Abstract
Recent studies have highlighted the impact of disrupted maternal gut microbiota on the colonization of offspring gut microbiota, with implications for offspring developmental trajectories. The extent to which offspring inherit the characteristics of altered maternal gut microbiota remains elusive. In this study, we employed a mouse model where maternal gut microbiota disruption was induced using non-absorbable antibiotics. Systematic chronological analyses of dam fecal samples, offspring luminal content, and offspring gut tissue samples revealed a notable congruence between offspring gut microbiota profiles and those of the perturbed maternal gut microbiota, highlighting the profound influence of maternal microbiota on early-life colonization of offspring gut microbiota. Nonetheless, certain dominant bacterial genera in maternal microbiota did not transfer to the offspring, indicating a bacterial taxonomy-dependent mechanism in the inheritance of maternal gut microbiota. Our results embody the vertical transmission dynamics of disrupted maternal gut microbiota in an animal model, where the gut microbiota of an offspring closely mirrors the gut microbiota of its mother.
1. Introduction
The microbial bond links mother and offspring in humans and other mammals.1) The early-life gut microbiota is primarily seeded by the environmental microbes that offspring encounter from the time of delivery, and the maternal microbiota plays a pivotal role in colonization of the gut microbiota of offspring2),3). Various factors, such as exposure to antibiotics or other microbe-affecting drugs, infection, stress, and diet during perinatal periods, can significantly alter the maternal microbiota.1),4) Perturbation of the maternal microbiota during perinatal periods can potentially affect the colonization of offspring gut microbiota, leading to long-term effects on the developmental outcomes of offspring.5)-10) Studies have suggested that factors that possibly alter the composition of maternal microbiota are associated with increased risks of several diseases in offspring, including neurodevelopmental disorders, allergies, and metabolic diseases.11)-14) Although previous studies have indicated the association between the perturbed maternal gut microbiota and affected colonization of offspring gut microbiota, it remains unclear to what extent the offspring inherit the profiles of the perturbed maternal gut microbiota, which directly influences the health outcomes of offspring.
To address these questions, we developed a mouse model, the ‘perturbed maternal gut microbiota’ (PMGM) model, in which the maternal gut microbiota is perturbed by administrating non-absorbable antibiotics (AB) to dams.7) Our previous study showed that the offspring born from the PMGM model exhibited abnormal behavior, including a spatial preference, which could be partially ameliorated by being nurtured by normal dams postnatally.7) In this study, we performed systematic chronological analyses of the gut microbiota of dams and their offspring in the PMGM model to investigate the vertical transmission of perturbed maternal gut microbiota to offspring and its impact on the postnatal colonization of gut microbiota. Through microbiome analyses of dam fecal samples, offspring luminal content, and offspring gut tissue samples, we discerned parallels and disparities between the profiles of offspring gut microbiota and those of the perturbed maternal gut microbiota. These findings demonstrated the vertical transmission of perturbed maternal gut microbiota and highlighted the influences of perturbation of maternal microbiota on early-life colonization of offspring gut microbiota.
2. Materials and methods
2.1. Ethics statement.
All animal procedures complied with the National Institute of Health Guide for the Care and Use of Laboratory Animals. Ethical approval for animal manipulations was approved by the Committee for the Ethics of Animal Research at Suzuka University of Medical Science and the Animal Research Committee of University of Fukui.
2.2. Animals and non-absorbable antibiotics treatment.
Specific pathogen-free timed-pregnant C57BL/6J mice were purchased from SLC Japan (Hamamatsu, Japan). Embryonic and postnatal stages were defined with the day of vaginal plug detection as E0 and the day of birth of offspring as P0. We perturbed the gut microbiota of mice by administering a combination of non-absorbable AB to pregnant mice in accordance with previously published protocols with minor modifications.7),15)-17) Non-absorbable AB becomes concentrated in the gastrointestinal tract, thereby prohibiting the growth of intestinal microbiota with minimal penetration into serum.16) Pregnant C57BL/6J mice received a solution of non-absorbable AB, which consisted of 4 mg/ml neomycin trisulfate salt hydrate (Sigma-Aldrich, St. Louis, MO, USA; N6386-25G), 300 U/ml bacitracin (Sigma-Aldrich; B0125-1250KU), 1.25 μ g/ml pimaricin (Sigma-Aldrich; P9703-25MG; 5 mg/ml solution in acetic acid used as stock), and 0.075% (v/v) acetic acid (Nacalai Tesque, Kyoto, Japan; 08885-45) dissolved in drinking water by voluntary drinking on E9–E16 (n = 26). Previous research showed that intraperitoneal administration of non-absorbable AB solution to SPF mice or oral administration of the non-absorbable AB solution to germ-free mice did not induce any apparent effects,16) suggesting that the non-absorbable AB solution used in this study had minimal collateral effects. Control mice were given normal drinking water (n = 26). Mice were fed a certified diet (MF, Oriental Yeast, Tokyo, Japan). On E17, the cages and lids were replaced to eliminate any residue of the AB solution, ensuring that the offspring would not be directly exposed to the AB. Control and AB-treated mice gave birth to pups and provided them with nurturing ad libitum. The number of pups was not intentionally regulated. The offspring were weaned from their mothers at P23, and 2 to 5 sex-matched mice from control or AB-treated mice were cohoused in a single cage. The offspring from control and AB-treated dams were never cohoused in the same cage to avoid cross-contamination of indigenous bacteria. Male mice pups were decapitated under deep anesthesia using isoflurane or ice anesthesia for luminal contents and large intestinal tissue removal. The large intestine (cecum, colon, and rectum) was then removed, and luminal contents were extracted by carefully incising the intestinal wall using Vannas-style spring micro scissors with curved blades (No. 15000-04, Fine Science Tools, Canada), where liquid and solid contents were present and gently extracting them with forceps (DUMONT No.5/45°, Fine Science Tools) into a microcentrifuge tube. Harsh squeezing of the tissue was avoided to minimize disruption of the mucosa-associated microbiota. Large intestinal tissue samples were rinsed in saline to remove the remaining luminal contents, placed into a microcentrifuge tube, and frozen in liquid nitrogen. The weight of each microcentrifuge tube was measured before and after sampling to investigate the weight of the tube contents. All samples were stored at -80℃ until homogenization using a bead-beating protocol before RNA extraction and sequencing.
2.3. Microbiota analyses.
Bacterial RNA, not DNA, was analyzed to identify more active bacteria for microbiota analyses.18) After bead beating of the fecal, luminal, and large intestinal tissue samples, bacterial RNA was extracted using QuickGene RNA tissue kit S (KURABO, Osaka, Japan) and treated with DNase using RNase-Free DNase set (Qiagen, Tokyo, Japan). cDNA was synthesized using ReverTra AceⓇ qPCR RT kit (TOYOBO, Osaka, Japan). RNA extraction and cDNA synthesis were conducted in accordance with the manufacturer’s instructions. Library preparation and deep sequencing were performed exactly as Inoue et al. described, using MiSeq (Illumina, Tokyo, Japan).19) The sequencing data were analyzed using QIIME2 (ver 2021.11) with SILVA 138 as a database instead of GreenGenes following the protocol described by Liyanage et al.20) Based on the quality check conducted in QIIME2, two offspring luminal content samples (one each from P2 Control and P4 Control) and six offspring intestinal tissue samples (one each from P2 Control, P7 Control, P11 Control, P4 Antibiotics, and P11 Antibiotics, and two from P7 Control) were excluded from the processed data. The relative abundances of bacteria across various stages and groups are detailed in Supplementary Data 1-3. Bacterial quantification was performed using the following method. Real-time PCR was used with cDNA synthesized with ReverTra AceⓇ qPCR RT kit (TOYOBO, Osaka, Japan). PCR amplification was performed with total bacteria primers 5'- GTGSTGCAYGGYTGTCGTCA-3' and 5'-ACGTCRTCCMCACCTTCCTC-3'.21) The PCR amplification protocol consisted of an initial denaturation for 10 s at 95℃, followed by 40 cycles of melting and annealing/extension at 95℃ for 20 s and 65℃ for 31 s, respectively. Total bacterial cell numbers were calculated using E.coli DH5a (Toyobo, Osaka, Japan) cell numbers as a reference. SYBR premix Ex Taq (Takara Bio, Shiga, Japan) was used in both methods for amplification in a 10 μL reaction volume containing 1 μL of cDNA extract and 0.2 μmol/L of each primer.
2.4. Statistical analysis.
Values were expressed as mean ± standard error of the mean (SE) unless stated otherwise. Statistical analyses were conducted using GraphPad Prism version 9.5.1 (GraphPad Software, Inc., La Jolla, CA, USA). We used either a two-tailed unpaired t test or multiple unpaired t tests to compare mean values between two groups in accordance with the GraphPad Prism instructions. P-values < 0.05 were considered to indicate statistical significance. We conducted principal coordinate analysis (PCoA) using UniFrac distances within the QIIME2 platform. Additionally, we calculated PERMANOVA p-values (9999 permutations) and generated PCoA plots based on Bray-Curtis dissimilarities in R (version 4.2.2) using packages qiime2R,22) Phyloseq,23) MicrobiotaProcess,24) microbiomeutilities,25) and vegan.26)
3. Results
3.1. The decreased diversity of maternal perturbed gut microbiota was reflected in the diversity of offspring gut microbiota during the early colonization period.
We administered non-absorbable AB to pregnant mice on embryonic day 9 (E9)–E16 in order to establish the PMGM model.7) To investigate the impact of perturbations in maternal gut microbiota on the postnatal colonization of offspring gut microbiota, we performed comparative analyses of gut microbiota profiles. This involved comparing control dams with AB-treated dams and offspring born from control dams (control offspring) with those born from AB-treated dams (AB offspring) using 16S rRNA sequencing. Maternal fecal samples were collected at various time points: E9 prior to AB administration, E14, E16, E18, postnatal day (P) 2, P4, P7, P9, P14, P18, and P23. We also collected gut contents and tissues of offspring at different postnatal stages, including P2, P4, P7, P11, P14, and P18, to analyze gut microbiota in the intestinal lumen (luminal microbiota: LM) and the microbiota present in the mucus surface of the colon and in proximity to intestinal epithelial cells (mucosa-associated microbiota: MAM).27) Offspring fecal samples were analyzed at P23 in place of the gut contents to analyze the bacterial components of the LM. The results showed that the number of total bacterial cells in feces significantly declined during AB administration, with a return to control levels and even exceeding control levels after cessation of AB administration. Moreover, the total bacterial cell numbers in AB-exposed dams tended to be even higher than those in the control dams at later stages (Fig. 1A). Notably, the bacterial cell numbers in the LM remained comparable between control and AB offspring, except for at P14, when the LM count was significantly higher in AB offspring (Fig. 1B). MAM bacterial cell numbers in AB offspring tended to exceed those in control offspring at P7-14 but showed a significant decrease at P18 (Fig. 1C). Alpha diversity assessment based on Chao1 and Shannon indices showed that the bacterial richness of maternal gut microbiota remained stable throughout the antenatal and postnatal periods in control dams, whereas diversity decreased significantly by AB administration. In contrast to the bacterial cell numbers, the decreased bacterial richness failed to revert to control levels after AB cessation and persisted across the stages examined in AB-treated dams (Fig. 1D and G). The bacterial richness of offspring LM tended to be greater in AB offspring compared with control offspring at P2-11. However, this trend reversed course, as control offspring exhibited significantly higher richness at P18 and P23 (Fig. 1E and H). This observation suggested a gradual increase in LM diversity in control offspring during postnatal stages, with a particularly steep increase after P18. Notably, this increase after P18 was attenuated in the LM of AB offspring (Fig. 1E and H). Chao1 and Shannon indices of control offspring MAM remained relatively stable from onset of the postnatal period, with a modest rise at P11. Notably, the Chao1 indices for MAM in AB offspring exhibited a modest decline after P11, culminating in a significantly reduced level compared with control offspring at P18 (Fig. 1F). In contrast, the MAM Shannon indices remained similar between control and AB offspring in the stages subsequent to P11 (Fig. 1I). Collectively, our results show that AB treatment causes a significant decrease in the alpha diversity of maternal gut microbiota, which influences the diversity trajectories of offspring LM and MAM.

Fig. 1
(Color online) E9-E16 perturbation of maternal gut microbiota exerts influences on the maternal microbiota (Dam), offspring luminal microbiota (LM), and offspring mucosa-associated microbiota (MAM) in the PMGM model. (A) Longitudinal alterations in estimated cell numbers in maternal fecal samples. Control dams (Control) and antibiotics (AB)-treated dams (Antibiotics); n=6 at each stage for both groups. (B) Longitudinal alterations in estimated cell numbers in the LM of offspring. Control offspring (Control): P2, n=7; P4, n=3; P7, n=3; P11, n=6; P14, n=6; P18, n=6. Antibiotics: P2, n=7; P4, n=4; P7, n=3; P11, n=5; P14, n=5; P18, n=5. (C) Longitudinal alterations in estimated cell numbers in the MAM of offspring. Control: P2, n=7; P4, n=3; P7, n=3; P11, n=6; P14, n=6; P18, n=6. Antibiotics: P2, n=7; P4, n=4; P7, n=3; P11, n=5; P14, n=5; P18, n=5. (D, G) Temporal alteration in alpha diversity of maternal gut microbiota based on the Chao1 index (D) and the Shannon index (G) throughout the experiment. n=6 at each stage for both groups. The alpha diversity was suppressed after exposure to non-absorbable AB. (E, H) Temporal alteration in alpha diversity of offspring LM based on the Chao1 index (E) and the Shannon index (H). The alpha diversity in Control samples increased at stages after P14, whereas the increase in alpha diversity in AB offspring was suppressed at these stages. Control: P2, n=6; P4, n=6; P7, n=8; P11, n=6; P14, n=6; P18, n=6; P23, n=7. Antibiotics: P2, n=7; P4, n=6; P7, n=5; P11, n=5; P14, n=6; P18, n=5; P23, n=3. Gut contents were analyzed for P2-18 LM. (F, I) Temporal alteration in alpha diversity of the MAM based on the Chao1 index (F) and the Shannon index (I). Control: P2, n=6; P4, n=7; P7, n=6; P11, n=5; P14, n=6; P18, n=6. Antibiotics: P2, n=6; P4, n=5; P7, n=5; P11, n=6; P14, n=5; P18, n=5. Data are shown as the mean ± SE; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 (Multiple unpaired t tests).
PCoA based on weighted UniFrac distances revealed significant disparities in the beta diversity of gut microbiota between control dams and AB-treated dams, persisting across nearly all stages after the onset of AB administration (Supplementary Fig. 1A). Comparative evaluations of the gut microbiota profiles between stages, as indicated by PERMANOVA p-values, demonstrated the general stability of control gut microbiota throughout the stages examined (Supplementary Fig. 1B). In contrast, the profiles of gut microbiota perturbed by AB administration continued to vary after the cessation of AB administration, with the effects particularly evident from E16 to P4 (Supplementary Fig. 1B). Significant differences in beta diversity in the LM were observed between control and AB offspring at each postnatal stage except for P2 (Supplementary Fig. 2A). The LM profiles of control offspring, based on PERMANOVA p-values, demonstrated marked alterations at P18 and P23 compared with the earlier stages. Conversely, the LM profiles in AB offspring exhibited sustained variability after birth, notably from P2 to P18 (Supplementary Fig. 2B). Beta diversity analyses also indicated significant disparities in the MAM bacterial profiles between control and AB offspring at all examined stages, with exceptions of P2 and P11 (Supplementary Fig. 3A). The MAM profiles in control offspring underwent apparent alterations at P11 and P18, whereas the profiles in AB offspring made a substantial shift at P7 (Supplementary Fig. 3B). Comparison of the LM and MAM profiles in control or AB offspring based on PERMANOVA p-values and PCoA plots based on Bray-Curtis dissimilarities at the genus level revealed significant differences in beta diversity between the LM and MAM profiles in both offspring groups from P2 to P11 (Fig. 2A and B). At P14, a significant difference in beta diversity persisted only in control offspring, with no statistical difference reached in AB offspring (Fig. 2A and B). By P18, the LM and MAM profiles converged in both groups (Fig. 2A and B). Variation in the developmental timing of convergence between the LM and MAM profiles in control and AB offspring may also reflect the difference in postnatal colonization patterns of gut microbiota between control and AB offspring. These results collectively suggest that perturbation of maternal gut microbiota exerted long-lasting influences on the early colonization of offspring gut microbiota.


Taxonomic analyses revealed the most notable impact of AB administration on the phylum Bacteroidota, whereas Firmicutes and Proteobacteria compensated for the decreased abundance of Bacteroidota in the gut microbiota of AB-treated dams (Fig. 3A). This trend persisted long after AB administration ceased (Fig. 3A). In both the LM and MAM of offspring born from AB-treated dams, elevation in the relative abundance of Bacteroidota after P14 was suppressed (Fig. 3B and C). In our subsequent taxonomic analyses, we identified 31 genera that demonstrated significant differences in relative abundance at one or more stages after E14 between control and AB-treated dams, each with more than 1% abundance at one or more stages. In addition to these 31 genera, we included Enterococcus, known as an early colonizer in mammalian gut microbiota,28) for further analysis. These 32 genera (including Enterococcus) are highlighted in Supplementary Data 1-3. We observed that the genera Bacteroides, an unclassified genus belonging to Muribaculaceae, Alistipes, and Parabacteroides were significantly affected by AB administration, and their suppressed occupancy continued through the later stages (Fig. 4A, D, G, and J). These genera emerged in the LM of control offspring at P14 or P18, whereas their appearance was delayed in the LM of AB offspring (Fig. 4B, E, H, and K). Similarly, these genera started to increase their occupancies in the MAM of control offspring at P14 or P18, but this increase was curtailed in the MAM of AB offspring (Fig. 4 C, F, I, and L). The relative abundances of the genera Lactobacillus, Desulfovibrio, ASF356, and unclassified Oscillospiraceae underwent significant reduction following exposure to AB in the microbiota of AB-treated dams (Fig. 5A, D, G, and J). In both the LM and MAM of AB offspring, the tendency for these genera to increase in prevalence was generally suppressed across the stages examined (Fig. 5B, C, E, F, H, I, K, and L). In the maternal microbiota, twelve other genera significantly affected by AB exposure exhibited a common pattern where the postnatal appearance and increase in abundance tended to be delayed in both the LM and MAM of AB offspring despite certain temporal variations (Supplementary Fig. 4-Fig. 6). In contrast, the genera Clostridium sensu stricto 1, Clostridium sensu stricto 13, and Marvinbryantia demonstrated notable occupancies after AB administration in the gut microbiota of AB-treated dams, despite their scarcity in the control gut microbiota (Fig. 6A, D, and G). With temporal variations, these elevated occupancies were also observed in both the LM and MAM of AB offspring, in contrast to their almost undetectable presence in the LM and MAM of control offspring (Fig. 6B, C, E, F, H, and I). The genus Turicibacter showed significant presence, specifically at P2, within the gut microbiota of AB-treated dams. At other stages, Turicibacter exhibited constant occupancies with no discernible differences between control and AB-treated dams (Fig. 6J). There were tendencies that the relative abundances of Turicibacter were elevated in both the LM and MAM of AB offspring compared with control offspring at some of the early postnatal stages prior to P18 (Fig. 6K and L). Notable elevations in the occupancies of the genera Bacillus, Sporosarcina, Enterococcus, and Staphylococcus were detected in the gut microbiota of AB-treated dams with variations in temporal patterns (Fig. 7A, D, G, and J). However, significant increases in the occupancies of these genera were not discerned within either the LM or MAM of AB offspring across the postnatal stages (Fig. 7B, C, E, F, H, I, K, and L). We also identified four genera (Atopostipes, Jeotgalicoccus, Anaerosporobacter, and an uncultured genus from the Lachnospiraceae family) whose relative abundances were higher in the gut microbiota of AB-treated dams than in control dams. Each genus exhibited a distinct temporal pattern of abundance (Supplementary Fig. 7A, D, G, and J). However, the abundances of these genera were not significantly elevated in the LM and MAM of AB offspring during postnatal colonization except for Anaerosporobacter at P7 (Supplementary Fig. 7B, C, E, F, H, I, K, and L). These results collectively suggest that the dominant genera in the perturbed maternal microbiota have a tendency to influence the early colonization of offspring microbiota, although not all bacteria with elevated dominance in the maternal gut microbiota are transmitted to the offspring.
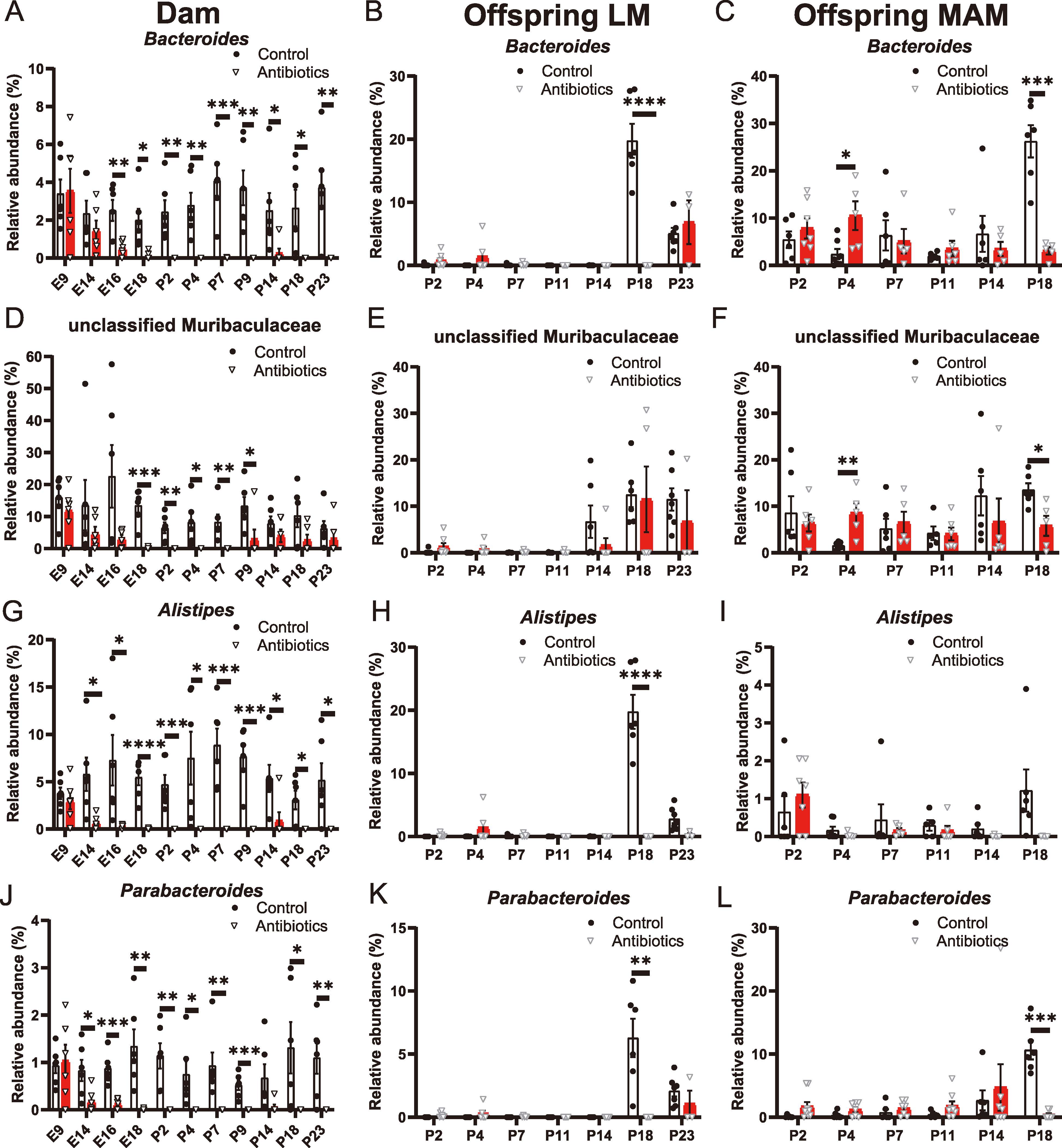
Fig. 4
(Color online) Longitudinal alterations in the relative abundances of dominant bacterial taxa affected in AB offspring within the PMGM model. The charts detail the longitudinal alterations in the relative abundances of the dominant genera belonging to the order Bacteroidales: Bacteroides, an unclassified genus belonging to Muribaculaceae, Alistipes, and Parabacteroides. (A, D, G, J) Maternal gut microbiota, n=6 at each stage for control (Control) and AB-treated dams (Antibiotics). (B, E, H, K) Offspring luminal microbiota (LM). Control offspring (Control): P2, n=6; P4, n=6; P7, n=8; P11, n=6; P14, n=6; P18, n=6; P23, n=7. AB offspring (Antibiotics): P2, n=7; P4, n=6; P7, n=5; P11, n=5; P14, n=6; P18, n=5; P23, n=3. Gut contents were analyzed for P2-18 LM. Fecal samples were analyzed for P23 LM. (C, F, I, L) Offspring mucosa-associated microbiota (MAM). Control offspring (Control): P2, n=6; P4, n=7; P7, n=6; P11, n=5; P14, n=6; P18, n=6. AB offspring (Antibiotics): P2, n=6; P4, n=5; P7, n=5; P11, n=6; P14, n=5; P18, n=5. Genera that are dominant in control but significantly affected by AB in maternal microbiota display distinct colonization patterns in offspring microbiota. Data are shown as the mean ± SE; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 (Multiple unpaired t tests).



Beta diversity analyses to evaluate differences between the maternal (control or AB-treated) and offspring microbiota (Control LM, Antibiotics LM, Control MAM, or Antibiotics MAM) revealed consistent and marked differences during the early postnatal period, from P2 to P14. Notably, at P23, the comparison between control dam and control offspring LM yielded a higher p-value, indicating less pronounced dissimilarity, compared with the comparison between control dam and AB offspring LM at the same stage (Supplementary Fig. 8A and C). Similarly, at P18 and P23 the comparison between AB-treated dams and AB offspring LM showed a higher p-value than those between AB-treated dams and control offspring LM (Supplementary Fig. 8). These findings may imply a potential trend toward convergence of microbial profiles between dams and their corresponding offspring groups in the later postnatal stages.
4. Discussion
This study investigated the vertical transmission of perturbed maternal microbiota to offspring using the PMGM model in mice. Our results demonstrated that the gut microbiota of offspring closely reflects the profiles of maternal gut microbiota, underscoring a robust influence of maternal microbiota on early-life colonization of offspring gut microbiota. The maternal microbiota is susceptible to perturbation by various environmental factors during the perinatal period, such as exposure to antibiotics or other microbiota-affecting drugs, infection, high-fat diets, and stress.1) Accumulating evidence suggests that inheriting perturbed maternal gut microbiota exerts lasting influences on the postnatal development of offspring.7),11)-13),29)-35) In the context of the transmission of perturbed maternal gut microbiota to offspring, Shulfer et al. demonstrated that germ-free adult pregnant mice, inoculated with the colon contents obtained from penicillin-exposed mice, transmitted their microbiota with specific profiles to their own offspring.36) Through longitudinal analyses of the profiles of dams and offspring utilizing the PMGM model, we observed that perturbed maternal gut microbiota significantly shapes the early colonization of offspring gut microbiota, highlighting the fidelity of the vertical transmission of maternal gut microbiota to offspring in mammalian mother-infant bonding.
Emerging studies consistently indicate the presence of microbial communities within the placenta, amniotic fluid, and meconium in mammals, pointing to the intriguing possibility of intrauterine colonization of offspring microbiota.37),38) Following the widely accepted view, the initial step in inheriting maternal microbiota is closely associated with parturition.38),39) As offspring pass through the birth canal, they encounter their initial microbial colonization from the vaginal flora, an essential process for the initial seeding of the gut microbiome.38),39) In experimental mammalian models, including mice, offspring are consistently exposed to maternal feces until the weaning period, which serves as a significant route for bacterial transmission from mother to offspring. Our research contributes to understanding the ‘priority effect’ in postnatal microbiota colonization, a phenomenon where the initial seed bacterial community shapes subsequent community composition through microbiome-microbiome interactions.40) Our comprehensive chronological analysis of control and AB offspring demonstrates the priority effects resulting from differences in seed bacterial communities inherited from the dams. We noted parallels between the microbiota properties of offspring and their mothers. However, it is also evident that certain dominant bacterial genera present in maternal microbiota are not transferred to the offspring, suggesting a taxonomy-specific mechanism governing the inheritance of maternal gut microbiota. The priority effects appear to underlie this pattern of bacterial classification-dependent colonization. Numerous studies have highlighted the critical roles of microbiota in determining host health and disease. Our findings suggest that the inheritance of perturbed maternal microbiota by offspring affects the postnatal colonization of offspring gut microbiota based on the priority effect, possibly leading to alterations in offspring development. Further exploration of this aspect could yield essential insights into the complexities of microbiota inheritance and its implications for health.
Our results showed that the disruption of maternal gut microbiota through AB administration during pregnancy exerts a long-lasting reduction in the alpha diversity of the gut microbiome, which coincides with the diminished alpha diversity of the offspring gut microbiome. In postnatal colonization, the phylogenic diversity of the gut microbiota tends to increase over time.39),41),42) As the infant grows and consumes solid foods, there is a noticeable increase in microbiota diversity.39) This changing pattern of human microbial diversity aligns with the pattern of LM in control offspring, where a rapid increase is evident at P14 (Fig. 1E and H) in our observation. Notably, in the LM of AB offspring, the influences of the decreased diversity of maternal microbiota of AB-treated dams manifested at stages later than P14 (Fig. 1E and H), coinciding with the onset of the postnatal rapid growth in microbial diversity. In both the LM and MAM of AB offspring, we observed the reduced levels of Bacteroides, an unclassified genus belonging to Muribaculaceae, Alistipes, Parabacteroides, Lactobacillus, Desulfovibrio, and ASF356 (Fig. 4 and Fig. 5). Conversely, Clostridium sensu stricto 1, Clostridium sensu stricto 13, and Marvinbryantia dominated the gut microbiota in the LM and MAM of AB offspring (Fig. 6). AB use during pregnancy is common during Caesarean section or assisted vaginal delivery as an intrapartum antibiotic prophylaxis (IAP).38) IAP or perinatal AB exposure typically reduces the diversity of the offspring gut microbiome.38),43)-45) IAP administration is typically associated with decreased levels of Bacteroidota, Bifidobacterium, and Lactobacillus, alongside increased levels of Firmicutes and Proteobacteria in the offspring gut microbiome.38),46) At the genus level, IAP leads to decreased Bacteroides and increased Clostridium abundances.46),47) These previous findings on the subsequent effects on human infant colonization after perinatal AB exposure of maternal gut microbiota are broadly consistent with our results.
Our chronological comparison of maternal microbiota and offspring microbiota effectively embodies the vertical transmission of the dominant bacterial species from the perturbed maternal gut microbiota to the offspring microbiota. However, we also observed limited inheritance of genera that exhibit abnormally high occupancies in the perturbed maternal gut microbiota, such as Bacillus, Sporosarcina, Enterococcus, and Staphylococcus, by the LM and MAM of AB-offspring (Fig. 7). Li et al. reported that approximately 72% of the gut microbiome population in control infants originated from their mothers, whereas only 25% of the microbial population of infants whose mothers received AB treatment during the perinatal period derived from their mothers.48) This observation suggests that perinatal perturbations of maternal microbiota shift the source of colonization of the offspring gut microbiota from vertical transmission of maternal microbiome towards horizontal transfer from the environment.48) Those bacteria abundant in the perturbed microbiota but less frequently inherited by offspring may face competition with bacteria from environmental sources. The competition with bacteria from environmental sources may be one of the mechanisms underlying the ’priority effect,’ which, as discussed above, involves the initial seed bacterial community shaping subsequent postnatal colonization through microbial interactions. The present study provides valuable insights into the early postnatal colonization of the LM and MAM in C57BL/6 mice. Beta diversity analyses revealed that significant dissimilarities between the LM and MAM are significant during the early postnatal period, from P2 to P14, in control offspring, as shown in Fig. 2. By a later stage (P18), the profiles of the LM and MAM begin to converge (Fig. 2). Notably, this convergence of the LM and MAM profiles occurred earlier in AB offspring (Fig. 2). These observations further support the idea that the inheritance of perturbed maternal microbiota has a profound effect on the postnatal colonization pattern of offspring microbiota. These findings highlight the necessity of conducting distinct analyses for the LM and MAM, particularly during the early postnatal colonization period. Such distinct analyses are crucial for gaining deeper insights into the dynamics and underlying mechanisms governing how maternal microbiota perturbations affect the postnatal colonization patterns of offspring microbiota.
Our results of taxonomic analyses revealed that, at the genus level, the LM of control mice is initially dominated by Enterococcus after birth, followed by subsequent Lactobacillus dominance (Fig. 5B and Fig. 7H). At the stages after pups start eating solid foods, Lactobacillus is outcompeted by members of the Bacteroidota phylum, including Bacteroides, an unclassified genus belonging to Muribaculaceae, Alistipes, and Parabacteroides (Fig. 4B, E, H, and K). These temporal patterns are consistent with prior findings from both mouse and human studies.6),49),50) In contrast to the LM, early colonization of Bacteroides, an unclassified genus belonging to Muribaculaceae, and Alistipes characterized the MAM in control mice, illustrating distinct bacterial composition patterns between postnatal colonization of the LM and MAM (Fig. 4C, F, and I). Notably, in the PMGM model, both the LM and MAM of AB offspring are dominated by the genera Clostridium sensu stricto 1, Clostridium sensu stricto 13, and Marvinbryantia (Fig. 6B, C, E, F, H, and I). A previous study by Galley et al. also reported significant differences between the LM and MAM communities in adult male mice.51) Additionally, they observed distinct responses of the LM and MAM communities to the prolonged restraint stress.51) These differences in postnatal colonization patterns, adult gastrointestinal bacterial components, and stress responses between the LM and MAM highlight the importance of analyzing gastrointestinal bacterial communities with distinct intestinal niches.
Our results offer visual insights into the status of the maternal gut microbiota during the perinatal period. During the first trimester of pregnancy, the gut microbial composition remains similar to that in healthy non-pregnant women.37),52) However, a significant shift occurs in the properties of gut microbiota from the first trimester to the third trimester.37),53) Nonetheless, the question of whether there are changes in the properties of the maternal gut microbiota before and after parturition remains. A significant change in hormone levels within the maternal body, including a decrease in estrogen levels, occurs during the postpartum period in mammals.37) Many gut microbiota species are capable of metabolizing estrogen by secreting β-glucuronidase to release the subsequent metabolites into the circulation system of the host.54) These facts suggest that properties of the gut microbiota might change in response to alterations in host physiology. Notably, our results encompassing alpha diversity (Fig. 1D and G), beta diversity between perinatal stages (Supplementary Fig. 1B), and bacterial composition at the phylum level (Fig. 3A) did not indicate a significant shift in the properties of the maternal gut microbiota before and after parturition. This observation contradicted our expectations. However, recent findings in sows have shown that the relative abundance of Prevotella significantly decreases, whereas the relative abundance of Lactobacillus significantly increases from late pregnancy to the postpartum period.55) Further analyses are needed to explore alterations in the gut microbiota profiles before and after parturition.
One notable limitation of our study was its narrow time frame. Our primary focus was on the early colonization period, extending until the weaning of offspring. In humans, the microbiota composition approaches an adult-like state with the introduction of solid foods by the end of the first year of life, eventually resembling adult microbial profiles by 2.5 years of age.37),39) The extent to which perturbed maternal microbiota influences the colonization of offspring microbiota until full maturation raises an important question for future research in mice. Another significant limitation in the interpretation of our study was the gender bias in sample collection. In order to perform specific analyses at particular developmental stages, we had to focus primarily on male offspring, which restricted the generalizability of our results. This aspect must be addressed in future studies to provide a more comprehensive analysis.
Overall, the results of this study underscore the critical importance of the mother-infant physiological bond mediated by gut microbiota. Furthermore, the results highlight the need for multidimensional research to fully comprehend the mechanism through which the inheritance of perturbed maternal gut microbiota leads to disturbance of the development of offspring.1),3),12),13) Future studies should investigate specific environmental factors and microbial species that contribute to these disturbances and explore potential interventions as well as the optimal timing of intervention that allows for prevention and amelioration of the effects on offspring development.
Acknowledgments
This work was supported by KAKENHI (Grant Numbers: 17K19225, 20K20320, 20K08242, and 23K07278 awarded to S. T.) from the Japan Society for the Promotion of Science (JSPS). We are also grateful for research grants from the OKASAN-KATO Foundation, Mie, Japan; the Morinaga Foundation for Health & Nutrition, Tokyo, Japan; and the Mishima Kaiun Memorial Foundation, Tokyo, Japan (awarded to S.T.). Our appreciation extends to Dr. Hideo Matsuzaki, the department head at University of Fukui, for his generous support. In preparing this manuscript, we utilized several web-based tools to enhance the quality of the writing. Specifically, we used Ludwig (https://ludwig.guru/) and THESAURUS (https://www.thesaurus.com/) to aid the writing process. We used Grammarly (https://www.grammarly.com/) and ChatGPT-3.5 and 4 (https://openai.com/blog/chatgpt/) for English editing to obtain suggestions to refine the readability and clarity of the manuscript. We meticulously reviewed the suggestions provided by these platforms and incorporated only those appropriate to the text.
Supplementary materials
Supplementary materials are available at https://doi.org/10.2183/pjab.100.020.
Conflict of interest
The authors declare no conflicts of interest.
Data availability statement
The sequencing data are deposited at NCBI Sequence Read Archive: PRJNA974821. The relative abundances of bacteria across various stages and groups are detailed in Supplementary Data 1-3. Detailed metadata and other relevant data are available for appropriate scientific purposes upon request.
Author contributions
S.T. conceived the study, analyzed the overall data, designed and performed the primary animal experiments, and wrote the manuscript. R. I. and T. T. designed and executed the experiments related to microbiota analyses.
Notes
Edited by Shizuo AKIRA, M.J.A.
Correspondence should be addressed to: S. Tochitani, Department of Radiological Technology, Faculty of Health Science, Suzuka University of Medical Science, 3500-3 Minamitamagaki, Suzuka, Mie 513-8670, Japan (e-mail:
tochitani.shiro@gmail.com).
Non-standard abbreviation list: AB: antibiotics; PMGM: perturbed maternal gut microbiota; LM: luminal microbiota; MAM: mucosa-associated microbiota; PCoA: Principal Coordinate Analyses; IPA: intrapartum antibiotic prophylaxis.
References
- 1) Tochitani, S. (2021). Vertical transmission of gut microbiota: Points of action of environmental factors influencing brain development. Neurosci. Res. 168, 83-94.
- 2) McDonald, B. and McCoy, K.D. (2019). Maternal microbiota in pregnancy and early life. Science 365, 984-985.
- 3) Tian, M., Li, Q., Zheng, T., Yang, S., Chen, F., Guan, W. et al. (2023). Maternal microbe-specific modulation of the offspring microbiome and development during pregnancy and lactation. Gut Microbes 15, 2206505.
- 4) Mesa, M.D., Loureiro, B., Iglesia, I., Fernandez Gonzalez, S., Llurba Olive, E., Garcia Algar, O. et al. (2020). The evolving microbiome from pregnancy to early infancy: A comprehensive review. Nutrients 12, 133.
- 5) Tormo-Badia, N., Hakansson, A., Vasudevan, K., Molin, G., Ahrne, S. and Cilio, C.M. (2014). Antibiotic treatment of pregnant non-obese diabetic mice leads to altered gut microbiota and intestinal immunological changes in the offspring. Scand. J. Immunol. 80, 250-260.
- 6) Deshmukh, H.S., Liu, Y., Menkiti, O.R., Mei, J., Dai, N., O'Leary, C.E. et al. (2014). The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat. Med. 20, 524-530.
- 7) Tochitani, S., Ikeno, T., Ito, T., Sakurai, A., Yamauchi, T. and Matsuzaki, H. (2016). Administration of non-absorbable antibiotics to pregnant mice to perturb the maternal gut microbiota is associated with alterations in offspring behavior. PloS one 11, e0138293.
- 8) Stewart, C.J., Ajami, N.J., O'Brien, J.L., Hutchinson, D.S., Smith, D.P., Wong, M.C. et al. (2018). Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562, 583-588.
- 9) Bokulich, N.A., Chung, J., Battaglia, T., Henderson, N., Jay, M., Li, H. et al. (2016). Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 8, 343ra382.
- 10) Uzan-Yulzari, A., Turta, O., Belogolovski, A., Ziv, O., Kunz, C., Perschbacher, S. et al. (2021). Neonatal antibiotic exposure impairs child growth during the first six years of life by perturbing intestinal microbial colonization. Nat. Commun. 12, 443.
- 11) Tamburini, S., Shen, N., Wu, H.C. and Clemente, J.C. (2016). The microbiome in early life: implications for health outcomes. Nat. Med. 22, 713-722.
- 12) O'Connor, R., Moloney, G.M., Fulling, C., O'Riordan, K.J., Fitzgerald, P., Bastiaanssen, T.F.S. et al. (2021). Maternal antibiotic administration during a critical developmental window has enduring neurobehavioural effects in offspring mice. Behav. Brain Res. 404, 113156.
- 13) Morel, C., Martinez Sanchez, I., Cherifi, Y., Chartrel, N. and Diaz Heijtz, R. (2023). Perturbation of maternal gut microbiota in mice during a critical perinatal window influences early neurobehavioral outcomes in offspring. Neuropharmacology 229, 109479.
- 14) Ji, C., Zhang, G., Xu, S., Xiang, Q., Huang, M., Zhao, M. et al. (2022). Antibiotic treatments to mothers during the perinatal period leaving hidden trouble on infants. Eur. J. Pediatr. 181, 3459-3471.
- 15) Verdu, E.F., Bercik, P., Verma-Gandhu, M., Huang, X.X., Blennerhassett, P., Jackson, W. et al. (2006). Specific probiotic therapy attenuates antibiotic induced visceral hypersensitivity in mice. Gut 55, 182-190.
- 16) Bercik, P., Denou, E., Collins, J., Jackson, W., Lu, J., Jury, J. et al. (2011). The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology 141, 599-609.e3.
- 17) Rakoff-Nahoum, S., Paglino, J., Eslami-Varzaneh, F., Edberg, S. and Medzhitov, R. (2004). Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118, 229-241.
- 18) Hamilton, T.L., Peters, J.W., Skidmore, M.L. and Boyd, E.S. (2013). Molecular evidence for an active endogenous microbiome beneath glacial ice. ISME J. 7, 1402-1412.
- 19) Inoue, R., Sakaue, Y., Sawai, C., Sawai, T., Ozeki, M., Romero-Perez, G.A. et al. (2016). A preliminary investigation on the relationship between gut microbiota and gene expressions in peripheral mononuclear cells of infants with autism spectrum disorders. Biosci. Biotechnol. Biochem. 80, 2450-2458.
- 20) Liyanage, G.S.G., Inoue, R., Fujitani, M., Ishijima, T., Shibutani, T., Abe, K. et al. (2021). Effects of soy isoflavones, resistant starch and antibiotics on polycystic ovary syndrome (PCOS)-like features in letrozole-treated rats. Nutrients 13, 3759.
- 21) Maeda, H., Fujimoto, C., Haruki, Y., Maeda, T., Kokeguchi, S., Petelin, M. et al. (2003). Quantitative real-time PCR using TaqMan and SYBR Green for Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis, Prevotella intermedia, tetQ gene and total bacteria. FEMS Immunol. Med. Microbiol. 39, 81-86.
- 22) GitHub-jbisanz/qiime2R https://github.com/jbisanz/qiime2R.
- 23) McMurdie, P.J. and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS one 8, e61217.
- 24) Xu, S., Zhan, L., Tang, W., Wang, Q., Dai, Z., Zhou, L. et al. (2023). MicrobiotaProcess: A comprehensive R package for deep mining microbiome. Innovation (Camb.) 4, 100388.
- 25) Shetty S, L.L. (2022). microbiomeutilities: Utilities for Microbiome Analytics. R package version 1.00.17. https://microsud.github.io/\\ microbiomeutilities/index.html.
- 26) Dixon, P. (2003). VEGAN, a package of R functions for community ecology. Journal of Vegetation Science 14, 927-930.
- 27) Paone, P. and Cani, P.D. (2020). Mucus barrier, mucins and gut microbiota: the expected slimy partners? Gut 69, 2232-2243.
- 28) Mitsuoka, T. (1996). Intestinal flora and human health. Asia Pac. J. Clin. Nutr. 5, 2-9.
- 29) Jasarevic, E., Rodgers, A.B. and Bale, T.L. (2015). A novel role for maternal stress and microbial transmission in early life programming and neurodevelopment. Neurobiol. Stress 1, 81-88.
- 30) Degroote, S., Hunting, D.J., Baccarelli, A.A. and Takser, L. (2016). Maternal gut and fetal brain connection: Increased anxiety and reduced social interactions in Wistar rat offspring following peri-conceptional antibiotic exposure. Prog. Neuropsychopharmacol. Biol. Psychiatry 71, 76-82.
- 31) Buffington, S.A., Di Prisco, G.V., Auchtung, T.A., Ajami, N.J., Petrosino, J.F. and Costa-Mattioli, M. (2016). Microbial reconstitution reverses maternal diet-induced social and synaptic deficits in offspring. Cell 165, 1762-1775.
- 32) Nyangahu, D.D., Lennard, K.S., Brown, B.P., Darby, M.G., Wendoh, J.M., Havyarimana, E. et al. (2018). Disruption of maternal gut microbiota during gestation alters offspring microbiota and immunity. Microbiome 6, 124.
- 33) Diaz Heijtz, R. (2016). Fetal, neonatal, and infant microbiome: Perturbations and subsequent effects on brain development and behavior. Semin. Fetal Neonatal Med. 21, 410-417.
- 34) Codagnone, M.G., Stanton, C., O'Mahony, S.M., Dinan, T.G. and Cryan, J.F. (2019). Microbiota and neurodevelopmental trajectories: Role of maternal and early-life nutrition. Ann. Nutr. Metab. 74 (Suppl. 2), 16-27.
- 35) Ratsika, A., Codagnone, M.C., O'Mahony, S., Stanton, C. and Cryan, J.F. (2021). Priming for life: Early life nutrition and the microbiota-gut-brain axis. Nutrients 13, 423.
- 36) Schulfer, A.F., Battaglia, T., Alvarez, Y., Bijnens, L., Ruiz, V.E., Ho, M. et al. (2018). Intergenerational transfer of antibiotic-perturbed microbiota enhances colitis in susceptible mice. Nat. Microbiol. 3, 234-242.
- 37) Nuriel-Ohayon, M., Neuman, H. and Koren, O. (2016). Microbial changes during pregnancy, birth, and infancy. Front Microbiol. 7, 1031.
- 38) Moore, R.E. and Townsend, S.D. (2019). Temporal development of the infant gut microbiome. Open Biol. 9, 190128.
- 39) Clemente, J.C., Ursell, L.K., Parfrey, L.W. and Knight, R. (2012). The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258-1270.
- 40) Jian, C., Carpen, N., Helve, O., de Vos, W.M., Korpela, K. and Salonen, A. (2021). Early-life gut microbiota and its connection to metabolic health in children: Perspective on ecological drivers and need for quantitative approach. EBioMedicine 69, 103475.
- 41) Koenig, J.E., Spor, A., Scalfone, N., Fricker, A.D., Stombaugh, J., Knight, R. et al. (2011). Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. U.S.A. 108 (Suppl 1), 4578-4585.
- 42) Yatsunenko, T., Rey, F.E., Manary, M.J., Trehan, I., Dominguez-Bello, M.G., Contreras, M. et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222-227.
- 43) Nogacka, A., Salazar, N., Suarez, M., Milani, C., Arboleya, S., Solis, G. et al. (2017). Impact of intrapartum antimicrobial prophylaxis upon the intestinal microbiota and the prevalence of antibiotic resistance genes in vaginally delivered full-term neonates. Microbiome 5, 93.
- 44) Korpela, K., Salonen, A., Virta, L.J., Kekkonen, R.A., Forslund, K., Bork, P. et al. (2016). Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nature Commun. 7, 10410.
- 45) Yassour, M., Vatanen, T., Siljander, H., Hamalainen, A.M., Harkonen, T., Ryhanen, S.J. et al. (2016). Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med. 8, 343ra381.
- 46) Tapiainen, T., Koivusaari, P., Brinkac, L., Lorenzi, H.A., Salo, J., Renko, M. et al. (2019). Impact of intrapartum and postnatal antibiotics on the gut microbiome and emergence of antimicrobial resistance in infants. Sci. Rep. 9, 10635.
- 47) Kim, H., Sitarik, A.R., Woodcroft, K., Johnson, C.C. and Zoratti, E. (2019). Birth mode, breastfeeding, pet exposure, and antibiotic use: Associations with the gut microbiome and sensitization in children. Curr. Allergy Asthma Rep. 19, 22.
- 48) Li, W., Tapiainen, T., Brinkac, L., Lorenzi, H.A., Moncera, K., Tejesvi, M.V. et al. (2021). Vertical transmission of gut microbiome and antimicrobial resistance genes in infants exposed to antibiotics at birth. J. Infect. Dis. 224, 1236-1246.
- 49) Pandey, U. and Aich, P. (2022). Postnatal intestinal mucosa and gut microbial composition develop hand in hand: A mouse study. Biomed. J. 46, 100519.
- 50) Tanaka, M. and Nakayama, J. (2017). Development of the gut microbiota in infancy and its impact on health in later life. Allergol. Int. 66, 515-522.
- 51) Galley, J.D., Yu, Z., Kumar, P., Dowd, S.E., Lyte, M. and Bailey, M.T. (2014). The structures of the colonic mucosa-associated and luminal microbial communities are distinct and differentially affected by a prolonged murine stressor. Gut Microbes 5, 748-760.
- 52) Collado, M.C., Isolauri, E., Laitinen, K. and Salminen, S. (2008). Distinct composition of gut microbiota during pregnancy in overweight and normal-weight women. Am. J. Clin. Nutr. 88, 894-899.
- 53) Koren, O., Goodrich, J.K., Cullender, T.C., Spor, A., Laitinen, K., Backhed, H.K. et al. (2012). Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 150, 470-480.
- 54) Song, J., Zhou, B., Kan, J., Liu, G., Zhang, S., Si, L. et al. (2022). Gut microbiota: Linking nutrition and perinatal depression. Front. Cell. Infect. Microbiol. 12, 932309.
- 55) Huang, X., Gao, J., Zhao, Y., He, M., Ke, S., Wu, J. et al. (2019). Dramatic remodeling of the gut microbiome around parturition and its relationship with host serum metabolic changes in sows. Front. Microbiol. 10, 2123.

