin{equation}
\text{CH$_{3}$OH} + \text{OH} \to \text{CH$_{3}$O} + \text{H$_{2}$O}.
\end{equation}
| [16] |
By including this reaction, the CH
3O/CH
2OH ratio is expected to become higher than that in the UV photolysis of pure solid methanol. As shown in
Fig. 8a, OH radicals are produced during the UV irradiation of methanol on ASW. The OH signal does not appear in the early stages of UV irradiation but starts to increase after the MM production is terminated, indicating that the OH radicals are rapidly consumed via Reaction
[16]. Subsequently, the MM-to-MF conversion pathway was investigated by irradiating the MM deposited on an Al substrate. The conversion of MM to MF under UV irradiation was clearly observed:
| \begin{align}
&\text{CH$_{3}$OCH$_{2}$OH (MM)} + h\nu \\
&\quad \to \text{HCOOCH$_{3}$ (MF)} + \text{2H (or H$_{2}$)}.
\end{align}
| [17] |
Through experimental determination of the reaction pathways, a formation path diagram was constructed for MF and related COMs formed via radical–radical reactions driven by CH3O radicals on the interstellar dust surface (Fig. 8c). After the production of trace amounts of methanol on icy grains, COMs—such as MF—can be synthesized in cold MCs. In realistic icy dust grains, where CH3 and HCO radicals also exist, the formation of dimethyl ether (CH3O + CH3 → CH3OCH3) and the direct formation of MF (CH3O + HCO → HCOOCH3) may occur. The COMs originating from CH3O radicals (that is, MM, MF, and dimethyl ether) are more abundant than those originating from CH2OH radicals (EG and GA) in various astronomical objects,17),115) which is consistent with the mechanism illustrated in Fig. 8c. Finally, OH radicals were confirmed to facilitate the formation of various COMs, particularly in cold MCs.21),102)
5. Other important processes
5.1. Ortho-to-para nuclear spin conversion of H2.
The H2 molecule has two nuclear spin isomers: ortho and para (total nuclear spin, 1 and 0, respectively).119) According to the Pauli exclusion principle, ortho- and para-H2 molecules are coupled with the rotational states of odd and even quantum numbers, respectively. The energy difference between the lowest energy state of the isomers is 14.7 meV. In the gas phase, interconversion between the ortho and para species is forbidden, and nuclear spin conversion (NSC) occurs only via proton or H-atom exchange on a conversion timescale of 105–107 years. Because of this remarkably slow conversion, ortho- and para-H2 are often considered different species in astrochemistry. The ortho-to-para abundance ratio (OPR) of H2 is an important parameter that influences the chemistry and physics of H2 in astrophysical environments. For example, the OPR affects chemical evolution processes, such as deuterium fractionation in the gas phase.120) Moreover, because of the different heat capacities of ortho- and para-H2, the OPR affects the gas dynamics of core formation during star formation.121)
The OPR at thermal equilibrium is determined using the rotational Boltzmann distribution, and it approaches the statistical value of 3 at temperatures above ∼200 K.119) In the warm gas of molecular shock having a kinetic temperature above 300 K, the OPR is lower than the statistical value (0.5–2.0),122) indicating that the OPR is off-equilibrium with the circumstances. In other words, the OPR in astronomical objects cannot be estimated from their temperature. Moreover, the OPRs of interstellar H2 in MCs cannot be readily determined through astronomical observations because of the absence of a dipole moment in H2. Therefore, to elucidate the implications of astronomically observed OPRs, the possible mechanisms underlying the NSC of H2—particularly in MCs—should be clarified.
Compared to the gas-phase mechanisms, the NSC on dust grain surfaces is less understood. By combining the sublimation method (PSD or TPD) and detection technique (REMPI), which can selectively ionize ortho- and para-H2, we determined the NSC timescale on the surfaces of ASW and bare grain surface models (amorphous silicate and DLC) as a function of surface temperature.123)–125) The surface temperature dependence of the NSC time constants is plotted in Fig. 9. The NSC of H2 occurred rapidly on these surfaces (within ∼103 s). The mechanisms to explain the temperature dependent NSC rates were proposed in original papers.123)–125) To determine the potential effects of the NSC on dust grains on the gas-phase OPR under astrophysical conditions, we performed numerical simulations considering the rate of collision between the gaseous ortho-H2 and the grain surface, sticking probability, NSC time constant, and residence time of adsorbed H2.124),126) The simulation results indicated that the H2-related NSC processes on the surfaces helped decrease the gaseous H2 OPR in MCs.

The OPRs of polyatomic molecules such as H2O and NH3 have also been investigated for various astronomical objects to infer the chemical and thermal history of these molecules.6),23) At thermal equilibrium, the OPR of H2O reaches its statistical value (3) at temperatures above 50 K. The OPR values lower than 3 have been reported for certain star-forming regions (0.1–2.4) and proto-planetary disks (0.8).6),23),127),128) Because thermal desorption of water molecules does not typically occur in cold regions (<50 K), photodesorption is considered a plausible process that leads to the observed gaseous abundance. However, the effects of photodesorption on the OPR of H2O molecules are poorly understood. Given the lack of understanding of this process, the observed OPR value should not be linked with the formation temperature of ice; therefore, the implications of the observed OPRs of various astronomical objects remain unclear despite extensive observational studies.
Hama et al. experimentally studied the OPR of photodesorbed H2O from water ice at 10 K.129) They desorbed H2O molecules from vapor-deposited ASW or ASW produced in situ through the nonenergetic hydrogenation of solid O2 and determined the OPR (spin temperature) by REMPI spectroscopy. Interestingly, the OPR of H2O photodesorbed from 10 K ASW had a statistical value of 3, regardless of the ASW preparation method. The authors suggested that the lowest energy levels of ortho- and para-H2O molecules nearly degenerated in ice because of rotational hindrance, leading to rapid NSC between the ortho- and para-H2O (timescale: 10–100 µs) for achieving dynamic equilibrium at a statistical ratio of 3. Because the photodesorption occurs considerably faster (500 fs), the NSC during a photodesorption event is negligible. These considerations are corroborated by the determination of OPR (= 3) for the photodesorption from ice prepared from para-H2O monomers.130)
These experimental results suggest that the OPR of gaseous H2O molecules present in astronomical objects cannot be used to deduce the surface temperature of the interstellar dust where H2O formation or condensation occurs. In cold regions, the OPR of gaseous H2O molecules may decrease from 3 to lower values upon desorption via gas-phase reactions with ions such as H+ and H3O+. Therefore, to simulate the OPR values in astronomical objects, the mechanism of gaseous NSC processes should be clarified and their rate coefficients should be accurately determined.
5.3. Structure of ice mantle.
The ice mantles surrounding dust grains in MCs and protoplanetary disk environments are implicitly assumed to have homogeneous layers, regardless of their composition or crystallinity. This layered structure is often considered “onion-like”. For example, Ehrenfreund et al. proposed that the icy grains toward massive protostars have spherical structures comprising a silicate core, an inner H2O-dominated polar ice mantle, and an outer apolar ice mantle dominated by CO, N2, and some O2.131) Similar layered structures—H2O-rich ice in the interior, a mixture of CO and CO2 ice in the middle layer, and CO-rich ice on the exterior—have been suggested to exist for ice in MCs by Pontoppidan et al., Boogert et al., and Öberg.7),132),133) These structures have been simply deduced based on the following sequence of ice formation processes: (i) dominant production of H2O ice (ASW) in the early stage, (ii) initiation of the reaction CO + OH → CO2 + H, and (iii) significant deposition of CO, referred to as CO freeze-out. However, the structure of the ice mantle should not be simply deduced from the sequence, but by considering the accretion rate (or formation rate) of molecules and their diffusion rate and wettability on the surface. The shape of the ice that uniformly wets the substrate or forms islands can be directly detected by transmission electron microscopy (TEM). Kouchi et al. developed an ultrahigh-vacuum low-temperature TEM apparatus134) for direct observation135) and studied the structure of astronomically relevant ice.108) They proposed a new model for the structure of icy dust grains.
The structure of the icy grains in MCs is briefly discussed herein. In conventional layered models (left image in Fig. 10), the grain comprises silicate and organic cores along with layers of ASW, amorphous CO2 (a-CO2), and amorphous CO (a-CO).136)–138) A markedly different model was suggested by Kouchi et al. (right image in Fig. 10), in which the core is covered with ASW containing nanocrystals of CO2 (CO2 I), and one CO crystal (α-CO) is attached to the ASW surface. The model established by Kouchi et al. has several astrochemical and astrophysical implications, particularly concerning chemical evolution; nonthermal desorption processes; and the collision, sticking, and sintering of dust grains. For example, in MCs at 10 K, different chemical evolutions occur simultaneously on the a-H2O and crystalline CO surfaces of a single icy grain. Successive CO hydrogenation9),10) can occur at the boundary between a-H2O and crystalline CO to produce H2CO and CH3OH. Additional information on the further thermal evolution of icy grains in MCs and protoplanetary disks can be found in the original article.108)

6. Future perspectives
Laboratory experiments simulating low-temperature high-vacuum conditions deepen our understanding of the chemical evolution occurring in MCs, which produce stars and, eventually, planetary systems. Chemical processes on icy dust surfaces have long been suggested to be responsible for the large variety and abundance of molecules in MCs. Improvements in experimental methods are essential for studying these processes. For example, the development of an H-atom source that can provide the H-atom fluence expected in the typical lifetime of an MC (106 years) within a laboratory timescale can reveal the formation mechanisms of abundant organic molecules such as H2CO and CH3OH.
As discussed in Section 2.1, molecular hydrogen (H2) is the most abundant molecule in the Universe and plays an important role in several astronomical processes. Experiments have indicated the efficient production of H2 molecules on the surface of low-temperature ice dust. Low temperatures (such as ∼10 K) allow H atoms to remain on the surface for a sufficiently long time, and the surface serves as a third body to dissipate the reaction energy (4.5 eV) of the H + H → H2 reaction. Notably, this mechanism requires extremely low temperatures. Diffuse clouds, which are the precursors of MCs and are at a temperature of ∼80 K, exist in interstellar media.139) H2 molecules are abundant in diffuse clouds also, and the formation mechanism in this relatively high-temperature environment remains unclear. Although we found that the formation of H2 on a DLC surface is remarkably efficient even at 20 K, this result is valid under laboratory conditions of a high H-atom flux; in other words, the residence time of H atoms does not need to be very long.35) Therefore, the diffusive recombination reaction of physisorbed H atoms (via the Langmuir–Hinshelwood mechanism) cannot be responsible for the production of H2; however, the reaction between the H atoms colliding with the dust and those chemisorbed on the dust surface (via the Eley–Rideal mechanism) can occur even at high temperatures. Indirect evidence for H2 formation via the Eley–Rideal mechanism has been provided for an amorphous carbon surface.140),141) Direct detection of nascent H2 molecules, possibly using the PSD-REMPI method, is desirable to clarify the exact mechanism.
As described in Section 3.1, deuterium enrichment in methanol occurs efficiently via H–D substitution reactions. Among the deuterated isotopologues of methanol, those deuterated in the hydroxyl group—such as CH3OD—have not been observed in laboratory experiments (simultaneously irradiating solid CO with H and D atoms).88) In contrast, CH3OD has been detected around low-mass and high-mass protostars.142) Moreover, the CH2DOH/CH3OD ratio differs between low- and high-mass protostars (>10 and ∼1, respectively), indicating different formation pathways. The issue concerning the absence of CH3OD in the experiments has been addressed based on the experimentally revealed fact that the H–D substitution does not occur for the hydroxyl group at 10 K, which indicates that the absence is likely related to the process of formation. In the successive hydrogenation reaction of CO ($\text{CO}\overset{\text{H}}{\to}\text{HCO}\overset{\text{H}}{\to}\text{H$_{2}$CO}\overset{\text{H}}{\to}\text{CH$_{3}$O}/\text{CH$_{2}$OH}\overset{\text{H}}{\to}\text{CH$_{3}$OH}$), the first and third steps are rate-limiting and occur via quantum mechanical tunneling, whereas the other two steps are barrierless. During the simultaneous H- and D-atom irradiation of solid CO, the first and third steps occur only with H atoms because of the significant isotope effect. Because H atoms are abundant near the position where these reactions occur, the second and fourth steps may occur either with H or D atoms (according to the H/D ratio of the irradiation). The absence of CH3OD in this experiment indicates that the product of the third step is CH2OH rather than CH3O. Under the realistic conditions of icy dust surfaces, hydrogen (and deuterium) atoms are deposited at a rate of approximately one per day, indicating the lack of competition between H and D atoms in the first and third steps. Therefore, the reaction H2CO + D → CH2OD likely occurs on icy dust surfaces, although it has been shown to be less efficient than the H-abstraction reaction.89) If an extremely low H(D)-atom flux is used, the reaction rate is determined by the H(D)-atom deposition rates, but the reaction product yields are too low to be detected by IR spectrometry. Therefore, the use of highly sensitive methods such as PSD-REMPI and Cs+-ion pickup under low H-atom-flux conditions may shed light on the CH3OD formation mechanism. A new mechanism for methanol formation, CH3O + H2CO → CH3OH + HCO, has recently been suggested experimentally; however, this mechanism cannot explain the formation of CH3OD, as flagged by the authors.143)
The formation mechanisms of COMs have attracted significant attention in recent years, particularly because of the significant advances in observational studies. In particular, the Atacama Large Millimeter/submillimeter Array (ALMA) has allowed us to trace chemical evolution with ultrahigh spatial resolution. The recently launched James Webb Space Telescope (JWST) shows promise for revealing the complexity of the molecular universe. COMs are believed to be formed during the temperature increase during star formation. Light radical species, such as OH, HCO, CH3, NH, and NH2, are expected to diffuse on the surface to form new molecules. However, quantitative information on the thermally activated diffusion of these species is lacking; in various chemical models, activation energies are systematically estimated using computationally determined binding energies. The PSD-REMPI method, which was introduced in Section 4.1 for the thermal diffusion of OH radicals on ice, has proven to be a powerful technique for investigating the behavior of radicals on ice; this method will hopefully be applied to other radicals in the near future. Moreover, Cs+-ion pickup can be highly effective for elucidating COM formation mechanisms. However, as mentioned earlier, the existing apparatus exhibits poor selectivity for structural isomers. We plan to incorporate a component to distinguish structural isomers. For example, after the picking-up event, the Cs+–M complex can be accumulated in an ion trap and photodissociated to distinguish isomers.
The origin of biomolecular homochirality is a remarkably fascinating mystery related to the origin of life. Asymmetric adsorption and asymmetric synthesis on chiral inorganic crystal surfaces have been proposed as possible chiral selection processes.144),145) Recently, we found that a chiral crystalline CO (α–CO) is formed on each icy grain (Fig. 10).108),109) This can be regarded as the first chiral crystal in chemical evolution. If icy grains are irradiated with one-handed circularly polarized light during the nucleation of CO, the formed crystals may have an enantiomeric excess. Moreover, the presence of metals or high-index nanoparticles on icy dust could enhance the enantiomeric excess through plasmon-assisted asymmetric nucleation.146) A similar process may occur during the crystallization of ASW to form chiral ice crystals; moreover, amorphous methanol and ammonia crystallize in protoplanetary disks, yielding α–CH3OH and NH3 I, respectively.147) These processes have not been scrutinized experimentally and are therefore worth investigating.
Acknowledgements
The authors are grateful to colleagues who contributed to the original studies cited in this review and the technical staff at the Institute of Low Temperature Science, who helped develop the experimental apparatus. The studies discussed herein were supported in part by JSPS KAKENHI grants (JP22H00159 and JP21H01139) and a JSPS Grant-in-Aid for Specially Promoted Research (JP17H06087).
Notes
Edited by Yoshio FUKAO, M.J.A.
Correspondence should be addressed to: N. Watanabe, Institute of Low Temperature Science, Hokkaido University, Kita 19, Nishi 8, Kita-ku, Sapporo, Hokkaido 060-0819, Japan (e-mail: watanabe@lowtem.hokudai.ac.jp).
References
- 1) Lombardi, M., Bouy, H., Alves, J. and Lada, C.J. (2014) Herschel-Planck dust optical-depth and column-density maps. Astron. Astrophys. 566, A45.
- 2) Tricco, T.S., Price, D.J. and Laibe, G. (2017) Is the dust-to-gas ratio constant in molecular clouds? Mon. Not. R. Astron. Soc. 471, L52–L56.
- 3) CDMS (The Cologne Database for Molecular Spectroscopy) classic documentation: Molecules in Space. https://cdms.astro.uni-koeln.de/classic/molecules.
- 4) Yamamoto, S. (2017) Introduction to Astrochemistry. Springer Tokyo, Tokyo.
- 5) Watanabe, N. and Kouchi, A. (2008) Ice surface reactions: A key to chemical evolution in space. Prog. Surf. Sci. 83, 439–489.
- 6) van Dishoeck, E.F., Herbst, E. and Neufeld, D.A. (2013) Interstellar water chemistry: From laboratory to observations. Chem. Rev. 113, 9043–9085.
- 7) Boogert, A.C.A., Gerakines, P.A. and Whittet, D.C.B. (2015) Observations of the icy universe. Annu. Rev. Astron. Astrophys. 53, 541–581.
- 8) Wakelam, V., Bron, E., Cazaux, S., Dulieu, F., Gry, C., Guillard, P. et al. (2017) H2 formation on interstellar dust grains: The viewpoints of theory, experiments, models and observations. Mol. Astrophys. 9, 1–36.
- 9) Watanabe, N. and Kouchi, A. (2002) Efficient formation of formaldehyde and methanol by the addition of hydrogen atoms to CO in H2O-CO ice at 10 K. Astrophys. J. Lett. 571, L173–L176.
- 10) Fuchs, G.W., Cuppen, H.M., Ioppolo, S., Romanzin, C., Bisschop, S.E., Andersson, S. et al. (2009) Hydrogenation reactions in interstellar CO ice analogues. Astron. Astrophys. 505, 629–639.
- 11) Millar, T.J. (2003) Deuterium fractionation in interstellar clouds. Space Sci. Rev. 106, 73–86.
- 12) Linsky, J.L. (2003) Atomic deuterium/hydrogen in the galaxy. Space Sci. Rev. 106, 49–60.
- 13) Robert, F., Gautier, D. and Dubrulle, B. (2000) The solar system d/h ratio: Observations and theories. Space Sci. Rev. 92, 201–224.
- 14) Lis, D.C., Bockelée-Morvan, D., Güsten, R., Biver, N., Stutzki, J., Delorme, Y. et al. (2019) Terrestrial deuterium-to-hydrogen ratio in water in hyperactive comets. Astron. Astrophys. 625, L5.
- 15) Geiss, J. and Reeves, H. (1981) Deuterium in the solar system. Astron. Astrophys. 93, 189–199.
- 16) Cuppen, H.M., Walsh, C., Lamberts, T., Semenov, D., Garrod, R.T., Penteado, E.M. et al. (2017) Grain surface models and data for astrochemistry. Space Sci. Rev. 212, 1–58.
- 17) Herbst, E. and van Dishoeck, E.F. (2009) Complex organic interstellar molecules. Annu. Rev. Astron. Astrophys. 47, 427–480.
- 18) Garrod, R.T., Widicus Weaver, S.L. and Herbst, E. (2008) Complex chemistry in star-forming regions: An expanded gas-grain warm-up chemical model. Astrophys. J. 682, 283–302.
- 19) Jin, M. and Garrod, R.T. (2020) Formation of complex organic molecules in cold interstellar environments through nondiffusive grain-surface and ice-mantle chemistry. Astrophys. J. Suppl. Ser. 249, 26.
- 20) Watanabe, N. and Tsuge, M. (2020) Experimental approach to physicochemical hydrogen processes on cosmic ice dust. J. Phys. Soc. Jpn. 89, 051015.
- 21) Tsuge, M. and Watanabe, N. (2021) Behavior of hydroxyl radicals on water ice at low temperatures. Acc. Chem. Res. 54, 471–480.
- 22) Vidali, G. (2013) H2 formation on interstellar grains. Chem. Rev. 113, 8762–8782.
- 23) Hama, T. and Watanabe, N. (2013) Surface processes on interstellar amorphous solid water: Adsorption, diffusion, tunneling reactions, and nuclear-spin conversion. Chem. Rev. 113, 8783–8839.
- 24) Duley, W.W. and Williams, D.A. (1984) Interstellar Chemistry. Academic Press, London.
- 25) Gould, R.J. and Salpeter, E.E. (1963) Interstellar abundance of hydrogen molecule. 1. Basic processes. Astrophys. J. 138, 393–407.
- 26) Perets, H.B., Biham, O., Manicó, G., Pirronello, V., Roser, J., Swords, S. et al. (2005) Molecular hydrogen formation on ice under interstellar conditions. Astrophys. J. 627, 850–860.
- 27) Matar, E., Congiu, E., Dulieu, F., Momeni, A. and Lemaire, J.L. (2008) Mobility of D atoms on porous amorphous water ice surfaces under interstellar conditions. Astron. Astrophys. 492, L17–L20.
- 28) Hama, T., Kuwahata, K., Watanabe, N., Kouchi, A., Kimura, Y., Chigai, T. et al. (2012) The mechanism of surface diffusion of H and D atoms on amorphous solid water: Existence of various potential sites. Astrophys. J. 757, 185.
- 29) Watanabe, N., Kimura, Y., Kouchi, A., Chigai, T., Hama, T. and Pirronello, V. (2010) Direct measurements of hydrogen atom diffusion and the spin temperature of nascent H2 molecule on amorphous solid water. Astrophys. J. Lett. 714, L233–L237.
- 30) Kuwahata, K., Hama, T., Kouchi, A. and Watanabe, N. (2015) Signatures of quantum-tunneling diffusion of hydrogen atoms on water ice at 10 K. Phys. Rev. Lett. 115, 133201.
- 31) Smoluchowski, R. (1979) Formation of H2 on amorphous ice grains and their importance for planetary atmospheres. Astrophys. Space Sci. 65, 29–38.
- 32) Smoluchowski, R. (1981) Rate of H2 formation on amorphous grains. Astrophys. Space Sci. 75, 353–363.
- 33) Smoluchowski, R. (1983) Adsorption and mobility on amorphous surfaces. Application to astrophysical problems. J. Phys. Chem. 87, 4229–4233.
- 34) Kimura, Y., Tsuge, M., Pirronello, V., Kouchi, A. and Watanabe, N. (2018) Measurements of the activation energies for atomic hydrogen diffusion on pure solid CO. Astrophys. J. Lett. 858, L23.
- 35) Tsuge, M., Hama, T., Kimura, Y., Kouchi, A. and Watanabe, N. (2019) Interactions of atomic and molecular hydrogen with a diamond-like carbon surface: H2 formation and desorption. Astrophys. J. 878, 23.
- 36) van Dishoeck, E.F., Kristensen, L.E., Benz, A.O., Bergin, E.A., Caselli, P., Cernicharo, J. et al. (2011) Water in star-forming regions with the Herschel space observatory (WISH). I. Overview of key program and first results. Publ. Astron. Soc. Pac. 123, 138.
- 37) Tielens, A.G.G.M. and Hagen, W. (1982) Model calculations of the molecular composition of interstellar grain mantles. Astron. Astrophys. 114, 245–260.
- 38) Cuppen, H.M. and Herbst, E. (2007) Simulation of the formation and morphology of ice mantles on interstellar grains. Astrophys. J. 668, 294.
- 39) Koussa, H., Bahri, M., Jaïdane, N. and Ben Lakhdar, Z. (2006) Kinetic study of the reaction H2O2 + H → H2O + OH by ab initio and density functional theory calculations. J. Mol. Struct. THEOCHEM 770, 149–156.
- 40) Baulch, D.L., Cobos, C.J., Cox, R.A., Esser, C., Frank, P., Just, T. et al. (1992) Evaluated kinetic data for combustion modelling. J. Phys. Chem. Ref. Data 21, 411–734.
- 41) Miyauchi, N., Hidaka, H., Chigai, T., Nagaoka, A., Watanabe, N. and Kouchi, A. (2008) Formation of hydrogen peroxide and water from the reaction of cold hydrogen atoms with solid oxygen at 10 K. Chem. Phys. Lett. 456, 27–30.
- 42) Oba, Y., Miyauchi, N., Hidaka, H., Chigai, T., Watanabe, N. and Kouchi, A. (2009) Formation of compact amorphous H2O ice by codeposition of hydrogen atoms with oxygen molecules on grain surfaces. Astrophys. J. 701, 464–470.
- 43) Keane, J.V., Tielens, A.G.G.M., Boogert, A.C.A., Schutte, W.A. and Whittet, D.C.B. (2001) Ice absorption features in the 5–8 µm region toward embedded protostars. Astron. Astrophys. 376, 254–270.
- 44) McClure, M.K., Rocha, W.R.M., Pontoppidan, K.M., Crouzet, N., Chu, L.E.U., Dartois, E. et al. (2023) An ice age JWST inventory of dense molecular cloud ices. Nat. Astron. https://doi.org/10.1038/s41550-022-01875-w.
- 45) Oba, Y., Watanabe, N., Hama, T., Kuwahata, K., Hidaka, H. and Kouchi, A. (2012) Water formation through a quantum tunneling surface reaction, OH + H2, at 10 K. Astrophys. J. 749, 67.
- 46) Atkinson, R., Baulch, D.L., Cox, R.A., Crowley, J.N., Hampson, R.F., Hynes, R.G. et al. (2004) Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I - gas phase reactions of Ox, HOx, NOx and SOx species. Atmos. Chem. Phys. 4, 1461–1738.
- 47) Charnley, S.B., Tielens, A.G.G.M. and Rodgers, S.D. (1997) Deuterated methanol in the orion compact ridge. Astrophys. J. 482, L203–L206.
- 48) Hiraoka, K., Ohashi, N., Kihara, Y., Yamamoto, K., Sato, T. and Yamashita, A. (1994) Formation of formaldehyde and methanol from the reactions of H atoms with solid CO at 10–20 K. Chem. Phys. Lett. 229, 408–414.
- 49) Hiraoka, K. and Sato, T. (2001) Laboratory simulation of tunneling reactions in interstellar ices. Radiat. Phys. Chem. 60, 389–393.
- 50) Hidaka, H., Kouchi, A. and Watanabe, N. (2007) Temperature, composition, and hydrogen isotope effect in the hydrogenation of CO on amorphous ice surface at 10–20 K. J. Chem. Phys. 126, 204707.
- 51) Hidaka, H., Watanabe, N., Shiraki, T., Nagaoka, A. and Kouchi, A. (2004) Conversion of H2CO to CH3OH by reactions of cold atomic hydrogen on ice surfaces below 20 K. Astrophys. J. 614, 1124–1131.
- 52) Watanabe, N., Shiraki, T. and Kouchi, A. (2003) The dependence of H2CO and CH3OH formation on the temperature and thickness of H2O-CO ice during the successive hydrogenation of CO. Astrophys. J. Lett. 588, L121–L124.
- 53) Watanabe, N., Nagaoka, A., Shiraki, T. and Kouchi, A. (2004) Hydrogenation of CO on pure solid CO and CO-H2O mixed ice. Astrophys. J. 616, 638–642.
- 54) Hidaka, H., Miyauchi, N., Kouchi, A. and Watanabe, N. (2008) Structural effects of ice grain surfaces on the hydrogenation of CO at low temperatures. Chem. Phys. Lett. 456, 36–40.
- 55) Tsuge, M., Hidaka, H., Kouchi, A. and Watanabe, N. (2020) Diffusive hydrogenation reactions of CO embedded in amorphous solid water at elevated temperatures ∼70 K. Astrophys. J. 900, 187.
- 56) Tsuge, M., Kouchi, A. and Watanabe, N. (2022) Penetration of nonenergetic hydrogen atoms into amorphous solid water and their reaction with embedded benzene and naphthalene. Astrophys. J. 933, 138.
- 57) Simons, M.A.J., Lamberts, T. and Cuppen, H.M. (2020) Formation of COMs through CO hydrogenation on interstellar grains. Astron. Astrophys. 634, A52.
- 58) Bacmann, A., Taquet, V., Faure, A., Kahane, C. and Ceccarelli, C. (2012) Detection of complex organic molecules in a prestellar core: a new challenge for astrochemical models. Astron. Astrophys. 541, L12.
- 59) Fredon, A., Radchenko, A.K. and Cuppen, H.M. (2021) Quantification of the role of chemical desorption in molecular clouds. Acc. Chem. Res. 54, 745–753.
- 60) Minissale, M., Congiu, E. and Dulieu, F. (2016) Direct measurement of desorption and diffusion energies of O and N atoms physisorbed on amorphous surfaces. Astron. Astrophys. 585, A146.
- 61) Chuang, K.-J., Fedoseev, G., Qasim, D., Ioppolo, S., van Dishoeck, E.F. and Linnartz, H. (2018) Reactive desorption of CO hydrogenation products under cold pre-stellar core conditions. Astrophys. J. 853, 102.
- 62) Oba, Y., Tomaru, T., Lamberts, T., Kouchi, A. and Watanabe, N. (2018) An infrared measurement of chemical desorption from interstellar ice analogues. Nat. Astron. 2, 228–232.
- 63) Oba, Y., Tomaru, T., Kouchi, A. and Watanabe, N. (2019) Physico-chemical behavior of hydrogen sulfide induced by reactions with H and D atoms on different types of ice surfaces at low temperature. Astrophys. J. 874, 124.
- 64) Nguyen, T., Oba, Y., Shimonishi, T., Kouchi, A. and Watanabe, N. (2020) An experimental study of chemical desorption for phosphine in interstellar ice. Astrophys. J. Lett. 898, L52.
- 65) Garrod, R.T., Wakelam, V. and Herbst, E. (2007) Non-thermal desorption from interstellar dust grains via exothermic surface reactions. Astron. Astrophys. 467, 1103–1115.
- 66) Vasyunin, A.I. and Herbst, E. (2013) A unified Monte Carlo treatment of gas-grain chemistry for large reaction networks. II. A multiphase gas-surface-layerd bulk model. Astrophys. J. 762, 86.
- 67) Wakelam, V., Loison, J.C., Mereau, R. and Ruaud, M. (2017) Binding energies: New values and impact on the efficiency of chemical desorption. Mol. Astrophys. 6, 22–35.
- 68) Cazaux, S., Cobut, V., Marseille, M., Spaans, M. and Caselli, P. (2010) Water formation on bare grains: When the chemistry on dust impacts interstellar gas. Astron. Astrophys. 522, A74.
- 69) Furuya, K., Oba, Y. and Shimonishi, T. (2022) Quantifying the chemical desorption of H2S and PH3 from amorphous water-ice surfaces. Astrophys. J. 926, 171.
- 70) Goldsmith, P.F. and Linke, R.A. (1981) A study of interstellar carbonyl sulfide. Astrophys. J. 245, 482–494.
- 71) Palumbo, M.E., Tielens, A.G.G.M. and Tokunaga, A.T. (1995) Solid carbonyl sulphide (OCS) in W33A. Astrophys. J. 449, 674–680.
- 72) Tsuge, M. and Lee, Y.-P. (2016) Infrared spectra of two isomers of protonated carbonyl sulfide (HOCS+ and HSCO+) and t-HOCS in solid para-hydrogen. J. Chem. Phys. 145, 164308.
- 73) Nguyen, T., Oba, Y., Sameera, W.M.C., Kouchi, A. and Watanabe, N. (2021) Successive H-atom addition to solid OCS on compact amorphous solid water. Astrophys. J. 922, 146.
- 74) Nguyen, T., Oba, Y., Sameera, W.M.C., Kouchi, A. and Watanabe, N. (2021) Experimental and computational studies on the physicochemical behavior of phosphine induced by reactions with H and D atoms on interstellar ice grains. Astrophys. J. 918, 73.
- 75) Sameera, W.M.C., Senevirathne, B., Nguyen, T., Oba, Y., Ishibashi, A., Tsuge, M. et al. (2022) Modelling the radical chemistry on ice surfaces: An integrated quantum chemical and experimental approach. Front. Astron. Space Sci. 9, 890161.
- 76) Molpeceres, G., Jiménez-Serra, I., Oba, Y., Nguyen, T., Watanabe, N., de la Concepción, J.G. et al. (2022) Hydrogen abstraction reactions in formic and thioformic acid isomers by hydrogen and deuterium atoms. Astron. Astrophys. 663, A41.
- 77) Pettersson, M., Lundell, J., Khriachtchev, L. and Rasanen, M. (1997) IR spectrum of the other rotamer of formic acid, cis-HCOOH. J. Am. Chem. Soc. 119, 11715–11716.
- 78) Tsuge, M. and Khriachtchev, L. (2015) Tunneling isomerization of small carboxylic acids and their complexes in solid matrixes: A computational insight. J. Phys. Chem. A 119, 2628–2635.
- 79) Cuadrado, S., Goicoechea, J.R., Roncero, O., Aguado, A., Tercero, B. and Cernicharo, J. (2016) Trans-cis molecular photoswitching in interstellar space. Astron. Astrophys. 596, L1.
- 80) Taquet, V., Wirström, E.S., Charnley, S.B., Faure, A., López-Sepulcre, A. and Persson, C.M. (2017) Chemical complexity induced by efficient ice evaporation in the Barnard 5 molecular cloud. Astron. Astrophys. 607, A20.
- 81) Khriachtchev, L. (2008) Rotational isomers of small molecules in noble-gas solids: From monomers to hydrogen-bonded complexes. J. Mol. Struct. 880, 14–22.
- 82) Cassam-Chenaï, P., Pauzat, F. and Ellinger, Y. (1994) Is stripping of polycyclic aromatic hydrocarbons a route to molecular hydrogen? AIP Conf. Proc. 312, 543–547.
- 83) Le Page, V., Snow, T.P. and Bierbaum, V.M. (2009) Molecular hydrogen formation catalyzed by polycyclic aromatic hydrocarbons in the interstellar medium. Astrophys. J. 704, 274–280.
- 84) Tsuge, M., Tseng, C.-Y. and Lee, Y.-P. (2018) Spectroscopy of prospective interstellar ions and radicals isolated in para-hydrogen matrices. Phys. Chem. Chem. Phys. 20, 5344–5358.
- 85) Hama, T., Ueta, H., Kouchi, A., Watanabe, N. and Tachikawa, H. (2014) Quantum tunneling hydrogenation of solid benzene and its control via surface structure. J. Phys. Chem. Lett. 5, 3843–3848.
- 86) Hama, T., Ueta, H., Kouchi, A. and Watanabe, N. (2015) Quantum tunneling observed without its characteristic large kinetic isotope effects. Proc. Natl. Acad. Sci. U.S.A. 112, 7438–7443.
- 87) Parise, B., Castets, A., Herbst, E., Caux, E., Ceccarelli, C., Mukhopadhyay, I. et al. (2004) First detection of triply-deuterated methanol. Astron. Astrophys. 416, 159–163.
- 88) Nagaoka, A., Watanabe, N. and Kouchi, A. (2005) H-D Substitution in interstellar solid methanol: A key route for D enrichment. Astrophys. J. Lett. 624, L29–L32.
- 89) Hidaka, H., Watanabe, M., Kouchi, A. and Watanabe, N. (2009) Reaction routes in the CO-H2CO-dn-CH3OH-dm system clarified from H(D) exposure of solid formaldehyde at low temperatures. Astrophys. J. 702, 291–300.
- 90) Oehlers, C., Wagner, H.G., Ziemer, H., Temps, F. and Dóbé, S. (2000) An investigation of the D/H addition–elimination and H atom abstraction channels in the reaction D + H2CO in the temperature range 296 K ≤ T ≤ 780 K. J. Phys. Chem. A 104, 10500–10510.
- 91) Nagaoka, A., Watanabe, N. and Kouchi, A. (2007) Effective rate constants for the surface reaction between solid methanol and deuterium atoms at 10 K. J. Phys. Chem. A 111, 3016–3028.
- 92) Kerkeni, B. and Clary, D.C. (2004) Kinetic isotope effects in the reactions of D atoms with CH4, C2H6, and CH3OH: Quantum dynamics calculations. J. Phys. Chem. A 108, 8966–8972.
- 93) Loinard, L., Castets, A., Ceccarelli, C., Tielens, A.G.G.M., Faure, A., Caux, E. et al. (2000) The enormous abundance of D2CO in IRAS 16293-2422. Astron. Astrophys. 359, 1169–1174.
- 94) Ceccarelli, C., Loinard, L., Castets, A., Tielens, A.G.G.M., Caux, E., Lefloch, B. et al. (2001) Extended D2CO emission: The smoking gun of grain surface-chemistry. Astron. Astrophys. 372, 998–1004.
- 95) Oba, Y., Osaka, K., Chigai, T., Kouchi, A. and Watanabe, N. (2016) Hydrogen–deuterium substitution in solid ethanol by surface reactions at low temperatures. Mon. Not. R. Astron. Soc. 462, 689–695.
- 96) Oba, Y., Watanabe, N. and Kouchi, A. (2016) Negative catalytic effect of water on the reactivity of hydrogen abstraction from the C–H bond of dimethyl ether by deuterium atoms through tunneling at low temperatures. Chem. Phys. Lett. 662, 14–18.
- 97) Glavin, D.P., Dworkin, J.P. and Sandford, S.A. (2008) Detection of cometary amines in samples returned by Stardust. Meteorit. Planet. Sci. 43, 399–413.
- 98) Lee, C.-W., Kim, J.-K., Moon, E.-S., Minh, Y.C. and Kang, H. (2009) Formation of glycine on ultraviolet-irradiated interstellar ice-analog and implications for intestellar amino acids. Astrophys. J. 697, 428.
- 99) Holtom, P.D., Bennett, C.J., Osamura, Y., Mason, N.J. and Kaiser, R.I. (2005) A combined experimental and theoretical study on the formation of the amino acid glycine (NH2CH2COOH) and its isomer (CH3NHCOOH) in extraterrestrial ices. Astrophys. J. 626, 940.
- 100) Oba, Y., Chigai, T., Osamura, Y., Watanabe, N. and Kouchi, A. (2014) Hydrogen isotopic substitution of solid methylamine through atomic surface reactions at low temperatures: A potential contribution to the D/H ratio of methylamine in molecular clouds. Meteorit. Planet. Sci. 49, 117–132.
- 101) Miyazaki, A., Watanabe, N., Sameera, W.M.C., Nakai, Y., Tsuge, M., Hama, T. et al. (2020) Photostimulated desorption of OH radicals from amorphous solid water: Evidence for interation of visible light with OH-ice complex. Phys. Rev. A 102, 052822.
- 102) Miyazaki, A., Tsuge, M., Hidaka, H., Nakai, Y. and Watanabe, N. (2022) Direct determination of the activation energy for diffusion of OH radicals on water ice. Astrophys. J. Lett. 940, L2.
- 103) Crawford, T.D., Abrams, M.L., King, R.A., Lane, J.R., Schofield, D.P. and Kjaergaard, H.G. (2006) The lowest 2A′ excited state of the water-hydroxyl complex. J. Chem. Phys. 125, 204302.
- 104) Watanabe, N., Sameera, W.M.C., Hidaka, H., Miyazaki, A. and Kouchi, A. (2019) Ultraviolet-photon exposure stimulates negative current conductivity in amorphous ice below 50 K. Chem. Phys. Lett. 737, 136820.
- 105) Kitajima, K., Nakai, Y., Sameera, W.M.C., Tsuge, M., Miyazaki, A., Hidaka, H. et al. (2021) Delivery of electrons by proton-hole transfer in ice at 10 K: Role of surface OH radicals. J. Phys. Chem. Lett. 12, 704–710.
- 106) Ruffle, D.P. and Herbst, E. (2000) New models of interstellar gas-grain chemistry — I. Surface diffusion rates. Mon. Not. R. Astron. Soc. 319, 837–850.
- 107) Kouchi, A., Furuya, K., Hama, T., Chigai, T., Kozasa, T. and Watanabe, N. (2020) Direct measurements of activation energies for surface diffusion of CO and CO2 on amorphous solid water using in situ transmission electron microscopy. Astrophys. J. Lett. 891, L22.
- 108) Kouchi, A., Tsuge, M., Hama, T., Oba, Y., Okuzumi, S., Sirono, S.-i. et al. (2021) Transmission electron microscopy study of the morphology of ices composed of H2O, CO2, and CO on refractory grains. Astrophys. J. 918, 45.
- 109) Kouchi, A., Tsuge, M., Hama, T., Niinomi, H., Nakatani, N., Shimonishi, T. et al. (2021) Formation of chiral CO polyhedral crystals on icy interstellar grains. Mon. Not. R. Astron. Soc. 505, 1530–1542.
- 110) Furuya, K., Hama, T., Oba, Y., Kouchi, A., Watanabe, N. and Aikawa, Y. (2022) Diffusion activation energy and desorption activation energy for astrochemically relevant species on water ice show no clear relation. Astrophys. J. Lett. 933, L16.
- 111) Kim, S., Park, E. and Kang, H. (2011) Segregation of hydroxide ions to an ice surface. J. Chem. Phys. 135, 074703.
- 112) Brown, R.D., Crofts, J.G., Gardner, F.F., Godfrey, P.D., Robinson, B.J. and Whiteoak, J.B. (1975) Discovery of interstellar methyl formate. Astrophys. J. 197, L29–L31.
- 113) Bottinelli, S., Ceccarelli, C., Williams, J.P. and Lefloch, B. (2007) Hot corinos in NGC 1333-IRAS4B and IRAS2A. Astron. Astrophys. 463, 601–610.
- 114) Arce, H.G., Santiago-García, J., Jørgensen, J.K., Tafalla, M. and Bachiller, R. (2008) Complex molecules in the L1157 molecular outflow. Astrophys. J. 681, L21.
- 115) Öberg, K.I., Garrod, R.T., van Dishoeck, E.F. and Linnartz, H. (2009) Formation rates of complex organics in UV irradiated CH3OH-rich ices. Astron. Astrophys. 504, 891–913.
- 116) Chuang, K.-J., Fedoseev, G., Qasim, D., Ioppolo, S., van Dishoeck, E.F. and Linnartz, H. (2017) Production of complex organic molecules: H-atom addition versus UV irradiation. Mon. Not. R. Astron. Soc. 467, 2552–2565.
- 117) Schneider, H., Caldwell-Overdier, A., Coppieters ‘t Wallant, S., Dau, L., Huang, J., Nwolah, I. et al. (2019) Detection of methoxymethanol as a photochemistry product of condensed methanol. Mon. Not. R. Astron. Soc. Lett. 485, L19–L23.
- 118) Ishibashi, A., Hidaka, H., Oba, Y., Kouchi, A. and Watanabe, N. (2021) Efficient formation pathway of methyl formate: The role of OH radicals on ice dust. Astrophys. J. Lett. 921, L13.
- 119) Fukutani, K. and Sugimoto, T. (2013) Physisorption and ortho–para conversion of molecular hydrogen on solid surfaces. Prog. Surf. Sci. 88, 279–348.
- 120) Millar, T.J. (2002) Modelling deuterium fractionation in interstellar clouds. Planet. Space Sci. 50, 1189–1195.
- 121) Vaytet, N., Tomida, K. and Chabrier, G. (2014) On the role of the H2 ortho:para ratio in gravitational collapse during star formation. Astron. Astrophys. 563, A85.
- 122) Maret, S., Bergin, E.A., Neufeld, D.A., Green, J.D., Watson, D.M., Harwit, M.O. et al. (2009) Spitzermapping of molecular hydrogen pure rotational lines in NGC 1333: A detailed study of feedback in star formation. Astrophys. J. 698, 1244–1260.
- 123) Ueta, H., Watanabe, N., Hama, T. and Kouchi, A. (2016) Surface temperature dependence of hydrogen ortho-para conversion on amorphous solid water. Phys. Rev. Lett. 116, 253201.
- 124) Tsuge, M., Namiyoshi, T., Furuya, K., Yamazaki, T., Kouchi, A. and Watanabe, N. (2021) Rapid ortho-to-para nuclear spin conversion of H2 on a silicate dust surface. Astrophys. J. 908, 234.
- 125) Tsuge, M., Kouchi, A. and Watanabe, N. (2021) Measurements of ortho-to-para nuclear spin conversion of H2 on low-temperature carbonaceous grain analogs: Diamond-like carbon and graphite. Astrophys. J. 923, 71.
- 126) Furuya, K., Aikawa, Y., Hama, T. and Watanabe, N. (2019) H2 ortho–para spin conversion on inhomogeneous grain surfaces. Astrophys. J. 882, 172.
- 127) Choi, Y., van der Tak, F.F.S., Bergin, E.A. and Plume, R. (2014) A non-equilibrium ortho-to-para ratio of water in the Orion PDR. Astron. Astrophys. 572, L10.
- 128) Hogerheijde, M.R., Bergin, E.A., Brinch, C., Cleeves, L.I., Fogel, J.K.J., Blake, G.A. et al. (2011) Detection of the water reservoir in a forming planetary system. Science 334, 338–340.
- 129) Hama, T., Kouchi, A. and Watanabe, N. (2016) Statistical ortho-to-para ratio of water desorbed from ice at 10 kelvin. Science 351, 65–67.
- 130) Hama, T., Kouchi, A. and Watanabe, N. (2018) The ortho-to-para ratio of water molecules desorbed from ice made from para-water monomers at 11 K. Astrophys. J. Lett. 857, L13.
- 131) Ehrenfreund, P., Dartois, E., Demyk, K. and D’Hendecourt, L. (1998) Ice segregation toward massive protostars. Astron. Astrophys. 339, L17.
- 132) Pontoppidan, K.M., Boogert, A.C.A., Fraser, H.J., van Dishoeck, E.F., Blake, G.A., Lahuis, F. et al. (2008) The c2d spitzer spectroscopic survey of ices around low-mass young stellar objects. II. CO2. Astrophys. J. 678, 1005–1031.
- 133) Öberg, K.I. (2016) Photochemistry and astrochemistry: Photochemical pathways to interstellar complex organic molecules. Chem. Rev. 116, 9631–9663.
- 134) Kouchi, A., Kimura, Y., Kitajima, K., Katsuno, H., Hidaka, H., Oba, Y. et al. (2021) UV-induced formation of ice XI observed using an ultra-high vacuum cryogenic transmission electron microscope and its implications for planetary science. Front Chem. 9, 799851.
- 135) Tsuge, M., Nguyen, T., Oba, Y., Hama, T., Kouchi, A. and Watanabe, N. (2020) UV-ray irradiation never causes amorphization of crystalline CO2: A transmission electron microscopy study. Chem. Phys. Lett. 760, 137999.
- 136) Musiolik, G., Teiser, J., Jankowski, T. and Wurm, G. (2016) Collisions of CO2 ice grains in planet formation. Astrophys. J. 818, 16.
- 137) Pinilla, P., Pohl, A., Stammler, S.M. and Birnstiel, T. (2017) Dust density distribution and imaging analysis of different ice lines in protoplanetary disks. Astrophys. J. 845, 68.
- 138) Okuzumi, S. and Tazaki, R. (2019) Nonsticky ice at the origin of the uniformly polarized submillimeter emission from the HL Tau disk. Astrophys. J. 878, 132.
- 139) Tielens, A.G.G.M. (2021) Diffuse clouds. In Molecular Astrophysics. Cambridge University Press, Cambridge, pp. 318–365.
- 140) Mennella, V. (2008) HD formation by abstraction of H/D chemisorbed in carbon grains with D/H atoms under simulated interstellar conditions. Astrophys. J. Lett. 684, L25–L28.
- 141) Mennella, V., Hornekær, L., Thrower, J. and Accolla, M. (2012) The catalytic role of coronene for molecular hydrogen formation. Astrophys. J. 745, L2.
- 142) Kulterer, B.M., Drozdovskaya, M.N., Antonellini, S., Walsh, C. and Millar, T.J. (2022) Fevering interstellar ices have more CH3OD. ACS Earth Space Chem. 6, 1171–1188.
- 143) Santos, J.C., Chuang, K.-J., Lamberts, T., Fedoseev, G., Ioppolo, S. and Linnartz, H. (2022) First experimental confirmation of the CH3O + H2CO → CH3OH + HCO reaction: Expanding the CH3OH formation mechanism in interstellar ices. Astrophys. J. Lett. 931, L33.
- 144) Hazen, R.M. and Sholl, D.S. (2003) Chiral selection on inorganic crystalline surfaces. Nat. Mater. 2, 367–374.
- 145) Soai, K. (2019) Asymmetric autocatalysis. Chiral symmetry breaking and the origins of homochirality of organic molecules. Proc. Jpn. Acad. Ser. B 95, 89–110.
- 146) Niinomi, H., Sugiyama, T., Tagawa, M., Murayama, K., Harada, S. and Ujihara, T. (2016) Enantioselective amplification on circularly polarized laser-induced chiral nucleation from a NaClO3 solution containing Ag nanoparticles. CrystEngComm 18, 7441–7448.
- 147) Kouchi, A., Shimonishi, T., Yamazaki, T., Tsuge, M., Nakatani, N., Furuya, K. et al. (2022) Chiral ice crystals in space. In Crystal Growth—Technologies and Applications (eds. Marzouki, R. and Akitsu, T.). IntechOpen, Rijeka. doi: 10.5772/intechopen.106708.
Non-standard abbreviation list
ASWamorphous solid water
COMcomplex organic molecule
DLCdiamond-like carbon
EGethylene glycol (HOCH2CH2OH)
IRinfrared
MCmolecular cloud
MFmethyl formate (HCOOCH3); MM: methoxymethanol (CH3OCH2OH)
NSCnuclear spin conversion
OPRortho-to-para ratio
PSDphotostimulated desorption
QMSquadrupole mass spectrometer
REMPIresonance-enhanced multiphoton ionization
TEMtransmission electron microscopy
TPDtemperature-programmed desorption
Profile

Masashi Tsuge was born in 1979 in Aichi Prefecture, Japan. In 2007, he received his Ph.D. degree in Chemistry from the Tokyo Institute of Technology for spectroscopic studies on free radicals and weakly bound complexes. He subsequently joined the Niigata University of Pharmacy and Applied Life Science in 2007, where he studied molecules in strong laser fields and the ionization mechanisms of the MALDI process. From 2011 to 2013, he was a postdoc in Prof. M. Räsänen’s group at the University of Helsinki, where he participated in studies on matrix isolation spectroscopy of noble-gas-containing species and cis–trans isomerism of carboxylic acids. From 2014 to 2017, he was a postdoc in Prof. Y.-P. Lee’s group at National Chiao Tung University (Taiwan), where he employed the para-hydrogen matrix isolation technique for spectroscopic investigation of free radicals and ions. Since joining a research group at the Institute of Low Temperature Science (ILTS; Hokkaido University) in 2017, his research has focused on the reaction dynamics relevant to chemical reactions on/in interstellar grains.

Naoki Watanabe received a Ph.D. degree in atomic and molecular physics from Tokyo Metropolitan University in 1993. As a postdoctoral researcher at RIKEN until 1996, he worked on the photoionization of highly charged ions and molecules using synchrotron radiation. Since becoming a staff member at ILTS in 1996, he has investigated the physics and chemistry of solid surfaces, particularly those of solid water, at extremely low temperatures that are relevant to chemical evolution in space. He is currently a professor at Hokkaido University, and his research interests also cover gas-phase reactions, including those in low-temperature clusters.
