Abstract
Inflammation is a host defense response to various invading stimuli, but an excessive and persistent inflammatory response can cause tissue injury, which can lead to irreversible organ damage and dysfunction. Excessive inflammatory responses are believed to link to most human diseases. A specific type of leukocyte infiltration into invaded tissues is required for inflammation. Historically, the underlying molecular mechanisms of this process during inflammation were an enigma, compromising research in the fields of inflammation, immunology, and pathology. However, the pioneering discovery of chemotactic cytokines (chemokines), monocyte-derived neutrophil chemotactic factor (MDNCF; interleukin [IL]-8, CXCL8) and monocyte chemotactic and activating factor (MCAF; monocyte chemotactic factor 1 [MCP-1], CCL2) in the late 1980s finally enabled us to address this issue. In this review, we provide a historical overview of chemokine research over the last 35 years.
1. Introduction
Inflammation is a fundamental host defense response to invading external stimuli, such as infecting pathogenic microorganisms, allergens, environmental chemicals, and UV/irradiation, as well as internal stimuli such as oxidized lipids, glycated proteins, nucleic acids, alarmins released from dead cells, and urate/cholesterol crystals, which occur due to the excessive intake of foods and aberrant metabolism. Excessive persistent inflammatory responses, however, can cause tissue injury leading to irreversible organ damage and dysfunction, such as that associated with fibrosis. Inflammatory diseases account for over 90% of human diseases, and they include arteriosclerosis, ischemia reperfusion injury (cardiac infarction and brain infarction), diabetes mellitus, organ fibrosis, infections, and cancer. Inflammation is widely reported to be associated with a specific type of leukocyte infiltration into affected tissues.1) However, the underlying molecular mechanisms of the specific type of leukocyte infiltration were historically an enigma, which limited research in the fields of inflammation, pathology, and immunology. This was changed, however, in the late 1980s with the pioneering discovery of chemotactic cytokines (chemokines), monocyte-derived neutrophil chemotactic factor (MDNCF; interleukin [IL]-8, CXCL8)2),3) and monocyte chemotactic and activating factor (MCAF; monocyte chemotactic factor 1 [MCP-1], CCL2).4),5) This review presents a historical overview of chemokine research, including the odyssey involved in the discovery of chemokines, the chemokine family and their receptors, and the pillars of chemokine research, which have enabled the elucidation of the roles of chemokines in inflammation, immunity, and infection. Finally, we present the role of chemokines in the era of cancer immunotherapy and single-cell transcriptome.
2. Odyssey to the discovery of IL-8
Leukocyte-derived chemotactic factors, such as neutrophil chemotactic factor and monocyte chemotactic factor, were reported in the 1970s6); however, their molecular nature remained uncertain at that time. Most immunologists were skeptical about the biological roles of these factors in inflammation and immunity; the chemotactic responses of leukocytes (directional migration of leukocytes in response to the chemoattractant gradient) was considered as an in vitro artifact. Early in the 1980s, J.J. Oppenheim and his colleagues at NIDR, NIH, U.S.A. reported that a partially purified preparation of human keratinocyte cell line-derived IL-1 exhibited neutrophil chemotactic activity.7) A. Mantovani and his colleagues from Italy reported that tumor necrosis factor alpha (TNFα) was a chemotactic factor for monocytes and neutrophils.8) On joining the Oppenheim laboratory in 1982, the author (KM) was first involved in the purification of human IL-1. After succeeding in purifying human IL-1β from lipopolysaccharide (LPS)-stimulated human peripheral blood mononuclear cells (PBMCs), KM confirmed several activities ascribed to IL-1 at that time, including thymocyte co-mitogenic, fibroblast proliferation, acute phase protein-inducing, and endogenous pyrogen activities.9) On the other hand, neutrophil and monocyte chemotactic activities were not detected using purified IL-1. However, these activities were detected in conditioned media of activated human PBMCs. In 1986, with T. Yoshimura, who joined a neighboring laboratory at NCI, Frederick as a post-doctoral fellow, we clearly distinguish the neutrophil chemotactic activity from IL-1 activity in LPS-stimulated human PBMCs conditioned media, using high-performance liquid chromatography gel filtration.10) Consequently, we succeeded in purifying 400 µg of neutrophil chemotactic factor from 4L of LPS-stimulated human PBMC conditioned media.2) These studies on the purification of neutrophil chemotactic factor were selected as pillars of immunology by American Association of Immunologists (AAI), because they were the first reports on the existence of cell-type specific leukocyte chemotactic factor. E. Appella at NCI, Bethesda, determined the amino acid (aa) sequence of the purified factor, up to 40 aa from the N-terminus. The author (KM) subsequently cloned the cDNA using degenerate oligo-DNA probes, designed based on the obtained aa sequence information. We initially named this molecule as monocyte-derived neutrophil chemotactic factor (MDNCF). However, there was a serious concern pertaining to its concentration: the amount of purified MDNCF was 100-fold greater than that of the IL-1 in similarly conditioned media. Therefore, 1% of an unidentified biologically active molecule may cause contamination. To establish that the cloned cDNA encoded MDNCF, Appella chemically synthesized the whole mature form of MDNCF consisting of 72 aa deduced from the cDNA sequence.11) M. Yamada from Dainippon Pharmaceutical Co., Ltd. (Osaka, Japan) expressed recombinant MDNCF in Escherichia coli,12) and Yoshimura confirmed that the chemically synthesized and recombinant proteins were fully chemotactic for human neutrophils in vitro. In addition, a monoclonal antibody prepared against purified MDNCF adsorbed the neutrophil chemotactic activity in conditioned media. Based on these results, we published a paper describing the cDNA cloning of MDNCF in J. Exp. Med. in 1988.3) In science, often simultaneous serious competition exists unnoticed among scientists exploring the same important issues. This was the case in the discovery of MDNCF. Several laboratories, including those of M. Baggiolini,13) J.M. Schröder,14) and J. Van Damme’s15) purified identical neutrophil chemoattractants from different sources just after our purification and cDNA cloning. We showed the robust induction of MDNCF mRNA by stimulating various types of cells with IL-1 and TNFα in vitro.3) This explained why IL-1 and TNFα induced neutrophil infiltration into injected tissues in vivo,16) despite lacking chemotactic activity. MDNCF was renamed as IL-8 based on its chemotactic effect on T-lymphocytes17) at the International Symposium on Novel Neutrophil Chemotactic Activating Polypeptides held in 1988 in London.
The IL-8 cDNA was 1.6 kb in size, including a 1.2 kb non-coding 3′-region with a TATTTATT sequence.3) The cDNA encoded a 99 aa precursor of IL-8, which contained a signal peptide region. The genomic sequence of human IL-8 was cloned from a human placenta library by N. Mukaida.18) The human IL-8 gene was assigned to the 4q12–21 chromosomal region, which was later identified to harbor numerous IL-8-related CXCL chemokine genes.19) The human IL-8 gene consists of 4 exons and 3 introns with a single CAT- and TATA-like structure. The 5′-flanking region contains binding sites for several nuclear factors, such as activation factor 1/2, interferon (IFN) regulatory factor-1, Oct-1, NRF, nuclear factor kappa B (NF-κB), and C/EBP (NF-IL6). We extensively examined the regulation of IL-8 gene transcription by IL-1, TNFα, TNFα + IFN gamma (IFNγ), and LPS in various human cell lines and established that NF-κB in combination with AP-1 or C/EBP synergistically activated IL-8 gene expression in response to various inflammatory stimuli. NF-κB is indispensable; however, the requirement for AP-1 or C/EBP depends on the cell type or stimuli. In addition, we established that NF-κB is an endo-target of the suppressive action of glucocorticoid and FK506 in regulating IL-8 gene expression.20)–25)
3. Protein processing and structure of IL-8
IL-8 is not expressed in normal physiological conditions; however, a massive induction of IL-8 mRNA occurs in various types of tissue cells and leukocytes in response to inflammation. IL-8 is first translated as a precursor consisting of 99 aa. The signal peptide (first 20 aa) is removed, and the processed 77 aa form is secreted.2),3) Biologically active IL-8 exists mainly as the 77 (residues 23–99) and 72 (residues 28–99) aa forms. Another cleaved form is the 69 aa form.11),26),27) The 72 aa form is processed from the precursor mostly by serine proteases, such as plasmin and thrombin, and by matrix metalloproteinases and cathepsins; it exists stably as the predominant form. The 72 aa form is several fold more active than the 77 aa form. The 69 aa form is most active in in vitro chemotactic assays.28) There are four cysteine residues forming two disulfide linkage (cysteine 7-cysteine 34 and cysteine 9-cysteine 50) and 14 basic aa residues (lysine and arginine), which confer the basicity required for the binding of IL-8 to heparan sulfate-proteoglycan without N-glycosylation sites.29) G.M. Clore and A.M. Gronenborn, NCI, solved the three-dimensional structure of IL-8 using nuclear magnetic resonance spectroscopy.30),31) This preceded the analysis by E.T. Baldwin at NCI using X-ray crystallography32) and presented for the first time that the three-dimensional structure of a protein had been initially determined using nuclear magnetic resonance instead of X-ray crystallography. The three-dimensional structure of IL-8 indicates a dimer with two symmetrical anti-parallel α-helices, approximately 24 Å long, separated by 14 Å, on top of a six-stranded anti-parallel β-sheet platform. The N-terminal region appears to be disordered and is thus structurally unsolved.
4. Discovery of MCAF
In the early 1970s, R. Snyderman et al. at NIDR, NIH first described that human peripheral blood leukocytes produce monocyte chemotactic factor (MCF) in response to a specific antigen (purified protein derivative [PPD]) or a nonspecific mitogen (phytohemagglutinin [PHA]).6),33) They claimed that the production of MCF in vitro correlated with delayed-type hypersensitivity. Therefore, it was interesting for immunologists to identify and characterize the molecular properties of MCF. We observed several peaks of MCF activity during the purification of IL-8 from LPS-stimulated human PBMC-conditioned media; however, the amount of MCF was less than that of IL-8. Therefore, we used conditioned media from a human myelomonocytic cell line THP-1 as a source of MCF. We purified the major peak of MCF using a heparin-affinity column, exploiting the basic property of MCF. The N-terminus of the purified MCF was blocked (later found to be due to pyro-glutamination); therefore, we cleaved the purified material using cyanogen bromide and obtained partial aa sequence information, which was sufficient for cloning the cDNA. The cloned cDNA encoded a MCF precursor consisting of a putative 23 aa signal peptide and a mature 76 aa MCF sequence, which showed 25–55% homology with an inducible putative family of inflammatory polypeptides, including JE, LD78, RANTES, and TCA-3 (which were later determined, both functionally and structurally, to belong to the chemokine family).4),5) Purified MCF stimulated human monocytes to be cytostatic to several tumor cell lines in vitro; therefore, we initially named this protein as monocyte chemotactic and activating factor (MCAF).4) Yoshimura purified34) and cloned35) the same protein independently and simultaneously. Most notably, similar to IL-8, MCAF was induced when various types of cells were stimulated with IL-1 or TNFα. In addition, it was stimulated directly in monocytes and THP-1 cells by PPD, arguing against the production of MCF by human peripheral blood leukocytes as an in vitro correlate of antigen-specific T cell-mediated delayed hypersensitivity.6)
5. Establishment of pivotal roles of IL-8 and MCAF in regulating leukocyte infiltration in inflammation: an answer to a longstanding enigma in inflammation research
Our purification and cDNA cloning of IL-8 and MCAF revealed the existence of leukocyte-specific chemotactic factors; however, pharmaceutical companies waited for proof that these chemotactic cytokines indeed regulate type-specific leukocyte infiltration in inflammation, in order to consider chemotactic cytokines as therapeutic targets for treating inflammatory diseases. On returning from NIH to Japan in 1990, we conducted in vivo experiments using a neutralizing antibody to prove that these factors indeed regulate leukocyte infiltration and that the intervention of IL-8 alleviates inflammatory diseases. This included LPS-induced skin inflammation, LPS/IL-1-induced acute arthritis, acute glomerulonephritis owing to excessive immune complex (serum sickness), acute lung injury associated with reperfusion injury, brain infarction, and swelling associated with delayed-type hypersensitivity.36)–41) We also proved the pivotal role of MCAF in regulating monocyte infiltration into inflammatory tissues using antibodies. This included chronic glomerulonephritis (Masugi type rat model), arteriosclerosis associated with carotid artery injury, and monocrotaline-induced pulmonary hypertension.42)–44) Our experiments using antibodies to show the pivotal roles of IL-8 and MCAF in inflammatory disease models were supported by later experiments using gene-targeted mice deficient in the receptor for IL-8 homologue (Cxcr2)45) and MCAF (Ccl2) or MCAF receptor (Ccr2).46) These studies convinced pharmaceutical researchers to consider chemotactic cytokines as novel targets for inflammatory diseases. Notably, our discovery of IL-8 and MCAF and in vivo proof of their roles in inflammation addressed the longstanding enigma and elucidated the molecular basis of type-specific leukocyte infiltration in inflammation. These findings, in conjunction with increased knowledge about leukocyte adhesion molecules, have led to our current understanding of the molecular mechanisms of leukocyte infiltration into inflamed tissue (see Fig. 1).

6. Chemokines and their receptors
The discovery of IL-8 prompted other groups to identify a large family of chemotactic cytokines based on biological activities, cDNA cloning using a signal trap methods, and eventually through screening for genes encoding similar proteins in databases containing fragments of transcripts and genome sequences. Now, nearly 50 chemotactic cytokines with conserved aa sequences have been identified.47) Among the chemokine receptors, CXCR1 and CXCR2 were first identified by W.E. Holmes et al.48) and P.M. Murphy and H.L. Tiffany,49) respectively, in 1991. Holmes et al. transfected a cDNA library from human neutrophils to mammalian cell lines and then screened the cells for binding to isotope-labeled IL-8, eventually identifying CXCR1. Murphy and Tiffany identified CXCR2 as the human homologue of a mouse receptor for fMLF of bacterial origin called F3R.49),50) Subsequently, a total of 18 receptors for chemokines were identified. IL-8 and CXCR1 genes are absent in mice, but CXCL1/KC and CXCL2/MIP-2 act as ligands for CXCR2 and regulate neutrophil migration in mice.51) Cxcr2−/− mice show increased susceptibility to Citrobacter rodentium and Streptococcus pneumoniae infection and decreased neutrophil recruitment to the infected site.52),53) Given that Cxcl1−/− mice show a similar reduction of neutrophil infiltration and increased susceptibility to Klebsiella pneumoniae infection,54) the CXCL1-CXCR2 axis is essential for protective immunity against bacterial infection. Notably, Cxcr2−/− mice have also been shown to decrease macrophage infiltration into infected lungs.53) Whether CXCR2 directly regulates macrophage infiltration or indirectly promotes them via inflammation by neutrophil infiltration remains to be elucidated. Furthermore, in LPS-induced lung injury, wild-type mice reconstituted with Cxcr2−/− bone marrow hematopoietic cells still showed about 50% neutrophil recruitment into bronchoalveolar lavage fluid and the lung interstitium. Conversely, Cxcr2−/− mice reconstituted with wild-type bone marrow hematopoietic cells showed a surprisingly large defect in neutrophil recruitment.55) These results suggested a significant role of CXCR2 in non-hematopoietic cells for neutrophil recruitment in inflammation and provided novel mechanistic insights into the chemokine field.
In addition to the pathological role, CXCR2 plays pivotal roles in the homeostasis of neutrophils and hematopoiesis; Cxcr2−/− mice showed neutrophilia and an increase in myeloid cells in bone marrow and peripheral blood and myeloid and erythroid cells in the spleen.56) Such abnormalities in Cxcr2−/− mice were ameliorated in germ-free conditions, suggesting that CXCR2-dependent neutrophil infiltration into peripheral tissues may regulate physiological inflammatory responses to the commensal bacteria and maintain homeostasis of myelopoiesis and hematopoiesis. Ccr2−/− mice show reduced mobilization of monocytes from the bone marrow to the blood and their infiltration into inflammatory tissues.57) This impairment of monocytes resulted in reduced defense against Listeria monocytogenes infection,46) whereas the formation of atherosclerosis foci in the early stages of atherosclerosis was reduced.58)
Chemokine receptors consist of a rhodopsin-like 7-transmembrane structure that is usually coupled with a Gi protein. Gi proteins are heterotrimers (composed of αi, β, and γ subunits) that are constitutively bound to the intracellular second and third loops of the receptor. Regarding IL-8-mediated neutrophil migration, once IL-8 binds to the receptor, the β and γ subunits are released from Gαi2 and activate phosphoinositide 3-kinase-γ and phospholipases (PLs) Cβ2/β3, A2, and D. The small GTPases Ras, Rac, Rho, Cdc42, and Rap1, as well as protein kinase C (PKC) and Akt (PKB), are downstream of the β and γ subunits and regulate cell adhesion, membrane protrusion, and cell migration. Meanwhile, PLCβ activation generates inositol triphosphate and induces Ca2+ mobilization and diacylglycerol generation, thereby activating PKC and inducing granule exocytosis and respiratory burst47),59) (Fig. 2). In 1992, most of the investigators who contributed to the discovery of these chemotactic cytokines and their receptors gathered in Baden, Austria, and agreed to name this family as “chemokines” (a contraction of chemotactic cytokines). CXCL/CCL (based on the first two cysteine residues split by one aa or neighbored) and CXCR/CCR nomenclature was discussed at the Gordon conference on the chemotactic cytokines in 1996, and O. Yoshie and A. Zlotnik proposed the systematic nomenclature of the chemokine ligands at the Keystone symposium on Chemotactic Cytokines in 1999.60) In addition, a subfamily of atypical chemokine receptors, including ACKR1 (Duffy antigen), ACKR2 (D6), ACKR3 (CXCR7), CCRL2, and ACKR4 that do not transduce chemotactic signals, but may signal through β-arrestins instead of G proteins to act as chemokine scavenger receptors, were identified.47) ACKR1 was discovered in 1950 as a blood group antigen and named Duffy. ACKR1 is constitutively expressed on various types of cells except for leukocytes, and it binds over 20 chemokines. Erythrocyte ACKR1 is considered to be a chemokine “sink” that adsorbs various types of CXCL and CCL chemokines in the circulation. ACKR1 polymorphism causes selective defects in its expression on erythroid cells. A Duffy-negative phenotype is associated with resistance to Plasmodium vivax malaria infection, particularly in West Africa.61) ACKR2 binds only the inflammatory chemokines from the CC subfamily, functioning as a scavenging receptor. ACKR2 lacks the canonical DRYLAIV motif in transmembrane domain 3; therefore, it is unable to transduce chemotactic signals. Ligand internalization and recycling of the receptor require the activation of β-arrestin.62) Chemokines and their receptors are also encoded by herpes and pox viruses; these are constitutively active or activated in response to host ligands. In addition, soluble non-7 transmembrane type chemokine binding proteins, which exhibit anti-inflammatory activity through neutralization by binding to several host chemokines, have been identified from herpes and pox viruses.47)

7. Subsequent pillars in chemokine research
a. The mechanisms of entry and cell tropisms of the human immunodeficiency virus (HIV) to human leukocytes remained unknown for a long time because the identification of CD4 as a receptor of HIV-1 infection in human leukocytes was not made until the mid-1980s. E.A. Berger and his colleagues at NIH first identified a chemokine receptor, later named CXCR4, as a co-receptor for T-tropic HIV-1 infection in CD4+ T lymphocytes in 1996.63) Subsequently, CCR5 was identified as a co-receptor for M-tropic HIV-1 infection in CD4+ monocytes/macrophages.64) Subsequently, an inactivating mutation (delta32) in the CCR5 gene in a homozygous state was discovered to confer strong resistance to HIV-1 infection.65) These studies led to the development of maraviroc (CCR5 antagonist) as an HIV entry antagonist and incidentally plerixafor (CXCR4 antagonist) as a stem cell mobilizing agent.47)
b. In 1996, R. Förster and M. Lipp reported that a putative chemokine receptor, BLR1 (later identified as CXCR5), is expressed on mature B cells and a subpopulation of helper T cells.66) Deficiency of CXCR5 in mice resulted in the lack of inguinal lymph nodes (LNs) and occasional abnormal Peyer’s patches. Activated B cells failed to migrate from the T cell-rich zone into B cell follicles and to form functional germinal centers. This study for the first time indicated that chemokine receptors are involved in B cell migration and localization within specific anatomic compartments of the lymphoid tissue. Later, the same group reported that CCR7 deficiency led to severely delayed kinetics of antibody responses, lack of contact sensitivity, and delayed-type hypersensitivity.67) This was because of the abnormal architecture of the paracortex area with few T cells. This can be attributed to impaired migration of naïve/central memory type T cells into LNs through the high endothelial venule (HEV) and defects in the migration of antigen-captured mature CCR7+ dendritic cells (DCs) from the inflamed peripheral tissue to the draining LNs. The ligands for CCR7 were identified as CCL19 (ELC) and CCL21 (SLC). CCL19 is produced by afferent lymphatic endothelial cells (LECs) and fibroblastic reticular cells (FRCs) in LNs, whereas CCL21 is produced by HEVs in addition to LECs and FRCs. The plt/plt (paucity of lymph node T cells) murine model,68) which is naturally deficient in CCL19 and CCL21 isoforms in secondary lymphoid organs, shows a phenotype similar to that of Ccr7−/− mice. CXCR5 is now known to be expressed also on CD4+ follicular helper T cells,69) which is a key regulator of germinal center reactions and CD8+ progenitor exhausted T cells.70)
c. In 1998, C. Caux described that in vitro cultured immature DCs derived from CD34+ hematopoietic progenitor cells and monocytes preferentially express CCR6 mRNA and respond to CCL20. CCR6 mRNA expression decreases progressively as DCs mature, whereas CCR7 mRNA is upregulated with maturation, and it responds to CCL19.71) This was the first indication that the chemokine-chemokine receptor system might regulate antigen-captured DC migration from the peripheral inflamed tissue to the draining LNs to initiate antigen-specific immune responses. This was confirmed in Ccr7−/− mice, and it contributed to the establishment of the linkage between the innate/inflammatory responses in the peripheral tissues and acquired immunity in the secondary lymphoid organ (draining LNs), and the spatio-temporal control by chemokine-regulated DCs.
d. CXCL12 was first cloned by K. Tashiro from the T. Honjo group using a signal trap method,72) and then identified by T. Nagasawa from the T. Kishimoto group as a stroma-cell derived pre-B cell growth-stimulating factor through expression cloning.73) Deficiency of Cxcl12 in mice manifested as defects in B cell lymphopoiesis in fetal liver and bone marrow, and also as severe reduction of myelopoiesis in bone marrow.74) Furthermore, Cxcl12−/− as well as its receptor Cxcr4−/− mice showed defective formation of large vessels that supply the gastrointestinal tract and cardiac ventricular septal defect, indicating a pivotal role of the CXCL12/CXCR4 axis in organ development as well as in hematopoiesis during embryogenesis.75) Nagasawa subsequently established CXCL12-abundant reticular cells (CAR cells), a population of bone marrow-specific mesenchymal stem cells, as the major cellular hematopoietic stem cell niche.76)
e. The Th1/Th2 dichotomy hypothesis proposed by T.R. Mosmann et al. has ruled the immunology field ever since.77) However, additional subsets of CD4 helper T cell populations, such as Th17, Tfh, and regulatory T (Treg) cells, were identified. Polarized effector T cell subsets infiltrate specific immune sites at specific times; however, the underlying molecular mechanism remained uncertain. In 1999, O. Yoshie and his colleagues, in collaboration with our team, identified that CCR4 is specifically expressed on a subset of the CD4+ memory T cell population from the peripheral blood of healthy volunteers.78) CCR4-positive cells were identified as Th2 cytokine-producing cells, whereas the CCR4-negative population preferentially produced Th1 cytokines. We also noticed that the CD25+ CD4+ memory population expressed CCR4; this population was later identified as FoxP3+ Treg cells.79) It is now well accepted that Th1 cells express CCR5, CXCR3, and CXCR6; Th2 cells express CCR4 and CCR8; Th17, CCR4 and CCR6; Treg cells express CCR4, CCR8, CCR7, and CXCR3; and Tfh cells express CXCR5.
f. In 2001, A. Zlotnik and his colleagues reported that CXCR4 and CCR7 are highly expressed in human breast cancer cells, and their respective ligands, CXCL12 and CCL21, are expressed in the metastasis target organs.80) These ligands induce actin polymerization and pseudopodia formation in cancer cells and subsequently chemotactic and invasive responses in vitro. In addition, in vivo neutralization of CXCL12/CXCR4 significantly impaired metastasis of breast cancer cells to regional LNs and the lungs. This study was followed by studies from numerous groups as reviewed by F. Balkwill.81) K. Yasumoto and his colleagues elucidated the role of the CXCL12/CXCR4 axis in the peritoneal carcinomatosis of gastric cancer.82) Chemokines such as CXCL8 and CXCL12 promote tumor growth, and CXCR1 is selectively expressed in cancer stem cells.83) Tumor cells themselves produce angiogenic chemokines with the ELR motif, such as CXCL8, in a Ras-dependent manner.84) These observations provide novel insights and are intriguing in cancer biology; however, their significance is not well-established in clinical settings.
g. O. Yoshie and his colleagues, in collaboration with us, reported the selective high expression of CCR4 in adult T-cell leukemia/lymphoma (ATLL) cells, which indicated that CCR4 can be used as a therapeutic target for ATLL.85) Kyowa Hakko Kogyo Co., Ltd. (now Kyowa Kirin Co., Ltd.), in collaboration with us, developed a monoclonal antibody against CCR4 with a high binding affinity but no neutralization activity. This antibody was then converted into the humanized antibody KW0761 using human IgG1 Fc, and it was defucosylated using Potelligent® technology.86) This enhanced the binding of the humanized antibody to FcγRIIIa on natural killer cells to show potent antibody-dependent cellular cytotoxicity activity. In 2006, a phase I clinical trial in patients with relapsed CCR4+ ATLL and peripheral T-cell lymphoma was conducted in Japan. Subsequently, a phase II study was started in 2010 for patients with relapsed CCR4+ ATLL using an infusion of 1.0 mg/kg once a week for 8 weeks. Objective responses were obtained in 13 of 26 evaluable patients with an overall objective response rate of 50%.87),88) Accordingly, the Japanese PMDA approved KW0761 (mogamulizumab) in March 2012 as a therapeutic agent for relapsed/refractory ATLL.89)
8. Chemokines in the era of cancer immunotherapy and single cell transcriptome
Recent developments in cancer immunotherapy, including immune checkpoint antibodies and chimeric antigen receptors T cell (CAR-T)/T cell receptor (TCR)-T cell therapy, have proven the existence of a cancer immune-surveillance system and thus revolutionized cancer therapy. In cancer, antigen-captured CCR7+ mature DCs migrate from the tumor tissue through lymphatic vessels to the draining LNs. CCR7+ naïve CD8+ T cells also enter the T cell areas of draining LNs through HEV in a CCR7-CCL21-dependent manner and communicate with each other to induce tumor antigen-specific clonal expansion.90),91) Tumor-specific CD8+ T cells differentiate into effector cells, exit from the LNs and then infiltrate the tumor via the circulation. In cancer immunotherapy, the efficient recruitment of tumor-specific CD8+ T cells to the tumor site is essential for successful treatment. Furthermore, CD8+ T cells recognize tumor antigens presented not only by tumor cells but also by various antigen-presenting immune cells and are reactivated or functionally modulated to exert their antitumor functions. In these processes, the chemokine system plays a pivotal role not only in immune cell migration but also in cell-cell interactions; thus, it is an important therapeutic target in cancer immunotherapy. Recent developments using single-cell RNA sequencing (scRNA-seq) technology have enabled precise analysis of the cellular composition of the tumor and helped to identify chemokine receptors and their ligands expressed by a variety of immune and non-immune cells in the tumor microenvironment. The results have helped to establish that chemokine-chemokine receptor interactions promote or regulate antitumor immune responses92) (Fig. 3).
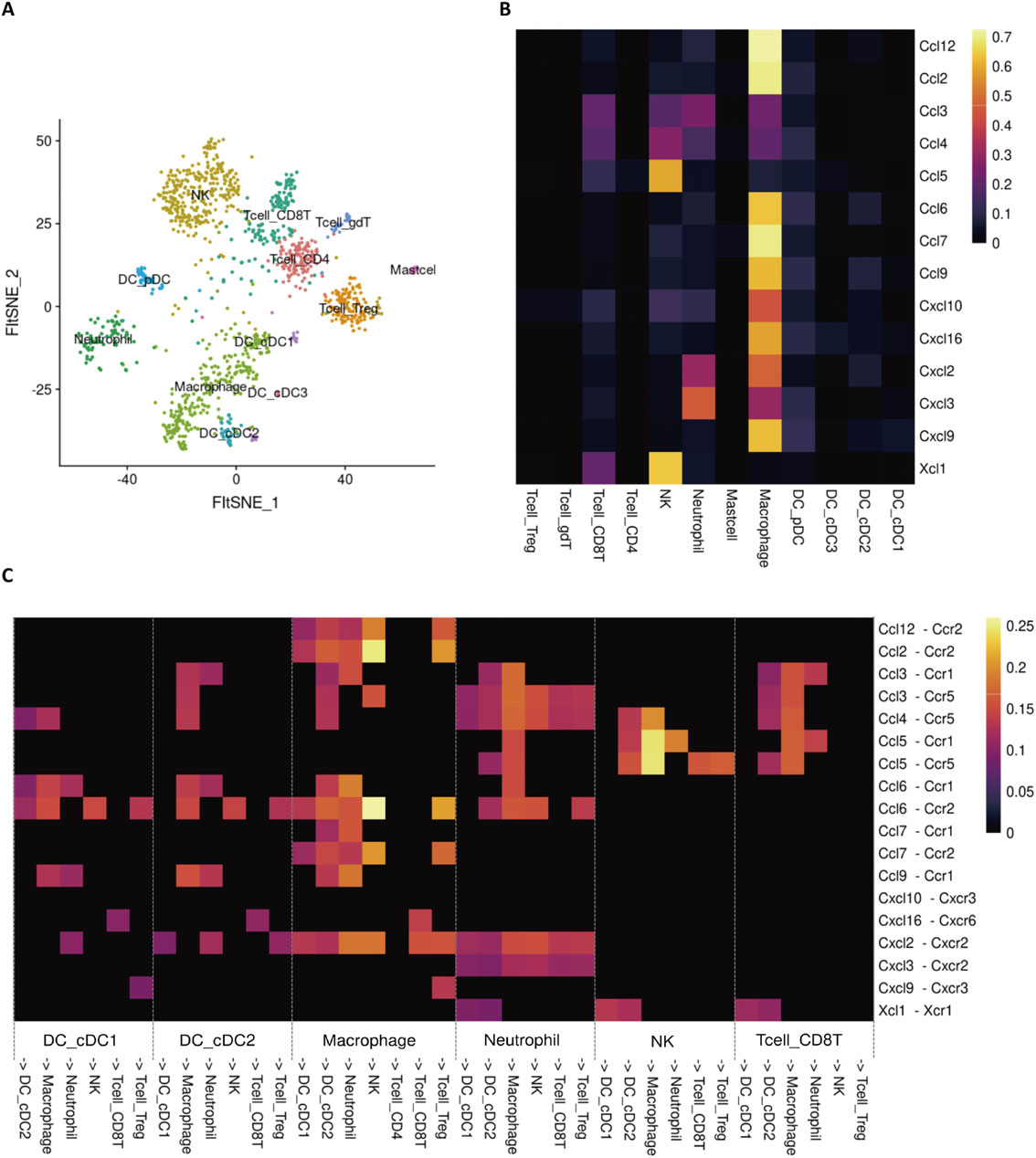
CXCR3 is highly expressed on CD8+ and CD4+ T cells that produce antitumor cytokines such as IFNγ. CXCL9 and CXCL10, which are CXCR3 ligands, are IFNγ-inducible genes. Their expression is induced by infiltration of IFNγ-producing T cells. Therefore, the CXCR3-ligand axis is believed to form a positive feedback loop that amplifies the mobilization of antitumor T cells into tumor tissues.93) In addition, IFNγ-stimulated DCs in tumors express high levels of CXCL9 and strongly interact with CXCR3+ CD8+ T cells, suggesting that the CXCR3 axis contributes to the reactivation and proliferation of IFNγ-producing T cells in the tumor and to their tumor invasion.94) CXCR6, like CXCR3, is a chemokine receptor highly expressed on a fraction of IFNγ-producing T cells. Its ligand, CXCL16, is highly expressed on mature regulatory DCs that express CCR7 within the tumor.92) Thus, the CXCR6-CXCL16 axis, like the CXCR3 axis, may contribute to the induction and maintenance of antitumor T cell responses.
b. Regulation of tumor infiltration and cell-cell interactions in immunosuppressive cells.
Tumor-infiltrating macrophages (TAMs), which are believed to promote immunosuppression, tissue remodeling, and angiogenesis, are derived from monocytes that infiltrate from the bone marrow into the blood and from the blood to the tumor in a CCR2-dependent manner.93),95) Inhibitors of CCR2 and its ligand, CCL2, are considered as potential TAM inhibitors. These inhibitors have been evaluated through several clinical trials; however, no clinical efficacy was reported. CCR2 contributes in part to the tumor recruitment of DCs. The reasons may be as follows. There is an immune-promoting subset within TAMs. TAMs themselves are important chemokine producers in the tumor field (Fig. 3B). These may limit the therapeutic efficacy of the CCR2-ligand axis inhibition. In addition, CD4+ Foxp3+ Treg cells, the key immunosuppressive cells in tumor immunity, highly express the chemokine receptors, CCR4 and CCR8, in the tumor.93) The expression of these receptors is low in Tregs in peripheral blood and lymphoid tissues; therefore, their expression increases with activation following tumor infiltration. This increased expression may contribute to interactions with TAMs that produce the CCR4 ligand, CCL22. Attempts were made to develop depleting antibodies targeting CCR4 and CCR8; however, clinical efficacy has not yet been reported.
c. Approaches to deliver engineered T cells to the tumor.
Induced expression of chemokine receptors on engineered T cells and induction of chemokine expression in tumors may be important approaches in CAR-T and TCR-T cell therapy, which may be useful against solid tumors. In the case of TCR-T cell therapy, the induction of CCR7 expression in engineered T cells may drive the further proliferation of the transferred cells in the draining LNs of patients. Induced expression of CXCR3 and CXCR6 may induce tumor infiltration and aid further activation of transfected T cells within the tumor. Induced expression of homing receptors of monocytes and neutrophils, such as CCR2, CXCR1, and CXCR2, in the engineered T cells may be useful against cancers with poor T cell infiltration. In the near future, it is expected that a resource of off-the-shelf T-cells equipped with various homing receptors will be established, enabling personalized therapy using engineered T cells with appropriate migratory capacity for each individual cancer.
In summary, the discovery of the chemokines IL-8 and MCAF solved the longstanding enigma and provided the molecular basis for subtype-specific leukocyte infiltration in inflammation. The subsequent discovery of the chemokine family revealed that homeostatic as well as pathological leukocyte migration and their interactions are regulated by various chemokines, and chemokines contribute even to the niche formation of hematopoiesis as well as lymphoid organogenesis. Furthermore, some of the chemokine receptors turned out to be co-receptors for HIV-1 infection. Chemokine research of the last 35 years has tremendously contributed not only to the fields of inflammation, immunology, and pathology but also to the broader life science. Chemokine research is still active now. We have also described the important role of chemokines and chemokine receptors in the era of cancer immunotherapy.
Acknowledgement
We would like to dedicate this review to Dr. Joost J. Oppenheim who passed away on May 14th, 2022. He provided KM with the opportunity to discover and study IL-8 and MCAF at NIH and continued with generous support of our studies on inflammation and immunological studies even after returning from NIH.
We would like to thank Editage (www.editage.com) for English language editing. Figures were created with BioRender.com.
Notes
Edited by Shizuo AKIRA, M.J.A.
Correspondence should be addressed to: K. Matsushima, Division of Molecular Regulation of Inflammation and Immune Diseases, Research Institute for Biomedical Sciences, Tokyo University of Science, 2669, Yamazaki, Noda, Chiba 278-0022, Japan (e-mail: koujim@rs.tus.ac.jp).
References
- 1) Anderson, W.A.D. (1971) Pathology (6th ed.). C. V. Mosby, Saint Louis, Mo.
- 2) Yoshimura, T., Matsushima, K., Tanaka, S., Robinson, E.A., Appella, E., Oppenheim, J.J. et al. (1987) Purification of a human monocyte-derived neutrophil chemotactic factor that has peptide sequence similarity to other host defense cytokines. Proc. Natl. Acad. Sci. U.S.A. 84, 9233–9237.
- 3) Matsushima, K., Morishita, K., Yoshimura, T., Lavu, S., Kobayashi, Y., Lew, W. et al. (1988) Molecular cloning of a human monocyte-derived neutrophil chemotactic factor (MDNCF) and the induction of MDNCF mRNA by interleukin 1 and tumor necrosis factor. J. Exp. Med. 167, 1883–1893.
- 4) Matsushima, K., Larsen, C.G., DuBois, G.C. and Oppenheim, J.J. (1989) Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J. Exp. Med. 169, 1485–1490.
- 5) Furutani, Y., Nomura, H., Notake, M., Oyamada, Y., Fukui, T., Yamada, M. et al. (1989) Cloning and sequencing of the cDNA for human monocyte chemotactic and activating factor (MCAF). Biochem. Biophys. Res. Commun. 159, 249–255.
- 6) Snyderman, R., Altman, L.C., Hausman, M.S. and Mergenhagen, S.E. (1972) Human mononuclear leukocyte chemotaxis: a quantitative assay for humoral and cellular chemotactic factors. J. Immunol. 108, 857–860.
- 7) Luger, T.A., Charon, J.A., Colot, M., Micksche, M. and Oppenheim, J.J. (1983) Chemotactic properties of partially purified human epidermal cell-derived thymocyte-activating factor (ETAF) for polymorphonuclear and mononuclear cells. J. Immunol. 131, 816–820.
- 8) Ming, W.J., Bersani, L. and Mantovani, A. (1987) Tumor necrosis factor is chemotactic for monocytes and polymorphonuclear leukocytes. J. Immunol. 138, 1469–1474.
- 9) Matsushima, K., Durum, S.K., Kimball, E.S. and Oppenheim, J.J. (1985) Purification of human interleukin 1 from human monocyte culture supernatants and identity of thymocyte comitogenic factor, fibroblast-proliferation factor, acute-phase protein-inducing factor, and endogenous pyrogen. Cell. Immunol. 92, 290–301.
- 10) Matsushima, K., Copeland, T.D., Onozaki, K. and Oppenheim, J.J. (1986) Purification and biochemical characteristics of two distinct human interleukins 1 from the myelomonocytic THP-1 cell line. Biochemistry 25, 3424–3429.
- 11) Tanaka, S., Robinson, E.A., Yoshimura, T., Matsushima, K., Leonard, E.J. and Appella, E. (1988) Synthesis and biological characterization of monocyte-derived neutrophil chemotactic factor. FEBS Lett. 236, 467–470.
- 12) Furuta, R., Yamagishi, J., Kotani, H., Sakamoto, F., Fukui, T., Matsui, Y. et al. (1989) Production and characterization of recombinant human neutrophil chemotactic factor. J. Biochem. 106, 436–441.
- 13) Walz, A., Peveri, P., Aschauer, H. and Baggiolini, M. (1987) Purification and amino acid sequencing of NAF, a novel neutrophil-activating factor produced by monocytes. Biochem. Biophys. Res. Commun. 149, 755–761.
- 14) Schröder, J.M., Mrowietz, U., Morita, E. and Christophers, E. (1987) Purification and partial biochemical characterization of a human monocyte-derived, neutrophil-activating peptide that lacks interleukin 1 activity. J. Immunol. 139, 3474–3483.
- 15) Van Damme, J., Van Beeumen, J., Opdenakker, G. and Billiau, A. (1988) A novel, NH2-terminal sequence-characterized human monokine possessing neutrophil chemotactic, skin-reactive, and granulocytosis-promoting activity. J. Exp. Med. 167, 1364–1376.
- 16) Utsunomiya, I., Ito, M., Watanabe, K., Tsurufuji, S., Matsushima, K. and Oh, S. (1996) Infiltration of neutrophils by intrapleural injection of tumour necrosis factor, interleukin-1, and interleukin-8 in rats, and its modification by actinomycin D. Br. J. Pharmacol. 117, 611–614.
- 17) Larsen, C.G., Anderson, A.O., Appella, E., Oppenheim, J.J. and Matsushima, K. (1989) The neutrophil-activating protein (NAP-1) is also chemotactic for T lymphocytes. Science 243, 1464–1466.
- 18) Mukaida, N., Shiroo, M. and Matsushima, K. (1989) Genomic structure of the human monocyte-derived neutrophil chemotactic factor IL-8. J. Immunol. 143, 1366–1371.
- 19) Nomiyama, H., Mera, A., Ohneda, O., Miura, R., Suda, T. and Yoshie, O. (2001) Organization of the chemokine genes in the human and mouse major clusters of CC and CXC chemokines: diversification between the two species. Genes Immun. 2, 110–113.
- 20) Mukaida, N., Mahe, Y. and Matsushima, K. (1990) Cooperative interaction of nuclear factor-kappa B- and cis-regulatory enhancer binding protein-like factor binding elements in activating the interleukin-8 gene by pro-inflammatory cytokines. J. Biol. Chem. 265, 21128–21133.
- 21) Mahé, Y., Mukaida, N., Kuno, K., Akiyama, M., Ikeda, N., Matsushima, K. et al. (1991) Hepatitis B virus X protein transactivates human interleukin-8 gene through acting on nuclear factor kB and CCAAT/enhancer-binding protein-like cis-elements. J. Biol. Chem. 266, 13759–13763.
- 22) Yasumoto, K., Okamoto, S., Mukaida, N., Murakami, S., Mai, M. and Matsushima, K. (1992) Tumor necrosis factor α and interferon γ synergistically induce interleukin 8 production in a human gastric cancer cell line through acting concurrently on AP-1 and NF-kB-like binding sites of the interleukin 8 gene. J. Biol. Chem. 267, 22506–22511.
- 23) Matsusaka, T., Fujikawa, K., Nishio, Y., Mukaida, N., Matsushima, K., Kishimoto, T. et al. (1993) Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc. Natl. Acad. Sci. U.S.A. 90, 10193–10197.
- 24) Mukaida, N., Morita, M., Ishikawa, Y., Rice, N., Okamoto, S., Kasahara, T. et al. (1994) Novel mechanism of glucocorticoid-mediated gene repression. Nuclear factor-kappa B is target for glucocorticoid-mediated interleukin 8 gene repression. J. Biol. Chem. 269, 13289–13295.
- 25) Okamoto, S., Mukaida, N., Yasumoto, K., Rice, N., Ishikawa, Y., Horiguchi, H. et al. (1994) The interleukin-8 AP-1 and kappa B-like sites are genetic end targets of FK506-sensitive pathway accompanied by calcium mobilization. J. Biol. Chem. 269, 8582–8589.
- 26) Clark-Lewis, I., Schumacher, C., Baggiolini, M. and Moser, B. (1991) Structure-activity relationships of interleukin-8 determined using chemically synthesized analogs. Critical role of NH2-terminal residues and evidence for uncoupling of neutrophil chemotaxis, exocytosis, and receptor binding activities. J. Biol. Chem. 266, 23128–23134.
- 27) Proost, P., Loos, T., Mortier, A., Schutyser, E., Gouwy, M., Noppen, S. et al. (2008) Citrullination of CXCL8 by peptidylarginine deiminase alters receptor usage, prevents proteolysis, and dampens tissue inflammation. J. Exp. Med. 205, 2085–2097.
- 28) Metzemaekers, M., Abouelasrar Salama, S., Vandooren, J., Mortier, A., Janssens, R., Vandendriessche, S. et al. (2021) From ELISA to immunosorbent tandem mass spectrometry proteoform analysis: The example of CXCL8/interleukin-8. Front. Immunol. 12, 644725.
- 29) Kuschert, G.S., Hoogewerf, A.J., Proudfoot, A.E., Chung, C.W., Cooke, R.M., Hubbard, R.E. et al. (1998) Identification of a glycosaminoglycan binding surface on human interleukin-8. Biochemistry 37, 11193–11201.
- 30) Clore, G.M., Appella, E., Yamada, M., Matsushima, K. and Gronenborn, A.M. (1989) Determination of the secondary structure of interleukin-8 by nuclear magnetic resonance spectroscopy. J. Biol. Chem. 264, 18907–18911.
- 31) Clore, G.M., Appella, E., Yamada, M., Matsushima, K. and Gronenborn, A.M. (1990) Three-dimensional structure of interleukin 8 in solution. Biochemistry 29, 1689–1696.
- 32) Baldwin, E.T., Franklin, K.A., Appella, E., Yamada, M., Matsushima, K., Wlodawer, A. et al. (1990) Crystallization of human interleukin-8. A protein chemotactic for neutrophils and T-lymphocytes. J. Biol. Chem. 265, 6851–6853.
- 33) Altman, L.C., Snyderman, R., Oppenheim, J.J. and Mergenhagen, S.E. (1973) A human mononuclear leukocyte chemotactic factor: characterization, specificity and kinetics of production by homologous leukocytes. J. Immunol. 110, 801–810.
- 34) Yoshimura, T., Robinson, E.A., Tanaka, S., Appella, E., Kuratsu, J. and Leonard, E.J. (1989) Purification and amino acid analysis of two human glioma-derived monocyte chemoattractants. J. Exp. Med. 169, 1449–1459.
- 35) Yoshimura, T., Yuhki, N., Moore, S.K., Appella, E., Lerman, M.I. and Leonard, E.J. (1989) Human monocyte chemoattractant protein-1 (MCP-1). Full-length cDNA cloning, expression in mitogen-stimulated blood mononuclear leukocytes, and sequence similarity to mouse competence gene JE. FEBS Lett. 244, 487–493.
- 36) Harada, A., Sekido, N., Kuno, K., Akiyama, M., Kasahara, T., Nakanishi, I. et al. (1993) Expression of recombinant rabbit IL-8 in Escherichia coli and establishment of the essential involvement of IL-8 in recruiting neutrophils into lipopolysaccharide-induced inflammatory site of rabbit skin. Int. Immunol. 5, 681–690.
- 37) Wada, T., Tomosugi, N., Naito, T., Yokoyama, H., Kobayashi, K., Harada, A. et al. (1994) Prevention of proteinuria by the administration of anti-interleukin 8 antibody in experimental acute immune complex-induced glomerulonephritis. J. Exp. Med. 180, 1135–1140.
- 38) Sekido, N., Mukaida, N., Harada, A., Nakanishi, I., Watanabe, Y. and Matsushima, K. (1993) Prevention of lung reperfusion injury in rabbits by a monoclonal antibody against interleukin-8. Nature 365, 654–657.
- 39) Matsumoto, T., Ikeda, K., Mukaida, N., Harada, A., Matsumoto, Y., Yamashita, J. et al. (1997) Prevention of cerebral edema and infarct in cerebral reperfusion injury by an antibody to interleukin-8. Lab. Invest. 77, 119–125.
- 40) Yokoi, K., Mukaida, N., Harada, A., Watanabe, Y. and Matsushima, K. (1997) Prevention of endotoxemia-induced acute respiratory distress syndrome-like lung injury in rabbits by a monoclonal antibody to IL-8. Lab. Invest. 76, 375–384.
- 41) Harada, A., Mukaida, N. and Matsushima, K. (1996) Interleukin 8 as a novel target for intervention therapy in acute inflammatory diseases. Mol. Med. Today 2, 482–489.
- 42) Wada, T., Yokoyama, H., Furuichi, K., Kobayashi, K.I., Harada, K., Naruto, M. et al. (1996) Intervention of crescentic glomerulonephritis by antibodies to monocyte chemotactic and activating factor (MCAF/MCP-1). FASEB J. 10, 1418–1425.
- 43) Kimura, H., Kasahara, Y., Kurosu, K., Sugito, K., Takiguchi, Y., Terai, M. et al. (1998) Alleviation of monocrotaline-induced pulmonary hypertension by antibodies to monocyte chemotactic and activating factor/monocyte chemoattractant protein-1. Lab. Invest. 78, 571–581.
- 44) Furukawa, Y., Matsumori, A., Ohashi, N., Shioi, T., Ono, K., Harada, A. et al. (1999) Anti-monocyte chemoattractant protein-1/monocyte chemotactic and activating factor antibody inhibits neointimal hyperplasia in injured rat carotid arteries. Circ. Res. 84, 306–314.
- 45) Cacalano, G., Lee, J., Kikly, K., Ryan, A.M., Pitts-Meek, S., Hultgren, B. et al. (1994) Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science 265, 682–684.
- 46) Kurihara, T., Warr, G., Loy, J. and Bravo, R. (1997) Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J. Exp. Med. 186, 1757–1762.
- 47) Bachelerie, F., Ben-Baruch, A., Burkhardt, A.M., Combadiere, C., Farber, J.M., Graham, G.J. et al. (2014) International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 66, 1–79.
- 48) Holmes, W.E., Lee, J., Kuang, W.J., Rice, G.C. and Wood, W.I. (1991) Structure and functional expression of a human interleukin-8 receptor. Science 253, 1278–1280.
- 49) Murphy, P.M. and Tiffany, H.L. (1991) Cloning of complementary DNA encoding a functional human interleukin-8 receptor. Science 253, 1280–1283.
- 50) Boulay, F., Tardif, M., Brouchon, L. and Vignais, P. (1990) Synthesis and use of a novel N-formyl peptide derivative to isolate a human N-formyl peptide receptor cDNA. Biochem. Biophys. Res. Commun. 168, 1103–1109.
- 51) Hol, J., Wilhelmsen, L. and Haraldsen, G. (2010) The murine IL-8 homologues KC, MIP-2, and LIX are found in endothelial cytoplasmic granules but not in Weibel-Palade bodies. J. Leukoc. Biol. 87, 501–508.
- 52) Spehlmann, M.E., Dann, S.M., Hruz, P., Hanson, E., McCole, D.F. and Eckmann, L. (2009) CXCR2-dependent mucosal neutrophil influx protects against colitis-associated diarrhea caused by an attaching/effacing lesion-forming bacterial pathogen. J. Immunol. 183, 3332–3343.
- 53) Herbold, W., Maus, R., Hahn, I., Ding, N., Srivastava, M., Christman, J.W. et al. (2010) Importance of CXC chemokine receptor 2 in alveolar neutrophil and exudate macrophage recruitment in response to pneumococcal lung infection. Infect. Immun. 78, 2620–2630.
- 54) Cai, S., Batra, S., Lira, S.A., Kolls, J.K. and Jeyaseelan, S. (2010) CXCL1 regulates pulmonary host defense to Klebsiella infection via CXCL2, CXCL5, NF-κB, and MAPKs. J. Immunol. 185, 6214–6225.
- 55) Reutershan, J., Morris, M.A., Burcin, T.L., Smith, D.F., Chang, D., Saprito, M.S. et al. (2006) Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J. Clin. Invest. 116, 695–702.
- 56) Broxmeyer, H.E., Cooper, S., Cacalano, G., Hague, N.L., Bailish, E. and Moore, M.W. (1996) Involvement of Interleukin (IL) 8 receptor in negative regulation of myeloid progenitor cells in vivo: evidence from mice lacking the murine IL-8 receptor homologue. J. Exp. Med. 184, 1825–1832.
- 57) Tsou, C.L., Peters, W., Si, Y., Slaymaker, S., Aslanian, A.M., Weisberg, S.P. et al. (2007) Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Invest. 117, 902–909.
- 58) Peters, W. and Charo, I.F. (2001) Involvement of chemokine receptor 2 and its ligand, monocyte chemoattractant protein-1, in the development of atherosclerosis: lessons from knockout mice. Curr. Opin. Lipidol. 12, 175–180.
- 59) Matsushima, K., Yang, D. and Oppenheim, J.J. (2022) Interleukin-8: An evolving chemokine. Cytokine 153, 155828.
- 60) Zlotnik, A. and Yoshie, O. (2000) Chemokines: a new classification system and their role in immunity. Immunity 12, 121–127.
- 61) Miller, L.H., Mason, S.J., Dvorak, J.A., McGinniss, M.H. and Rothman, I.K. (1975) Erythrocyte receptors for (Plasmodium knowlesi) malaria: Duffy blood group determinants. Science 189, 561–563.
- 62) Borroni, E.M., Cancellieri, C., Vacchini, A., Benureau, Y., Lagane, B., Bachelerie, F. et al. (2013) β-arrestin-dependent activation of the cofilin pathway is required for the scavenging activity of the atypical chemokine receptor D6. Sci. Signal 6, ra30. Correction: Sci. Signal 6, er5.
- 63) Feng, Y., Broder, C.C., Kennedy, P.E. and Berger, E.A. (1996) HIV-1 entry cofactor: Functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272, 872–877.
- 64) Trkola, A., Dragic, T., Arthos, J., Binley, J.M., Olson, W.C., Allaway, G.P. et al. (1996) CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature 384, 184–187.
- 65) Dean, M., Carrington, M., Winkler, C., Huttley, G.A., Smith, M.W., Allikmets, R. et al. (1996) Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 273, 1856–1862.
- 66) Förster, R., Mattis, A.E., Kremmer, E., Wolf, E., Brem, G. and Lipp, M. (1996) A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell 87, 1037–1047.
- 67) Förster, R., Schubel, A., Breitfeld, D., Kremmer, E., Renner-Müller, I., Wolf, E. et al. (1999) CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 99, 23–33.
- 68) Nakano, H. and Gunn, M.D. (2001) Gene duplications at the chemokine locus on mouse chromosome 4: multiple strain-specific haplotypes and the deletion of secondary lymphoid-organ chemokine and EBI-1 ligand chemokine genes in the plt mutation. J. Immunol. 166, 361–369.
- 69) Breitfeld, D., Ohl, L., Kremmer, E., Ellwart, J., Sallusto, F., Lipp, M. et al. (2000) Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 192, 1545–1552.
- 70) Im, S.J., Hashimoto, M., Gerner, M.Y., Lee, J., Kissick, H.T., Burger, M.C. et al. (2016) Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421.
- 71) Dieu, M.C., Vanbervliet, B., Vicari, A., Bridon, J.M., Oldham, E., Ait-Yahia, S. et al. (1998) Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med. 188, 373–386.
- 72) Tashiro, K., Tada, H., Heilker, R., Shirozu, M., Nakano, T. and Honjo, T. (1993) Signal sequence trap: a cloning strategy for secreted proteins and type I membrane proteins. Science 261, 600–603.
- 73) Nagasawa, T., Kikutani, H. and Kishimoto, T. (1994) Molecular cloning and structure of a pre-B-cell growth-stimulating factor. Proc. Natl. Acad. Sci. U.S.A. 91, 2305–2309.
- 74) Nagasawa, T., Hirota, S., Tachibana, K., Takakura, N., Nishikawa, S., Kitamura, Y. et al. (1996) Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 382, 635–638.
- 75) Tachibana, K., Hirota, S., Iizasa, H., Yoshida, H., Kawabata, K., Kataoka, Y. et al. (1998) The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393, 591–594.
- 76) Sugiyama, T., Kohara, H., Noda, M. and Nagasawa, T. (2006) Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25, 977–988.
- 77) Mosmann, T.R., Cherwinski, H., Bond, M.W., Giedlin, M.A. and Coffman, R.L. (1986) Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136, 2348–2357.
- 78) Imai, T., Nagira, M., Takagi, S., Kakizaki, M., Nishimura, M., Wang, J. et al. (1999) Selective recruitment of CCR4-bearing Th2 cells toward antigen-presenting cells by the CC chemokines thymus and activation-regulated chemokine and macrophage-derived chemokine. Int. Immunol. 11, 81–88.
- 79) Yuan, Q., Bromley, S.K., Means, T.K., Jones, K.J., Hayashi, F., Bhan, A.K. et al. (2007) CCR4-dependent regulatory T cell function in inflammatory bowel disease. J. Exp. Med. 204, 1327–1334.
- 80) Müller, A., Homey, B., Soto, H., Ge, N., Catron, D., Buchanan, M.E. et al. (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410, 50–56.
- 81) Balkwill, F. (2004) The significance of cancer cell expression of the chemokine receptor CXCR4. Semin. Cancer Biol. 14, 171–179.
- 82) Yasumoto, K., Koizumi, K., Kawashima, A., Saitoh, Y., Arita, Y., Shinohara, K. et al. (2006) Role of the CXCL12/CXCR4 axis in peritoneal carcinomatosis of gastric cancer. Cancer Res. 66, 2181–2187.
- 83) Chen, L., Fan, J., Chen, H., Meng, Z., Chen, Z., Wang, P. et al. (2014) The IL-8/CXCR1 axis is associated with cancer stem cell-like properties and correlates with clinical prognosis in human pancreatic cancer cases. Sci. Rep. 4, 5911.
- 84) Ancrile, B.B., O’Hayer, K.M. and Counter, C.M. (2008) Oncogenic ras-induced expression of cytokines: a new target of anti-cancer therapeutics. Mol. Interv. 8, 22–27.
- 85) Yoshie, O., Fujisawa, R., Nakayama, T., Harasawa, H., Tago, H., Izawa, D. et al. (2002) Frequent expression of CCR4 in adult T-cell leukemia and human T-cell leukemia virus type 1-transformed T cells. Blood 99, 1505–1511.
- 86) Niwa, R., Sakurada, M., Kobayashi, Y., Uehara, A., Matsushima, K., Ueda, R. et al. (2005) Enhanced natural killer cell binding and activation by low-fucose IgG1 antibody results in potent antibody-dependent cellular cytotoxicity induction at lower antigen density. Clin. Cancer Res. 11, 2327–2336.
- 87) Yamamoto, K., Utsunomiya, A., Tobinai, K., Tsukasaki, K., Uike, N., Uozumi, K. et al. (2010) Phase I study of KW-0761, a defucosylated humanized anti-CCR4 antibody, in relapsed patients with adult T-cell leukemia-lymphoma and peripheral T-cell lymphoma. J. Clin. Oncol. 28, 1591–1598.
- 88) Ishida, T., Joh, T., Uike, N., Yamamoto, K., Utsunomiya, A., Yoshida, S. et al. (2012) Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J. Clin. Oncol. 30, 837–842.
- 89) Yoshie, O. and Matsushima, K. (2015) CCR4 and its ligands: from bench to bedside. Int. Immunol. 27, 11–20.
- 90) Roberts, E.W., Broz, M.L., Binnewies, M., Headley, M.B., Nelson, A.E., Wolf, D.M. et al. (2016) Critical role for CD103+/CD141+ dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell 30, 324–336.
- 91) Förster, R., Davalos-Misslitz, A.C. and Rot, A. (2008) CCR7 and its ligands: balancing immunity and tolerance. Nat. Rev. Immunol. 8, 362–371.
- 92) Chen, C.Y., Ueha, S., Ishiwata, Y., Shichino, S., Yokochi, S., Yang, D. et al. (2021) Combining an alarmin HMGN1 peptide with PD-L1 blockade results in robust antitumor effects with a concomitant increase of stem-like/progenitor exhausted CD8+ T cells. Cancer Immunol. Res. 9, 1214–1228.
- 93) Kohli, K., Pillarisetty, V.G. and Kim, T.S. (2022) Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 29, 10–21.
- 94) Chow, M.T., Ozga, A.J., Servis, R.L., Frederick, D.T., Lo, J.A., Fisher, D.E. et al. (2019) Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity 50, 1498–1512.e5.
- 95) Sawanobori, Y., Ueha, S., Kurachi, M., Shimaoka, T., Talmadge, J.E., Abe, J. et al. (2008) Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood 111, 5457–5466.
Profile

Kouji Matsushima was born in 1952 in Takaoka, Toyama, Japan. He graduated from the School of Medicine, Kanazawa University in 1978 (M.D.), and obtained his Ph.D. from the Graduate School of Medicine, Kanazawa University in 1982. He joined as a post-doctoral fellow the Laboratory of Dr. Joost J. Oppenheim at National Institute of Dental Research, National Institutes of Health (NIH) in the fall of 1982 and moved to the National Cancer Institute (NCI), NIH with Dr. Oppenheim in the spring of 1983. His major scientific achievements whilst at NIH were: 1) Purification of human interleukin-1 (IL-1) α and β, and identification of the amino-terminus of mature/active form of human IL-1β (cleavage site of caspase-1) and 2) Discovery of chemokines, interleukin-8 (IL-8, CXCL8) and MCAF (MCP-1, CCL2). He became a visiting scientist and was offered a tenure position at NCI in 1989. He returned to the Cancer Research Institute, Kanazawa University as a professor in 1990, and examined the pivotal roles of IL-8 in regulating neutrophils in acute inflammation models and MCAF in chronic inflammation models, resolving a longstanding enigma regarding the molecular mechanism of subset-specific leukocyte infiltration in inflammation. He moved to the Department of Molecular Preventive Medicine, The University of Tokyo in 1996, and developed an anti-CCR4 antibody for the treatment of adult T cell leukemia and lymphoma in collaboration with Kyowa Hakko Kirin Co., Ltd. in 2012. He moved to the Research Institute for Biomedical Sciences, Tokyo University of Science in 2018. He founded the Japanese Society of Molecular Cell Biology of Macrophages and also acted as a senior council member in several academic societies, including the International Association of Inflammation Societies (President, 2011–2013), The Japanese Society for Immunology and The Japanese Society for Inflammation and Regeneration (President, 2010–2012). He has been awarded the Public Health Special Recognition Award from DHHS, U.S.A. in 1991, Japanese Cancer Association-CHAAO Award in 2012, Japanese Society of Pharmacological Science-Science Award for Drug Development in 2015, The Commendation for Science and Technology by the Minister of Education, Culture, Sports, Science and Technology of Japan in 2016, Japanese Society of Immunology-Human Immunology Research Award in 2016, International Cytokine Interferon Society-The Life Time Honorary Membership Award in 2019, Toray Science and Technology Award in 2020, and Takeda Medical Award in 2021.
