Abstract
Enhancing the thermostability of cellulose-degrading enzymes is pivotal for establishing an efficient bioconversion system from cellulosic materials to value-added compounds. Here, by introducing random and saturation mutagenesis into the Thermoascus aurantiacus β-glucosidase gene, we generated a hyperthermostable mutant with five amino acid substitutions. Analysis of temperature-induced unfolding revealed the involvement of each replacement in the increased Tm value. Structural analysis showed that all replacements are located at the periphery of the catalytic pocket. D433N replacement, which had a pronounced thermostabilizing effect (ΔTm = 4.5°C), introduced an additional hydrogen bond with a backbone carbonyl oxygen in a long loop structure. The mutant enzyme expressed in Kluyveromyces marxianus exhibited a Tm of 82°C and hydrolyzed cellobiose with kcat and Km values of 200 s-1 and 1.8 mM, respectively. When combined with a thermostable endoglucanase, the mutant enzyme released 20% more glucose than wild-type enzyme from cellulosic material. The mutant enzyme is therefore a noteworthy addition to the existing repertoire of thermostable β-glucosidases.
1. Introduction
The efficient bioconversion of cellulose, the most abundant renewable bioresource, into biofuel or other value-added materials can be a key for replacing fossil fuels and maintaining a sustainable ecosystem.1),2) However, cellulose is recalcitrant to enzymatic degradation, hampering its efficient conversion into fermentable sugars, such as glucose. Developing a cost-effective saccharification process is essential to promoting an environment-oriented society.2),3) Hydrolysis of cellulose requires three classes of cellulolytic enzymes: endoglucanase (EC 3.2.1.4), cellobiohydrolase (EC 3.2.1.91 and EC 3.2.1.176), and β-glucosidase (EC 3.2.1.21).1),2) Recent studies also showed the importance of lytic polysaccharide monooxygenases for the efficient degradation of cellulose.4) Endoglucanases and lytic polysaccharide monooxygenases cleave internal glycosidic linkages, whereas cellobiohydrolases hydrolyze cellulose chains from the reducing or non-reducing ends. Thus, endoglucanase and cellobiohydrolase (cellulases) act synergistically on cellulose to generate cellooligosaccharides and cellobiose, which are further hydrolyzed by β-glucosidase to glucose.
One strategy for more efficient enzyme-based saccharification is to make the cellulolytic enzymes thermostable. Thermostable enzymes possess certain advantages such as improved compatibility with lignocellulose pretreatment for cellulose extraction, increased catalytic activity, and reduced contamination risk.5)-8) Despite continuous efforts to enhance thermostability through search for natural sources or random, targeted, and/or structure-based mutations, a cost-effective method for cellulose degradation to solve the global energy crisis has not yet been achieved yet.9),10)
In the cellulolytic enzyme cocktail, β-glucosidase is the central rate-determining component. It catalyzes the final step of cellulose conversion of cellobiose and/or short cellodextrins into glucose and relieves the inhibition effect of cellobiose on cellulases.11) The cellulolytic enzyme cocktail of Tricoderma reesei exhibits low β-glucosidase activity12)-14); thus, β-glucosidases from different fungal origins are generally added to the cocktail depending on the requirements of an experiment.15),16) Most fungal β-glucosidases belong to glycosyl hydrolase families 1 (GH1) and 3 (GH3), according to their classification in the Carbohydrate-Active enZymes database (CAZy) (
http://www.cazy.org)17) and are commonly used for saccharification of cellulosic materials both in the laboratory and commercially.18)-20) Among them, GH3 β-glucosidase from Aspergillus aculeatus (AaBgl1) is industrially important due to its high cellobiase activity.20)-22) However, its temperature stability (55°C) is not as high as those of the enzymes contained in cellulolytic cocktail of T. reesei (60°C).23),24) As a result, the cellulose digestion system has not been fully optimized.
Thermoascus aurantiacus is a filamentous fungus known to produce thermostable cellulolytic enzymes.25)-27) We previously isolated the gene for β-glucosidase (TaBglI) and expressed the enzyme in the yeast Pichia pastoris.28) As the primary sequences of TaBglI and AaBgl1 share relatively high identity (71%), TaBglI also exhibited cellobiase activity. Nonetheless, in our previous study, TaBglI was found to retain 70% of its activity after heat treatment for 1 h at 60°C, indicating its potential as a model enzyme for increasing thermostability.
In this study, we first established the Escherichia coli-based TaBglI expression system. Then, using this system combined with random and saturation mutagenesis, we screened for thermostable mutant enzymes and characterized their physicochemical properties. Furthermore, we determined the crystal structures of E. coli-expressed wild-type and thermostable quintuple mutant enzymes and evaluated their structural differences. Finally, the mutant enzyme was expressed in Kluyveromyces marxianus, and its usefulness in the saccharification of cellulosic materials was evaluated.
2. Materials and methods
2.1. Expression and purification of TaBglI in E. coli.
The expression vector contained the DNA coding for amino acid residues 21-861 of TaBglI (GenBank DQ114396) after genetically removing the signal peptide. The region was amplified using high-fidelity PCR with PrimeStar (Takara-Bio, Shiga, Japan) and primers 5′-AGGAGATATACCATGGATGACTTGGCCTACTCGCC-3′ (TaBglI-Ec-f) and 5′-GTTAGCAGCCGGATCTCAGTAAGGGGGAAGCGGTG-3′ (TaBglI-Ec-r), and the TaBglI expression plasmid pPICZαA/bglI as the template.28) The amplified fragment was inserted into the NcoI-BamHI site of pET16b (Novagen, Inc., Madison, WI) using In-Fusion methodology (Clontech, Palo Alto, CA), and sequenced to ensure accuracy. The resulting plasmid, pET16b-TaBglI, was used to transform Rosetta-gami B (DE3) cells (Novagen, Inc.).
The transformant was grown in 500 ml of LB medium containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol at 18°C. On reaching an optical density of 0.5 at 600 nm, IPTG was added to the final concentration of 50μM to induce protein expression. After 2 days, the cells were harvested, suspended in 20 mM sodium phosphate buffer (pH 6.0), and disrupted by sonication. The supernatant was fractionated using ammonium sulfate (20-40% saturation), and the precipitates were dialyzed against 20 mM sodium phosphate buffer (pH 6.0). The proteins in the sample were purified using DEAE-Sepharose Fast Flow column chromatography (Cytiva, Marlborough, MA) with a linear gradient of 0-0.5 M NaCl. The active fractions were combined and further purified using Mono Q 5/50 GL (0-0.4 M NaCl in 20 mM sodium phosphate buffer (pH 6.0)) (Cytiva) and Superdex 200 10/300 GL (150 mM NaCl in 20 mM sodium phosphate buffer (pH 6.0)) (Cytiva) column chromatography. The purified protein was dialyzed against 20 mM sodium phosphate buffer (pH 6.0) and concentrated using a Vivaspin 20-50K (Sartorius AG, Göttingen, Germany). Protein concentration was determined using a BCA Protein Assay kit (Thermo Fisher Scientific, Waltham, MA) with bovine serum albumin as a standard. Purified enzymes were analyzed by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie Brilliant Blue R-250 (CBB) staining. Molecular mass was analyzed by gel filtration chromatography on a Superdex 200 10/300 GL column using a Gel Filtration Calibration Kit (Cytiva, Thyroglobin: 669 kDa, Ferritin: 440 kDa, Aldolase: 158 kDa, Conalbumin: 75 kDa, and Ovalbumin: 44 kDa) as a standard.
2.2. Screening of mutant TaBglI proteins with increased thermostability..
Error-prone PCR was employed to construct a randomly mutagenized library of the TaBglI gene.29) The reaction mixture contained 10 mM Tris-HCl (pH 8.3), 0.4 mM dNTPs, 2 mM MgCl2, 0.25 mM MnCl2, 4 U of Taq polymerase, a 500 nM primer pair (TaBglI-Ec-f and TaBglI-Ec-r, see section 2.1), and 800 ng of pPICZαA/bglI in a total volume of 80 μl. The amplified fragments were ligated with pET16b and introduced into Origami B (DE3) cells (Novagen). Transformants (ca. 4,000) were grown at 30°C in 200 μl of LB medium containing 100 μg/ml ampicillin and 50 μM IPTG in 96-well plates. The cells were harvested by centrifugation and lysed in 20 μl of BugBuster Master Mix (Novagen). After incubating for 20 min, 80 μl of 20 mM sodium phosphate buffer (pH 6.0) was added and centrifuged to obtain clear lysates. Aliquots (20 μl) were transferred to new plates and incubated at 55°C for 1 h, in conditions in which the wild-type enzyme was inactivated. The lysates were then screened for residual ability after incubation at 55°C to hydrolyze 1 mM 4-nitrophenyl-β-D-glucoside (pNP-Glc) to assess the thermostability.
2.3. Site-directed mutagenesis and saturation mutagenesis.
Amino acid substitutions (K119R, M279V, F306L, T308S, K361R, D433N, and N514S/C) were generated using a QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) with pET16b-TaBglI as a template. The primers used are listed in Table S1. These replacements were combined to generate TaBglI variants,M279V/T308S, M279V/T308S/D433N, M279V/T308S/D433N/N514C, and M279V/T308S/K361R/D433N/N514C (referred to as M5). All the entire genes used for subsequent manipulations were sequenced. Mutant proteins were expressed and purified similarly as described for the E. coli expressing the wild-type enzyme.
Saturation mutagenesis was performed at the codons for M279, T308, K361, D433, and N514, using pET16b-TaBglI as a template. The primers used are listed in Table S2. Introduction of mutations at each target site was confirmed by sequencing 20 randomly selected plasmids. Two hundred colonies were selected per each mutagenesis and were used for the thermostability assay, as described in section 2.2.
2.4. Crystallography.
The wild-type and M5 enzymes expressed in E. coli (referred to as WTEc and M5Ec, respectively) were crystallized. Crystallization was performed using the hanging-drop vapor-diffusion method with 48-well VDX plates (Hampton Research, Aliso Viejo, CA) at 293 K. The initial conditions were screened using the Crystal Screen Cryo formulation (Hampton Research). Each drop was prepared by mixing 1 μl of purified protein solution (4 mg/ml in 5 mM HEPES pH 7.0) with 1 μl of reservoir solution, then equilibrated against 200 μl of reservoir solution. Crystals were obtained in drops containing 0.1 M HEPES with pH 7.0, 0.14 M magnesium chloride, 20% 2-propanol, and 0.5 mM 1-deoxynojirimycin (DNJ) for WTEc, and 0.1 M HEPES with pH 7.0, 0.16 M magnesium chloride, 25% 2-propanol, and 0.5 mM DNJ for M5Ec.
2.5. Data collection.
Diffraction datasets were collected from the crystals using a charge-coupled device camera on the beamline BL-5A of the Photon Factory (Tsukuba, Japan). The crystals were flash-cooled under a nitrogen stream at 100 K. The reservoir solution contained 20% (v/v) glycerol as the cryoprotectant. Diffraction images were indexed, integrated, and scaled using the HKL-2000 program suite30) (Table 1).
Table 1
Data collection statistics
|
WTEc
|
M5Ec
|
| Beamline |
Photon Factory BL-5A |
|
| Wavelength (Å) |
1.00000 |
|
| Detector |
CCD ADSC unsupported-q210 |
|
| Space group |
P 21 21 21
|
P 21 21 21
|
| a, b, c (Å) |
138.945, 149.752, 175.529 |
138.195, 150.165, 174.892 |
| α, β, γ (°) |
90.00, 90.00, 90.00 |
90.00, 90.00, 90.00 |
| Resolution range (Å) |
50.00-2.15 (2.19-2.15) |
50.00-1.94 (2.34-2.30) |
| Total no. of reflections |
827,725 |
669,844 |
| No. of unique reflections |
198,186 |
160,623 |
| Completeness (%) |
99.8 (100.0) |
88.6 (91.5) |
| Redundancy |
2.2 (2.1) |
2.6 (2.5) |
| mean I/σ (I) |
11.6 (3.2) |
11.6 (2.9) |
| R-merge (%)
|
12.9 (45.1) |
12.4 (47.8) |
Numbers in parentheses correspond to the shell of data at the highest resolution.
The structure was solved by molecular replacement using MOLREP31) in the CCP4 package, with 4IID chain A (crystal structure of AaBgl1; β-glucosidase 1 from Aspergillus aculeatus in complex with DNJ) as reference. Structural model building was performed using COOT.32) Water molecules were added using the built-in find-water function of COOT and individually checked for significant signals and consistent H-bond donor/acceptor contact. The structure was refined using REFMAC5.33) Molecular graphic images were generated with PyMOL software (www.pymol.org) (Table 2).
Table 2
Refinement statistics
|
WTEc
|
M5Ec
|
| Protein data bank code |
8Y0L |
8Y0M |
| Resolution range (Å) |
37.13-2.15 (2.207-2.151) |
40.20-2.3 (2.359-2.300) |
| σ Cutoff |
F > 0.000σ(F)
|
F > 0.000σ(F)
|
| No. of reflections, working set |
187801 (13698) |
153281 (11174) |
| No. of reflections, test set |
9967 (694) |
8008 (531) |
| Final Rcryst
|
0.16775 (0.208) |
0.16186 (0.200) |
| Final Rfree
|
0.20110 (0.244) |
0.18024 (0.214) |
| No. of non-H atoms |
|
|
| Total |
27252 |
27014 |
| Protein |
23908 |
23904 |
| Ligands |
44 (4 DNJ) |
44 (4 DNJ) |
| Ions |
12 |
10 |
| Solvent |
3288 |
3056 |
| R.M.S. deviations |
|
|
| Bond lengths (Å) |
0.012 |
0.012 |
| Angles (°) |
1.8 |
1.8 |
| Average B factors (Å2)
|
|
|
| Protein |
18.19791 |
22.31172 |
| Ligands |
12.77864 |
16.46364 |
| Ions |
27.88417 |
20.353 |
| Solvent |
27.97682 |
31.28422 |
| Ramachandran plot |
|
|
| In favored regions (%) |
95.29 |
95.32 |
| Outliers (%) |
0.68 |
0.52 |
| Model built |
|
|
| Chain A-D |
23-302, 312-686, 736-861 |
23-302, 312-686, 736-861 |
Numbers in parentheses correspond to the shell of data at the highest resolution.
DNJ; 1-deoxynojirimycin
TaBglI variants were also expressed in K. marxianus under the control of the inulinase promoter.34) The genes coding for wild-type and M5 proteins were PCR-amplified using 5′-CCGGGATCCGATGACTTGGCCTACTCG-3′ and 5′-GGGGAGCCCTCAGTAAGGGGGAAGCGGT-3′ (BamHI and BanII sites are underlined) for primers and E. coli expression plasmids for templates. The fragments were digested with restriction enzymes and inserted into the corresponding sites of pMC101,35) which contained the promoter and signal sequences of the inulinase gene of K. marxianus. After sequence confirmation, the terminator region of the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene and the URA3 marker gene were inserted into the BanII-SphI site and SphI (blunt-ended) site, respectively. The GAPDH terminator was amplified using primers 5′-CCGGGCTCAGTGAATTTACTTTAAATCTTGCA-3′ and 5′-CCGCATGCTCAATCAATGAATCGAAAATGTC-3′ (BanII and SphI sites are underlined) with YEGAp/cbh1, a plasmid for expression of the cellobiohydrolase 1 (cbh1) gene under the control of GAPDH promoter and terminator, as the template. The K. marxianus URA3-containing fragment was excised via EcoRI digestion from pKmURA3 and blunt-ended.36),37) The resulting plasmids were linearized through SphI digestion and introduced into the ura3 derivative of K. marxianus37) to allow integration into the genome. Transformants were selected on uracil dropout plates.36)
Recombinant K. marxianus was grown in 400 ml of basal medium consisting of 2% peptone, 1% yeast extract, and 2% sucrose at 37°C for a week. The supernatant was recovered by centrifugation and saturated with ammonium sulfate (90%). The protein precipitates were dissolved, dialyzed against 20 mM sodium phosphate buffer (pH 6.0), and purified using DEAE-Sepharose Fast Flow column chromatography, with a linear gradient of 0-0.5 M NaCl. The active fractions were collected, dialyzed against 20 mM citrate-phosphate buffer (pH 3.0), and further purified using Mono S 5/50 GL column chromatography (0-0.5 M NaCl in 20 mM citrate-phosphate buffer (pH 3.0)) (Cytiva). Purified wild-type and M5 proteins expressed in K. marxianus (referred to as WTKm and M5Km, respectively) were dialyzed against 20 mM sodium phosphate buffer (pH 6.0) and concentrated using a Vivaspin 20-50K (Sartorius AG). Protein concentration was determined using a BCA Protein Assay kit (Thermo Fisher Scientific) with bovine serum albumin as a standard. Purified enzymes were deglycosylated through endo-β-N-acetylglucosaminidase H (Endo-H) (New England Biolabs, Ipswich, MA) treatment for 12 h at 37°C and analyzed using 10% SDS-PAGE, followed by CBB staining. Molecular mass was analyzed via gel filtration chromatography, similar to that of E. coli-expressed TaBglIs.
2.8. Mass spectrometry (MS) analysis of N-linked glycans of TaBglI expressed in K. marxianus.
Purified WTKm and M5Km (100 μg) were digested with trypsin/chymotrypsin in 100 mM Tris-HCl buffer (pH 8.2) containing 10 mM CaCl2. The digested peptides were purified using a C18 cartridge column (Waters, Milford, MA), followed by sequential elution with 20% and 40% 2-propanol in 5% acetic acid. The eluents were combined, dried, and dissolved in 100 mM sodium phosphate buffer (pH 7.5), and incubated with peptide N-glycanase F (glycerol-free, New England Biolabs) to release N-glycans. The sugar chains were collected in the flow-through fractions of the C18 cartridge column and the eluent was lyophilized. Permethylation of N-glycans was carried out according to the method of Anumula and Taylor,38) and subsequently analyzed using an UltrafleXtreme matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) instrument (Bruker Daltonics, Billerica, MA) in the positive ion and a reflector modes, with 2,5-dihydroxybenzoic acid (Sigma-Aldrich, St. Louis, MO) as a matrix.
2.9. Enzyme assay.
The standard reaction mixture contained 50 mM citrate-phosphate buffer (pH 4.5). pNP-Glc, 4-methylumbelliferyl-β-D-glucoside (4MU-Glc), and cellooligosaccharides (Seikagaku Corporation, Tokyo, Japan) were used as substrates. The reaction was initiated by adding the enzyme and the mixture was incubated at 40°C for an appropriate time, during which the linearity of the reaction rate was observed. The amount of liberated p-nitrophenolate was determined from the absorbance at 405 nm. Fluorescence of 4-methylumbelliferone was detected at excitation and emission wavelengths of 360 and 460 nm, respectively. The amount of glucose liberated from cellobiose was measured using a Glucose Hexokinase Assay kit (Sigma-Aldrich) and estimated to be half of the total glucose concentrations in the reaction mixture. The enzyme concentrations used in the kinetic analysis were 0.044 nM (for 4MU-Glc) and 11 nM (for cellobiose). The kinetic parameters were calculated by curve fitting the experimental data with the Michaelis-Menten equation using Kaleidagraph (version 4.0; Synergy Software, Reading, PA). The substrate concentrations varied from 0.3 to 3 times the respective Km values. The assays were performed in triplicate. One unit was defined as the amount of enzyme to hydrolyze 1 μmol of the above substrate per minute.
The action of enzymes on cellooligosaccharides was examined using thin-layer chromatography (TLC; Silica gel 60, Merck, Darmstadt, Germany). Reaction mixtures containing 20 mM citrate-phosphate buffer (pH 4.5) and 5 mM cellooligosaccharides were incubated at 40°C for 1 h in the presence of the purified enzyme (23 nM) and developed using a solvent system of chloroform/methanol/water (3/3/1). Carbohydrates were visualized using diphenylamine-aniline-phosphoric acid reagent.39)
The temperature and pH stabilities were examined by measuring the residual pNP-Glc (1 mM)-hydrolyzing activity following enzyme incubation (0.34 mg/ml) in 20 mM sodium phosphate buffer (pH 6.0) for 1 h at various temperatures and overnight dialysis against various 10 mM buffers at 4°C. The buffers used were citrate-phosphate (pH 3.0-6.0), sodium phosphate (pH 6.0-8.0), and Tris-HCl (pH 8.0-9.0). The optimum temperature was determined by incubating the reaction mixture containing 50 mM citrate-phosphate buffer (pH 4.5) and 1 mM pNP-Glc at various temperatures for a short period of time (<1 min). The optimum pH for the hydrolysis of pNP-Glc (1 mM) was determined using various 50 mM buffers as described earlier.
Synergistic action of exo-glucosidase (TaBglI) and endo-glucanase was examined by using ground and sieved-pretreated corn stover (GS-PCS, Novozymes, Bagsvaerd, Denmark) as the substrate. Before starting the reactions, GS-PCS was washed twice with the same volume of water to remove pre-existing glucose. The reaction was carried out in a total volume of 150 μl at 75°C in the presence of 0.025 μg/ml (4.5 munit/ml for WT and 3.6 munit/ml for M5; unit is for pNP-Glc) and/or 0.15 unit/ml of recombinant thermostable endoglucanase (derived from Pyrococcus horikoshii, which has a Tm value of 96°C and retains >80% activity after 3 h of incubation at 97°C;40),41) Wako Pure Chemical Industries, Ltd., Osaka, Japan). Samples were collected at the indicated times and immediately placed in ice-water to stop the reaction. The glucose concentrations in the mixtures were determined using a Glucose Hexokinase Assay kit (Sigma-Aldrich). The reaction mixtures were also analyzed using TLC as described earlier.
2.10. Thermal inactivation half-life (t1/2).
Thermal inactivation half-life (t1/2) of the partially purified TaBglI variant was determined by plotting the common (base-10) logarithms of the residual activity against incubation periods. The partially purified enzyme (0.15 mg/ml, purified by ammonium sulfate fractionation and DEAE-Sepharose Fast Flow column chromatography) was incubated in sodium phosphate (pH 6.0) at 55°C or 60°C at the indicated times and immediately cooled in ice-water to prevent refolding. The remaining activity was examined using pNP-Glc (1 mM) as the substrate.
2.11. CD spectroscopy.
Temperature-induced unfolding was monitored using circular dichroism (CD) spectroscopy at 222 nm with a Jasco J-720 system (Jasco Co., Tokyo, Japan). The purified TaBglI variants (0.1 mg/ml in 20 mM sodium phosphate buffer (pH 6.0)) were heated using a Peltier-type temperature controller PTC-343 at a rate of 1°C/min. The unfolding curve was fitted to Eq. [1]42)-44) and the midpoint of the thermal unfolding curve (Tm) and the change in the enthalpy of protein unfolding (ΔHm) were calculated.
| \begin{aligned}
y & = \frac{a_{\rm N} + b_{\rm N}T}{1 + \exp((-\Delta H_{\rm m}/T + \Delta H_{\rm m}/ T_{\rm m})/R)}\notag\\
& \mathrel{\phantom{=}}{}\! + \frac{(a_{\rm U} + b_{\rm U}T)(\exp((-\Delta H_{\rm m}/T + \Delta H_{\rm m}/T_{\rm m})/R))}{1 + exp((-\Delta H_{\rm m}/T + \Delta H_{\rm m}/T_{\rm m})/R)}\end{aligned} | [1] |
where aN, bN, aU, bU, ΔHm, and Tm are the fitting parameters, while aN + bNT and aU + bUT represent pre- and post-transition regions, respectively. Measurements were performed in triplicate.
2.12. Phylogenetic analysis of protein sequences.
The amino acid sequences of β-glucosidases were obtained from the National Center for Biotechnology Information database. These sequences were aligned using CLUSTAL 2.1 and used to build a phylogenetic tree, visualized using BOX-SHADE 3.21. Evolutionary analyses were conducted using MEGA X.45) The evolutionary history was inferred using the neighbor-joining method.46) Bootstrap analysis based on 1,000 replications was performed to evaluate tree topologies. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown.47) The evolutionary distances, computed using the maximum composites likelihood method,48) are expressed as the numbers of amino acid substitutions per site.
3. Results
3.1. Isolation of mutant TaBglI proteins with increased thermostability.
Initially, we attempted to express TaBglI as a fusion protein with maltose-binding protein or thioredoxin, or as a tagged protein with hexahistidine- or N-utilization substance A. However, the addition of any sequences resulted in the formation of inclusion bodies in E. coli. Therefore, we expressed the protein in its non-tagged form. The recombinant protein (WTEc) showed activity that was comparable to that purified from T. aurantiacus.27) Using the constructed expression plasmid, we created a randomly mutagenized library of the TaBglI gene in E. coli. The average number of introduced mutations was 2 per kbp, as revealed by sequence analysis. Among the ca. 4,000 transformants examined, cell-free extracts of five clones tested positive for pNP-Glc hydrolysis after heat treatment at 55°C without any marked reduction in activity compared with non-treated ones. The introduced mutations were found to cause amino acid substitutions: M279V, T308S, N514S, K119R/D433N, and F306L/K361R. Next, we constructed K119R, M279V, F306L, T308S, K361R, D433N, and N514S single mutants (Table 3). Using crude preparations (20-40% ammonium sulfate fractionation only), we determined their t1/2 at 55°C (Table 3 and Fig. 1a). Five amino acid substitutions (M279V, T308S, K361R, D433N, and N514S) contributed to the prolonged t1/2 of the enzyme (34, 25, 23, 46, and 13 min, respectively), whereas the other two (K119R and F306L) were dispensable or rather detrimental to thermostability. Saturation mutagenesis was performed on amino acid residues 279, 308, 361, 433, and 514. After checking the mutation frequency, we screened 200 clones from each of the saturated sites. Except for residue 514, where cysteine was found to be better than serine in increasing thermostability, the best amino acid replacement had already occurred at all sites. Consequently, we identified the M279V, T308S, K361R, D433N, and N514C substitutions as mutations that enhance TaBglI thermostability.
Table 3
Amino acid substitutions that enhance thermostability of TaBglI
| Variants |
Mutation(s) |
Thermal inactivation half-life (t1/2)
(min)3
|
| 55°C |
60°C |
| Wild-type (WT) |
|
6.7 |
nd |
| K119R |
AAG to AGG1
|
6.2 |
nd |
| M279V |
ATG to GTG1
|
34 |
nd |
| F306L |
TTT to CTT1
|
1.8 |
nd |
| T308S |
ACC to TCC1
|
25 |
nd |
| K361R |
AAG to AGG1
|
23 |
nd |
| D433N |
GAC to AAC1
|
46 |
nd |
| N514S |
AAC to AGC1
|
13 |
nd |
| N514C |
AAC to TGC2
|
20 |
nd |
| M279V/T308S |
|
92 |
1.5 |
| M279V/T308S/D433N |
|
nd |
3.2 |
| M279V/T308S/D433N/N514C |
|
nd |
53 |
| M279V/T308S/K361R/D433N/N514C (M5) |
|
nd |
200 |
1Random mutagenesis.
2Saturation mutagenesis.
3Values are means of duplicate experiments (see Figs. 1a and 1b).
nd:not determined.

These five mutants were expressed in E. coli, purified to homogeneity, and used to determine the Tm by monitoring the temperature-induced unfolding via CD spectroscopy (Table 4). The unfolding process of these mutants was essentially reversible (>70%), and the curves fitted well with a two-state unfolding model (see Equation [1], section 2.11). As expected from the results of t1/2, these five mutants showed higher Tm values than WTEc, with the D433N substitution being the most effective (ΔTm = 4.5°C).
Table 4
Thermodynamic parameters for unfolding of TaBglI variants expressed in E. coli and K. marxianus
| Variants |
Tm(°C)
|
ΔTm(°C)
|
| E. coli
|
|
|
|
|
WTEc
|
60.7 (± 0.5) |
― |
|
M279V |
62.6 (± 0.5) |
1.9 |
|
T308S |
62.0 (± 0.2) |
1.3 |
|
K361R |
62.8 (± 0.5) |
2.1 |
|
D433N |
65.2 (± 0.1) |
4.5 |
|
N514C |
63.3 (± 0.5) |
2.6 |
|
M5Ec
|
69.9 (± 0.5) |
9.2 |
| K. marxianus
|
|
|
|
|
WTKm
|
75.6 (± 0.6) |
― |
|
M5Km
|
82.0 (± 0.8) |
6.4 |
Anticipating a synergistic effect, we combined amino acid replacements to generate M279V/T308S, M279V/T308S/D433N, M279V/T308S/D433N/N514C, and M279V/T308S/K361R/D433N/N514C (M5) mutants. The t1/2 values at 55°C and/or 60°C were determined using partially purified enzymes (purified by ammonium sulfate fractionation and DEAE-Sepharose Fast Flow column chromatography) (Table 3 and Fig. 1a, b). The accumulation of the mutations synergistically increased the t1/2 values in all cases, although the effects varied among the substitutions. The most elevated thermostability was observed for M5 mutant with t1/2 value of 200 min at 60°C.
3.2.2. Characterization of M5 mutant protein expressed in E. coli.
We expressed the M5 mutant in E. coli (M5Ec), purified it by three-step chromatography, and examined its enzymatic properties. The WTEc enzyme was similarly prepared and used for comparison. The Tm value of M5Ec enzyme was increased by 9.2°C compared with WTEc (Table 4). When the molecular mass was analyzed by SDS-PAGE, both proteins migrated as a single band with an apparent molecular mass of 95 kDa, consistent with the calculated mass (92 kDa) (Fig. 2a). Gel filtration chromatography revealed a native molecular mass of 170 kDa, indicating dimer formation in solution (Fig. 2b). The optimum pH was 4.5 for both WTEc and M5Ec (Fig. 2c), and the optimum temperatures were 65°C and 80°C, respectively (Fig. 2d). M5Ec was stable in a pH range of 3.0-9.0, whereas it showed instability at pH > 8.0 (Fig. 2e). WTEc activity was lost after incubation for 1 h at 55°C, M5Ec retained 94% of its activity (Fig. 2f). Incubation at 65°C for 1 h was required for M5Ec to lose its activity, reflecting the higher Tm value of M5Ec than that of WTEc.

The kinetic parameters of WTEc and M5Ec were determined using 4-MU-Glc and cellobiose as substrates (Table 5). The reactions were performed at 40°C to ensure the stability of WTEc. Although the kcat values of M5Ec for both substrates were decreased (by 31% for 4-MU-Glc and 35% for cellobiose), the Km values were also decreased (by 15% for 4-MU-Glc and 27% for cellobiose), resulting in M5Ec retaining 92% of the catalytic efficiency (kcat/Km) of WTEc for cellobiose. In addition, WTEc and M5Ec equally hydrolyzed cellotriose, cellotetraose, and cellopentaose (Fig. 3). Thus, the five amino acid substitutions introduced into TaBglI neither altered the chain-length specificity nor affected its intrinsic catalytic activity towards cellooligosaccharides.
Table 5
Kinetic parameters of TaBglI WT and M5 proteins expressed in E. coli and K. marxianus
| Variants |
4-MU-Glc |
|
Cellobiose |
|
Km (mM)
|
kcat (s-1)
|
kcat/Km (s-1mM-1)
|
|
Km (mM)
|
kcat (s-1)
|
kcat/Km (s-1mM-1)
|
Vmax (unit/mg)
|
| WTEc
|
0.039 (± 0.007) |
190 (± 11) |
5,000 (± 680) |
|
1.5 (± 0.3) |
200 (± 15) |
130 (± 20) |
130 (± 20) |
| M5Ec
|
0.033 (± 0.006) |
130 (± 7) |
3,800 (± 610) |
|
1.1 (± 0.1) |
130 (± 5) |
120 (± 9) |
90 (± 3) |
| WTKm
|
0.075 (± 0.005) |
220 (± 23) |
3,000 (± 110) |
|
1.7 (± 0.1) |
210 (± 5) |
120 (± 5) |
140 (± 3) |
| M5Km
|
0.052 (± 0.007) |
210 (± 12) |
4,100 (± 420) |
|
1.8 (± 0.1) |
200 (± 13) |
110 (± 5) |
130 (± 8) |
The kinetic parameters were determined at 40°C in 50 mM citrate-phosphate buffer (pH 4.5). The enzyme was used with 0.044 nM 4MU-Glc and 11 nM cellobiose.
TaBglI was originally a fungal secretory enzyme with 11 possible N-glycosylation sites (N-X-T/S), among which the NetNGlyc 1.0 server (
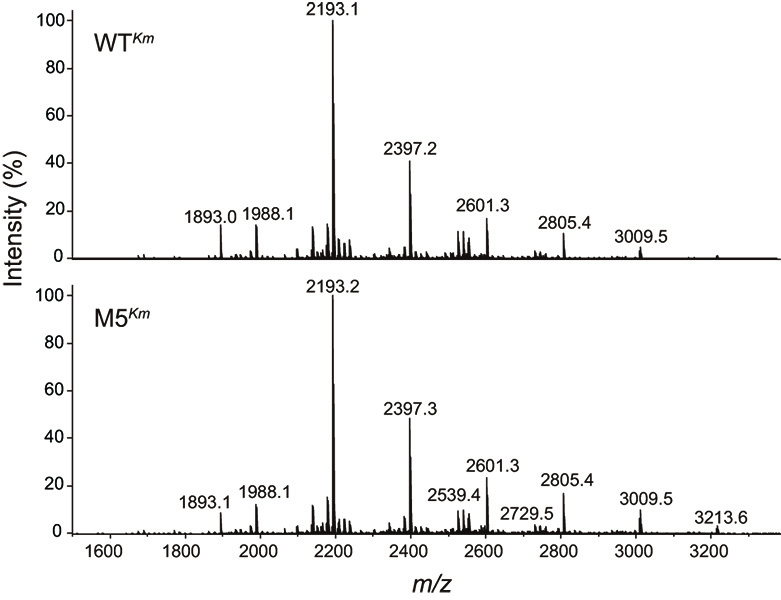
http://www.cbs.dtu.dk/services/NetNGlyc/)49) predicted that residues 62, 316, 442, and 565 may be glycosylated. We expressed the enzyme extracellularly using the yeast expression system developed in this study (see Materials and Methods 2.7.) and purified it from the culture medium using two-step chromatography following ammonium sulfate fractionation. WTKm and M5Km produced single bands with a molecular mass of 130 kDa in SDS-PAGE analysis. Digestion by Endo-H, which acts on N-acetylchitobiose core in high-mannose-type N-glycans, rendered the molecular size of both proteins smaller (ca. 100 kDa) than that of the non-treated preparations (Fig. 2a). N-Glycans of WTKm and M5Km proteins were released by peptide N-glycanase F, permethylated, and analyzed by MALDI-TOF MS. The glycan spectra and their estimated glycosyl compositions were fundamentally identical (Fig. 4 and Table S3), indicating no influence of the amino acid substitutions, quantitatively and qualitatively, on the glycosylation pattern of TaBglI.
Gel filtration chromatography indicated that yeast-expressed TaBglI also formed a dimer in solution (Fig. 2b). The optimum pH of WTKm and M5Km was 4.5, similar to that of E. coli-expressed WTEc and M5Ec (Fig. 2c), and the optimum temperatures were estimated to be >90°C for both enzymes (Fig. 2d). Both WTKm and M5Km were similarly stable over a broad pH range of 3.0-9.0 (Fig. 2e). Although WTKm activity decreased to 48% after 1 h of incubation at 65°C, M5Km activity remained unchanged (Fig. 2f). Incubation for 1 h at 70°C diminished most WTKm activity but did not significantly affect M5Km activity (80% activity retained). Thus, enzymes expressed in yeast were significantly more thermostable than those expressed in E. coli. The increased thermostability of WTKm and M5Km enzymes was demonstrated by Tm values 75.6°C and 82.0°C for WTKm and M5Km, respectively (Table 4). The Tm values of the yeast-expressed proteins were thus increased by 14.9°C and 12.1°C, respectively, compared with those of E. coli-expressed variants.
The kinetic parameters for 4-MU-Glc and cellobiose did not differ between WTKm and M5Km (Table 5), similar to that observed with WTEc and M5Ec. However, the Km values of WTKm and M5Km for 4-MU-Glc (0.075 and 0.052 mM, respectively) were slightly higher than those of E. coli-expressed enzymes. The kcat values of WTKm and M5Km for 4-MU-Glc were also slightly increased, which made the catalytic efficiency comparable between E. coli-expressed and yeast-expressed WT and M5 enzymes. The catalytic efficiency of the yeast-expressed TaBglI for cellobiose was also comparable to that of E. coli-expressed enzymes, with slightly higher Km values and kcat values. Thus, the amino acid substitutions and N-glycosylation did not affect the cellobiase activity of TaBglI.
3.3. Crystal structure analysis of TaBglI.
3.3.1. Overall structure.
To elucidate the structural differences underlying the increased thermal stability, we determined the crystal structures of E. coli-expressed TaBglIs (WTEc and M5Ec). The asymmetric unit contained four TaBglI subunits. TaBglI is dimeric in solution, and the dimer formed a dimer along with the non-crystallographic symmetry. The dimer structure was similar to that of AaBgl1 (PDB ID: 4IIB).50) The overall structures of the four subunits (chains A to D) of WTEc and M5Ec are shown in Fig. S1a. The root means square deviation of all Cα atoms among them was less than 0.170 Å for WTEc and 0.174 Å for M5Ec. The monomers of WTEc and M5Ec have identical domain architectures (root means square deviation = 0.124 Å), composed of three domains: a catalytic TIM (triosephosphate isomerase) barrel-like domain (L23-D357; Fig. 5a, blue), an α/β sandwich domain (R385-G589; Fig. 5a, green), and a FnIII (fibronectin type III) domain (Y655-Y861; Fig. 5a, yellow). These domains were connected by two linker regions (residues 358-384 and 590-654; Fig. 5a, gray). The E510 and D281 residues are equivalent to the acid/base (E509) and nucleophile (D280) residues in A. aculeatus AaBgl1, respectively. Two regions (residues 303-311 and 687-735) were excluded from the model because of disorder. In AaBgl1, residues (W68, Y248, W281, F305, W358, and Y511) forming a long cleft extending from subsites + 1 to + 4 are essential for high activity against cellooligosaccharides. These residues were all conserved in TaBglI (W69, Y249, W282, F306, W359, and Y512, respectively); however, F306 was invisible due to disorder in both the WTEc and M5Ec crystal structures (Fig. S1b). The structural comparison between WTEc and M5Ec revealed that the mutations occurred at the periphery of the catalytic pocket (Fig. 5a and Figs. S2a-d), although residue 308 was not visualized due to disorder in both WT and M5 proteins. In the following sections, we detail the structural changes introduced by the four mutations.

In the WTEc enzyme, the side chain of K361 forms a salt bridge with the side chain of D455 and makes hydrogen bonds with the carbonyl oxygens of the main chains of F100 and A101; however, in the M5Ec mutant, the side-chain guanidino group of R361 is rearranged to form salt bridges with the carboxyl groups of D102 and N379 and makes hydrogen bonds with the carbonyl oxygens of the main chains of F100 and D357 (Fig. 5b). The side-chain rearrangement at position 361 also changed the dihedral angle of F100 (Fig. S3a).
3.3.3. Amino acid replacement D433N.
In the WTEc enzyme, the side-chain carbonyl oxygen of D433 forms a hydrogen bond with the backbone amide nitrogen of F456 (Fig. 5c). In the M5Ec protein, in which the amino acid at position 433 was changed from aspartic acid to asparagine, the side-chain carboxamide can act as both a donor and an acceptor of hydrogen bonds,51)-53) resulting in an additional hydrogen bond between the side chain of N433 and the backbone peptide bond of F456.
3.3.4. Amino acid replacement M279V.
In the WTEc enzyme, M279 was surrounded by the hydrophobic side chains of L91, V140, L142, C188, F277, M246, W282, and M298 (Fig. 5d). The distances from the side chain of M279 to those of L91, C188, and F277 were 3.8, 3.7, and 3.8 Å, respectively. By changing methionine to valine at position 279 in the M5Ec mutant, these distances increased to 4.7, 4.0, and 4.4 Å, respectively, reducing the buried volume around the position.
3.3.5. Amino acid replacement N514C.
In the WTEc enzyme, the carbonyl oxygen in the N514 side chain formed hydrogen bonds with the backbone amide nitrogen atoms of N518 and G517 (Fig. 5e). In the M5Ec mutant, the change in the amino acid from asparagine to cysteine at position 514 resulted in the loss of hydrogen bonds with the backbone amide nitrogen.
3.4. Application of the enzymes for the saccharification of cellulosic materials.
We evaluated the efficiency of TaBglI (WTKm and M5Km) in synergistic action with a thermostable endoglucanase from P. horikoshii using a natural resource (GS-PCS). The reactions were performed at 75°C with the expectation that, at this temperature, the difference in stability between WTKm and M5Km would affect glucose-liberating ability (Fig. 2f). The addition of thermostable cellulase alone to the reaction mixture moderately stimulated glucose liberation from the substrate (Fig. 6a); however, the rate of glucose liberation decreased considerably after 1 h of incubation, probably because of the accumulation of cellobiose, which acts as an inhibitor of cellulase (Fig. 6b). TaBglI alone barely liberated glucose from the insoluble substrate GS-PCS. In contrast, the addition of both TaBglI and the thermostable cellulase significantly stimulated substrate degradation. When WTKm was added with thermostable cellulase, the glucose concentration was increased by 1.5-fold compared with that of thermostable cellulase alone (0.79 mM vs. 0.53 mM at 3 h). When M5Km was included, the glucose concentration reached 0.96 mM at 3 h, which was about 2-fold higher than the glucose liberated in the presence of cellulase only. The increased glucose-liberating activity observed for M5Km may have resulted from the increased enzyme thermostability, because the accumulation of cellobiose was apparent in the TLC analysis when WTKm was included in the mixture (Fig. 6b). In either case, although apparently being denatured, TaBglI was found to remain active during the incubation period (approx. 3 h) at 75°C, despite its significantly loss of activity in the thermal inactivation experiment conducted at 75°C (Fig. 2f). This discrepancy may be caused by the presence of the substrate (mainly cellobiose) in the mixtures, because enzyme-substrate complexes are known to be more stable than free enzymes.

4. Discussion
To the best of our knowledge, there have been no reports describing the expression of fungal GH3 β-glucosidases in active forms using E. coli as a host, which has hampered elucidation of the structure-function relationship of these enzymes. In this study, we established a TaBglI expression system in E. coli, screened mutant enzymes with high thermostability, and determined the structures of the wild-type and mutant enzymes. The thermostabilizing amino acid substitutions were located around the catalytic pocket of TaBglI. Long et al.54) reported the significant contribution of amino acid residues adjacent to the catalytic cavity to enzyme thermostability. However, a negative correlation has been frequently reported between thermostabilizing mutations around the catalytic pocket and the enzyme activity of the resulting mutants.55) Nonetheless, possibly owing to the screening strategy, we found that thermostabilizing mutations were introduced around the catalytic pocket without considerable attenuation of their intrinsic activity. TLC analysis indicated no effect of the thermostabilizing amino acid substitutions on the activity for hydrolyzing cellooligosaccharides (Fig. 3).
In the phylogenetic tree of fungal GH 3 enzymes, Treebupachatsakul et al.22) reported that β-glucosidases with high cellobiase activity form a cluster consisting of TaBglI, Rasamsonia emersonii Cel3a (ReCel3a), A. niger Bgl1 (AnBgl1), A. luchuensis BglA (AlBglA), A. aculeatus Bgl1 (AaBgl1), A. oryzae Bgl1 (AoBgl1), and T. reesei Cel3B (TrCel3B). The amino acid sequence identities of TaBglI with ReCel3a, AnBgl1, AlBglA, AaBgl1, AoBgl1, and TrCel3B were 75%, 70%, 70%, 71%, 72%, and 59%, respectively. Among the β-glucosidases in this cluster, TaBglI and ReCel3a are thermostable, with 72% residual activity retained after incubation at 60°C for 60 min (Fig. 2f) and t1/2 of 62 min at 65°C,25),26),56) respectively. On the other hand, the thermal stabilities of the others (AaBgl1, AnBgl1, TrCel3B, AoBgl1, and AlBglA) were below 60°C,22) 60°C,57) 50°C,22) 45°C,58) and 30°C,59) respectively. We performed multiple sequence alignment to compare the amino acids at the positions of thermostabilizing substitutions (M279V, T308S, K361R, D433N, and N514C) of TaBglI (Fig. 7). Among the five amino acid replacements, three (T308S, K361R, and D433N) reverted to the conserved amino acids, which are not unique to thermostable β-glucosidases, indicating these substitutions that have occurred in TaBglI during evolution are assumed to be unfavorable. The alignment of amino acid sequences of target enzymes with thermophilic counterparts is one of the strategies used for engineering thermostability of enzymes60); however, it might not be applicable to TaBglI. Instead, by applying random and saturation mutagenesis to TaBglI, we have succeeded in identifying the vulnerable points (T308S, K361R, and D433N) for protein stability among β-glucosidases in this cluster. None of the homologs adopt valine residues at the position of M279 of TaBglI. Among the GH3 β-glucosidases, V279 appeared in BGL2 from Saccharomycopsis fibuligera, which possesses relatively low thermostability (below 50°C).61) None of the homologs adopted cysteine residues at position N514 of TaBglI. To convert asparagine to cysteine, at least two bases must be changed. In this study, the codon for asparagine (ACC) was changed to that for cysteine (TGC) in the saturated mutagenesis. It should be unpredictable to obtain M279V and N514C replacements based on amino acid sequence alignment and random mutagenesis, respectively. Besides tolerance to high temperatures, thermostable enzymes frequently exhibit conformational stability over a broad pH range.62),63) Notably, M5Ec acquired greater stability than WTEc in the high pH range (Fig. 2e), indicating its superior conformational stability.

To evaluate the practical potential of TaBglI, we compared its temperature-dependent cellobiase activity to that of other β-glucosidases reported in the literature. β-Glucosidases from R. emersonii,56) Paecilomyces thermophila,64) and Myceliophthora thermophila65) showed high thermostabilities (50% at 65°C for 62 min, 85% at 60°C for 30 min, and 43.2% at 65°C for 120 min, respectively), but their catalytic activities against cellobiose (69.2 unit/mg for 1 mM substrate at 72°C, 113.1 unit/mg for 10 mg/ml substrate at 65°C, and 62.2 unit/mg for 10 mg/ml substrate at 50°C, respectively) were lower than those of mesophilic fungi, such as A. aculeatus Bgl1 (AaBgl1; 132 unit/mg for 5 mg/ml substrate at 37°C),21) and T. aurantiacus BglI (Vmax value of 140 unit/mg for WTKm and 130 unit/mg for M5Km at 40°C). GH1 β-glucosidase from a Gram-negative thermophilic anaerobe, Thermoanaerobacterium aotearoense, possesses high activity against cellobiose (Vmax 740.39 unit/mg at 60°C); however, the optimum temperature is 60°C.66) In contrast, the Tm value (82.0°C) of M5Km was still below the optimum temperature (>90°C) and the activity at 80°C was 9.8-fold higher than the activity at 40°C.
The mechanisms underlying the thermostability of enzymes vary and depend on their nature; nevertheless, some common features can be identified that contribute to stability. These features include more interactions (i.e., hydrogen bonds, salt bridges, hydrophobic interactions, and disulfide bonds) compared with those in less stable enzymes, and superior conformational structures (i.e., more rigidity, a stable α-helix, and conformational strain release).67) The importance of the surface salt bridges for protein stability has been reported by Alsop et al.,68) who showed a clear preference for stabilizing acid-base pairs on the surface of thermophilic proteins by comparing mesophilic-thermophilic orthologous proteins. Crystal structure analysis of TaBglI revealed possible explanations for the thermostability of each thermostabilizing mutations. The K361R mutation introduced an additional salt bridge and hydrogen bond on the surface of TaBglI, resulting in an increase of 2.1°C in the Tm value of the enzyme (Fig. 5b and Table 4). Furthermore, Ramachandran plot analysis (Fig. S3b) revealed that the side-chain rearrangement at position 361 changed the dihedral angle of F100 from allowed (WTEc) to favored (M5Ec). Main-chain stabilization on the protein surface around K361R (Fig. S2a) and the improved dihedral angle of the peptide bond at F100 might have contributed to thermostability. The D433N mutation introduced an additional hydrogen bond with F456 in the G426-T461 loop (Fig. S2b), which might increase thermostability by 4.5°C. The loop regions of proteins unfold more quickly at high temperatures; thus, rigidification of loop regions may an effective strategy for increasing thermostability.69),70) As mentioned above, the K361R and D433N replacements were reversions to residues conserved among GH3 β-glucosidases with high cellobiase activity. As expected, when we compared the structures between TaBglI and AaBgl1 (PDB ID: 4IIB), the K361R and D433N residues of the M5 mutant interacted with the surrounding residues in a manner very similar to that observed at the respective sites in AaBgl1.
Regarding the M279V mutation, we observed reductions in the buried volume (Fig. 5d). Ventura et al.71) reported that, using mutants of the spectrin SH3 domain, a reduction in the buried volume led to a deceleration in unfolding, which was assumed to be caused by the release of conformational strain. The overpacked structures around M279 in WTEc may cause conformational strain, and reduction in the buried volume in M5Ec increased the Tm value by 1.9°C. At the position of N514C mutation, two hydrogen bonds that are formed in WTEc are lost (Fig. 5e), suggesting the negative contribution of hydrogen bonds to thermal stability in this specific site. Hydrogen bond formation generally contributes favorably to protein stability; however, it sometimes destabilizes the protein by introducing intramolecular strain. Campos et al.72) estimated that the destabilizing contribution of the hydrogen bonding between D96 and N128 in apoflavodoxin was ~ +1.1 kcal/mol and showed the conformational strain of N128 by superimposing the protein structure with the structural equivalent protein lacking hydrogen bonding at this position. In our random mutagenesis screening, the N514S replacement was initially identified because it exhibited a prolonged t1/2 value at 55°C (Table 3). Notably, N514S also represents a reversion to the residue conserved among GH3 β-glucosidases with high cellobiase activity (Fig. 7). As mentioned earlier, the carbonyl oxygen of the N514 side chain in WTEc forms hydrogen bonds with the backbone nitrogen atoms of G517 and N518 (Fig. 5e). In contrast, the hydroxyl oxygen of S513 in AaBgl1 does not form any hydrogen bonds except to a water molecule. Similarly, in the M5Ec crystals, the sulfur atom of N514C is not positioned within hydrogen-bond distance of any residues. Therefore, the thermostabilizing effect observed for both N514S and N514C substitutions might be attributed to the release from adverse hydrogen bonding strain present in the WT enzyme.
Generally, glycosylated proteins expressed in yeast are more resistant to unfolding compared with non-glycosylated protein synthesized in E. coli.73)-76) The higher stability of glycosylated enzymes can be attributed to a less stable unfolded state and a more stable folded state.74) Consistent with this, TaBglI expressed in K. marxianus showed enhanced thermostability. In addition, M5Km showed a 6.4°C higher Tm value than WTKm. The folding of proteins expressed in yeast may differ from that of proteins expressed in E. coli due to post-translational modifications such as disulfide bond formation, subunit association, and glycosylation.77) In the crystal structure of TaBgl1, three disulfide bond are formed, as we used Rosetta-gami B as an expression host, which carries mutations in the thioredoxin reductase and glutathione reductase genes. Gel filtration analysis showed that TaBgl1 forms a dimer regardless of whether it is expressed in E. coli or yeast (Fig. 2b). The catalytic activity of the purified recombinant enzyme was comparable between the E. coli-expressed and yeast-expressed versions. Thus, overall tertiary and quaternary structures may not differ between the two proteins, and the increased thermostability of TaBgl1 expressed in yeast may be primarily attributed to its glycosylation.
The efficiency of the five amino acid mutations in TaBglI for natural biomass saccharification was evaluated by the formation of glucose from GS-PCS. The synergistic action of enzymes in the cellulase cocktails is critical for the effective saccharification of cellulosic biomass.78) Although many studies have demonstrated the advantages of thermal stabilization of β-glucosidase,79) the efficiency of cellulosic biomass degradation in synergistic conditions does not always align with stability because of the different affinities for natural substrates.80) Thus, we evaluated the effect of thermal stabilization of TaBglI on cellulolytic activity in synergy with endoglucanase. In the enzymatic degradation process of GS-PCS, M5Km released 1.2 times more glucose than WTKm after incubation for 3 h. The lower amount of residual cellobiose in the reaction mixture indicated a mitigation of the inhibitory effect on cellulases.11)
In the present study, we thermostabilized an industrially important GH3 β-glucosidase (TaBglI) and examined how and to what extent each of the amino acid replacements contributed to the thermostability through biochemical and structural analysis. Step-by-step improvement and understanding of efficient enzymatic saccharification of cellulosic materials will aid in utilizing renewable biomass as an alternative to fossil fuels, thereby ultimately supporting the sustainability of our ecosystem.
Acknowledgments
The authors are deeply grateful for collaborations with previous and current colleagues and students at the Graduate School of Agriculture and Graduate School of Biostudies, Kyoto University, and Ishikawa Prefectural University. The names of these people have been provided in the references.28),29),35)-37) The authors thank the staff of the Photon Factory for X-ray data collection.
The authors also appreciate the financial support offered by the Japan Society for the Promotion of Science (JP16380062 and JP24580120 to HK).
Author contributions
C. Matsuzaki and Y. Nakashima analyzed and interpreted the data and C. Matsuzaki also wrote the first draft of the manuscript. M. Hidaka performed X-ray data collection and structure determination. Y. Honda provided information for the analysis of CD spectroscopy and participated in data analysis. T. Koyanagi analyzed and interpreted the data. K. Ishikawa assisted in data interpretation and participated in revision of the manuscript. T. Katoh performed MS analysis and wrote the methodology. T. Katayama and H. Kumagai conceived and supervised the project and participated in critical revision of the manuscript. All authors have read and approved the final version of the manuscript.
Notes
Edited by Akira ISOGAI, M.J.A.
Correspondence should be addressed to: H. Kumagai, Research Institute for Bioresources and Biotechnology, Ishikawa Prefectural University, 1-308, Suematsu, Nonoichi, Ishikawa 921-8836, Japan (e-mail:
hidekuma@ishikawa-pu.ac.jp).
References
- 1) Peplow, M. (2014) Cellulosic ethanol fights for life. Nature 507, 152-153.
- 2) Dadwal, A., Sharma, S. and Satyanarayana, T. (2021) Thermostable cellulose saccharifying microbial enzymes: Characteristics, recent advances and biotechnological applications. Int. J. Biol. Macromol. 188, 226-244.
- 3) Himmel, M.E., Ding, S.Y., Johnson, D.K., Adney, W.S., Nimlos, M.R., Brady, J.W. et al. (2007) Biomass Recalcitrance: engineering plants and enzymes for biofuels production. Science 315, 804-807.
- 4) Uchiyama, T., Uchihashi, T., Ishida, T., Nakamura, A., Vermaas, J.V., Crowley, M.F. et al. (2022) Lytic polysaccharide monooxygenase increases cellobiohydrolases activity by promoting decrystallization of cellulose surface. Sci. Adv. 8, eade5155.
- 5) Burton, S.G., Cowan, D.A. and Woodley, J.M. (2002) The search for the ideal biocatalyst. Nature Biotechnol. 20, 37-45.
- 6) Szijártó, N., Siika-aho, M., Tenkanen, M., Alapuranen, M., Vehmaanperä, J., Réczey, K. et al. (2008) Hydrolysis of amorphous and crystalline cellulose by heterologously produced cellulases of Melanocarpus albomyces. J. Biotechnol. 136, 140-147.
- 7) Viikari, L., Alapuranen, M., Puranen, T., Vehmaanperä, J. and Siika-aho, M. (2007) Thermostable enzymes in lignocellulose hydrolysis. In Olsson, L. (Ed.), Biofuels (Vol. 108, pp. 121-145). Berlin, Heidelberg: Springer Berlin Heidelberg Retrieved from http://link.Springer.com/10.1007/10_2007_065.
- 8) Turner, P., Mamo, G. and Karlsson, N. (2007) Potential and utilization of thermophiles and thermostable enzymes in biorefining. Microb. Cell Fact. 6, 9.
- 9) Singhania, R.R., Patel, A.K., Pandey, A. and Ganansounou, E. (2017) Genetic modification: A tool for enhancing beta-glucosidase production for biofuel application. Bioresour. Technol. 245, 1352-1361.
- 10) Davidson, S. (2008) Sustainable bioenergy: Genomics and biofuels development. Nat. Edu. 1, 175.
- 11) Gruno, M., Väljamäe, P., Pettersson, G. and Johansson, G. (2004) Inhibition of the Trichoderma reesei cellulases by cellobiose is strongly dependent on the nature of the substrate. Biotechnol. Bioeng. 86, 503-511.
- 12) Barnett, C.C., Berka, R.M. and Fowler, T. (1991) Cloning and amplification of the gene encoding an extracellular β-glucosidase from Trichoderma reesei: evidence for improved rates of saccharification of cellulosic substrates. Nat. Biotechnol. 9, 562-567.
- 13) Martinez, D., Berka, R., Henrissat, B., Saloheimo, M., Arvas, M., Baker, S.E. et al. (2008) Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat. Biotechnol. 26, 553-1193.
- 14) Margeot, A., Hahn-Hagerdal, B., Edlund, M., Slade, R. and Monot, F. (2009) New improvements for lignocellulosic ethanol. Curr. Opin. Biotechnol. 20, 372-380.
- 15) Calsavara, L.P.V., De Moraes, F.F. and Zanin, G.M. (2001) Comparison of catalytic properties of free and immobilized cellobiase Novozym 188. Appl. Biochem. Biotechnol. 91-93, 615-626.
- 16) Sørensen, A., Lübeck, P.S., Lübeck, M., Teller, P.J. and Ahring, B.K. (2011) β-Glucosidase from a new Aspergillus species can substitute commercial β-glucosidase for saccharification of lignocellulosic biomass. Can. J. Microbiol. 57, 638-650.
- 17) Cantarel, B.L., Coutinho, P.M., Rancurel, C., Bernard, T., Lombard, V. and Henrissat, B. (2009) The carbohydrate-active enzymes database (CAZy): an expert resource for glycogenomics. Nucleic Acid Res. 37, D233-D238.
- 18) Malathi, V. and Devegowda, G. (2001) In vitro evaluation of nonstarch polysaccharide digestibility of feed ingredients by enzymes. Poultry Science, 80, 302-305.
- 19) Kim, H.W. and Ishikawa, K. (2010) Complete saccharification of cellulose at high temperature using endocellulase and β-glucosidase from Pyrococcus sp. J. Microbiol. Biotechnol. 20, 889-892.
- 20) Nakazawa, H., Kawai, T., Ida, K., Shida, Y., Kobayashi, Y., Okada, H. et al. (2012) Construction of a recombinant Trichoderma reeseistrain expressing Aspergillus aculeatus β-glucosidase 1 for efficient biomass conversion. Biotech. Bioeng. 109, 92-99.
- 21) Sakamoto, R., Arai, M. and Murao, S. (1985) Enzymic properties of three β-glucosidases from Aspergillus aculeatus No. F-50. Agric. Biol. Chem. 49, 1283-1290.
- 22) Treebupachatsakul, T., Nakazawa, H., Shinbo, H., Fujikawa, H., Nagaiwa, A., Ochiai, N. et al. (2016) Heterologously expressed Aspergillus aculeatus β-glucosidase in Saccharomyces cerevisiae is a cost-effective alternative to commercial supplementation of β-glucosidase in industrial ethanol production using Trichoderma reeseicellulases. J. Biosc. Bioeng. 121, 27-35.
- 23) Kupski, L., Pagnussatt, F.A., Buffon, J.G. and Furlong, E.B. (2014) Endoglucanase and total cellulase from newly isolated Rhizopus oryzae and Trichoderma reesei: production, characterization, and thermal stability. Appl Biochem Biotechnol, 172, 458-468.
- 24) Boer, H., Teeri, T.T., & Koivula, A. (2000). Characterization of Trichoderma reesei cellobiohydrolase Cel7A secreted from Pichia pastoris using two different promoters. Biotechnol. Bioeng. 69, 486-494.
- 25) Ajeje, S.B., Hu, Y., Song, G., Peter, S.B., Afful, R.G., Sun, F. et al. (2021) Thermostable cellulases/xylanases from thermophilic and hyperthermophilic microorganisms: current perspective. Front. Bioeng. Biotechnol. 9, 794304.
- 26) Patel, A.K., Singhania, R.R., Sim, S.J. and Pandey, A. (2019) Thermostable cellulases: Current status and perspectives. Bioresour. Technol. 279, 385-392.
- 27) Parry, N.J., Beever, D.E., Owen, E., Vandenverghe, I., Van Beeumen, J. and Bhat, M.K. (2001) Biochemical characterization and mechanism of action of a thermostable β-glucosidase purified from Thermoascus aurantiacus. Biochem. J. 353 117-127.
- 28) Hong, J., Tamaki, H. and Kumagai, H. (2007) Cloning and functional expression of thermostable β-glucosidase gene from Thermoascus aurantiacus. Appl. Microbiol. Biotechnol. 73, 1331-1339.
- 29) Katayama, T., Suzuki, H., Koyanagi, T. and Kumagai, H. (2000) Cloning and random mutagenesis of the Erwinia herbicola tyrR gene for high-level expression of tyrosine phenol-lyase. Appl. Environ. Microbiol. 66, 4764-4771.
- 30) Otwinowski, Z. and Minor, W. (1997) [20] Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology. Academic press, Vol. 276, pp. 307-326.
- 31) Vagin, A. and Teplyakov, A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22-25.
- 32) Emsley, P., Lohkamp, B., Scott, W.G. and Cowtan, K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486-501.
- 33) Murshudov, G.N., Skubák, P., Lebedev, A.A., Pannu, N.S., Steiner, R.A., Nicholls, R.A. et al. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355-367.
- 34) Bergkamp, R.J.M., Bootsman T.C., Toschka H.Y., Mooren A.T.A., Kox, L., Verbakel, J.M.A. et al. (1993) Expression of an α-galactosidase gene under control of the homologous inulinase promoter in Kluyveromyces marxianus, Appl. Microbiol. Biotechnol. 40, 309-317.
- 35) Matsuzaki, C., Nakagawa, A., Koyanagi, T., Tanaka, K., Minami, H., Tamaki, H. et al. (2012) Kluyveromyces marxianus-based platform for direct ethanol fermentation and recovery from cellulosic materials under air-ventilated conditions. J. Biosci. Bioeng. 113, 604-607.
- 36) Hong, J., Tamaki, H., Yamamoto, K. and Kumagai, H. (2003) Cloning of a gene encoding thermostable cellobiohydrolase from Thermoascus aurantiacus and its expression in yeast. Appl. Microbiol. Biotechnol. 63, 42-50.
- 37) Hong, J., Wang, Y., Kumagai, H., and Tamaki, H. (2007) Construction of thermotolerant yeast expressing thermostable cellulase genes. J. Biotechnol. 130, 114-123.
- 38) Anumula, K.R. and Taylor, P.B. (1992) A comprehensive procedure for preparation of partially methylated alditol acetates from glycoprotein carbohydrates. Anal. Biochem. 203, 101-108.
- 39) Anderson, K., Li, S.C. and Li, Y.T. (2000) Diphenylamine-aniline-phosphoric acid reagent, a versatile spray reagent for revealing glycoconjugates on thin layer chromatography plates. Anal. Biochem. 287, 337-339.
- 40) Ando, S., Ishida, H., Kosugi, Y. and Ishikawa, K. (2002) Hyperthermostable endoglucanase from Pyrococcus horikoshii. Appl. Environ. Microbiol. 68, 430-433.
- 41) Kashima, Y., Mori, K., Fukada, H. and Ishikawa, K. (2005) Analysis of the function of a hyperthermophilic endoglucanase from Pyrococcus horikoshii that hydrolyzes crystalline cellulose. Extremophiles 9, 37-43.
- 42) Eftink, M.R. (1995) Use of multiple spectroscopic methods to monitor equilibrium unfolding of proteins. Methods Enzymol. 259, 487-512.
- 43) Van Nuland, N.A.J., Meijberg, W., Warner, J., Forge, V., Scheek, R.M., Robillard, G.T. et al. (1998) Slow cooperative folding of a small globular protein HPr. Biochemistry 37, 622-637.
- 44) Pace, C.N. and Scholtz, J.M. (1997) Measuring the conformational stability of a protein, in Protein Structure. A Practical Approach, (Creighton, T.E., ed.), Oxford University Press Inc., New York, NY, pp. 299-321.
- 45) Kumar, S., Stecher, G., Li, M., Knyaz, C. and Tamura, K. (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35(6), 1547-1549.
- 46) Saitou, N. and Nei M. (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406-425.
- 47) Felsenstein, J. (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evol. 39, 783-791.
- 48) Tamura, K., Nei, M. and Kumar, S. (2004) Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 101, 11030-11035.
- 49) Gupta, R. and Brunak, S. (2002) Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. 7, 310-322.
- 50) Suzuki, K., Sumitani, J., Nam, Y.W., Nishimaki, T., Tani, S., Wakagi, T. et al. (2013) Crystal structures of glycoside hydrolase family 3 β-glucosidase 1 from Aspergillus aculeatus. Biochem. J. 452, 211-221.
- 51) Ippolito, J.A., Alexander, R.S. and Christianson, D.W. (1990) Hydrogen bond stereochemistry in protein structure and function. J. Mol. Biol. 215, 457-471.
- 52) Cuff, A.L., Janes, R.W. and Martin, A.C. (2006) Analysing the ability to retain sidechain hydrogen-bonds in mutant proteins. Bioinform. 22, 1464-1470.
- 53) Vennelakanti, V., Qi, H.W., Mehmood, R. and Kulik, H.J. (2021) When are two hydrogen bonds better than one? Accurate first-principles models explain the balance of hydrogen bond donors and acceptors found in proteins. Chem. Sci. 12, 1147-1162.
- 54) Long, S., Zhang, X., Rao, Z., Chen, K., Xu, M., Yang, T. et al. (2016) Amino acid residues adjacent to the catalytic cavity of tetramer L-asparaginase II contribute significantly to its catalytic efficiency and thermostability. Enzyme Microb. Technol. 82,15-22.
- 55) Cerdobbel, A., De Winter, K., Aerts, D., Kuipers, R., Joosten, H.J., Soetaert, W. et al. (2011) Increasing the thermostability of sucrose phosphorylase by a combination of sequence-and structure-based mutagenesis. Protein Eng. Des. Sel. 24, 829-834.
- 56) Murray, P., Aro, N., Collins, C., Grassick, A., Penttilä, M., Saloheimo, M. et al. (2004) Expression in Trichoderma reesei and characterisation of a thermostable family 3 β-glucosidase from the moderately thermophilic fungus Talaromyces emersonii. Protein Expres. Purif. 38, 248-257.
- 57) Yan, T.R. and Lin, C.L. (1997) Purification and characterization of a glucose-tolerant β-glucosidase from Aspergillus niger CCRC 31494. Biosci. Biotechnol. Biochem. 61, 965-970.
- 58) Riou, C., Salmon, J.M., Vallier, M.J. and Gunata, Z. (1998) Barre, P9758774: Purification, characterization, and substrate specificity of a novel highly glucose-tolerant β-glucosidase from Aspergillus oryzae. Appl. Environ. Microbiol. 64, 3607-3614.
- 59) Iwashita, K., Nagahara, T., Kimura, H., Takano, M., Shimoi, H. and Ito, K. (1999) The bglA gene of Aspergillus kawachii encodes both extracellular and cell wall-bound β-glucosidases. Appl. Environ. Microbiol. 65, 5546-5553.
- 60) Xiao, Z., Bergeron, H., Grosse, S., Beauchemin, M., Garron, M.L., Shaya, D. et al. (2008) Improvement of the thermostability and activity of a pectate lyase by single amino acid substitutions, using a strategy based on melting-temperature-guided sequence alignment. Appl. Environ. Microbiol. 74, 1183-1189.
- 61) Machida, M., Ohtsuki, I., Fukui, S. and Yamashita, I. (1998) Nucleotide sequences of Saccharomycopsis fibuligera genes for extracellular β-glucosidases as expressed in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 54, 3147-3155.
- 62) Kumar, S., Dangi, A.K., Shukla, P., Baishya, D. and Khare, S.K. (2019) Thermozymes: adaptive strategies and tools for their biotechnological applications. Bioresour. Technol. 278, 372-382.
- 63) Feller, G. (2018) Protein folding at extreme temperatures: Current issues. In Seminars in Cell & Developmental Biology. Academic Press, Vol. 84, pp. 129-137.
- 64) Yan, Q., Hua, C., Yang, S., Li, Y. and Jiang, Z. (2012) High level expression of extracellular secretion of a β-glucosidase gene (PtBglu3) from Paecilomyces thermophila in Pichia pastoris. Protein Expres. Purif. 84, 64-72.
- 65) Zhao, J., Guo, C., Tian, C. and Ma, Y. (2015) Heterologous expression and characterization of a GH3 β-glucosidase from thermophilic fungi Myceliophthora thermophila in Pichia pastoris. Appl. Biochem. Biotechnol. 177, 511-527.
- 66) Yang, F., Yang, X., Li, Z., Du, C., Wang, J. and Li, S. (2015) Overexpression and characterization of a glucose-tolerant β-glucosidase from T. aotearoense with high specific activity for cellobiose. Appl. Microbiol. Biotechnol. 99, 8903-8915.
- 67) Li, W.F., Zhou, X.X. and Lu, P. (2005) Structural features of thermozymes. Biotechnol. Adv. 23, 271-281.
- 68) Alsop, E., Silver, M. and Livesay, D.R. (2003) Optimized electrostatic surfaces parallel increased thermostability: a structural bioinformatic analysis. Protein Eng. 16, 871-874.
- 69) Daggett, V. and Levitt, M. (1993) Protein unfolding pathways explored through molecular dynamics simulations. J. Mol. Biol. 232, 600-619.
- 70) Yu, H., Yan, Y., Zhang, C. and Dalby, P.A. (2017) Two strategies to engineer flexible loops for improved enzyme thermostability. Sci. Rep. 7, 41212.
- 71) Ventura, S., Cristina Vega, M., Lacroix, E., Angrand, I., Spagnolo, L. and Serrano, L. (2002) Conformational strain in the hydrophobic core and its implications for protein folding and design. Nat. Struct. Mol. Biol. 9, 485-493.
- 72) Campos, L.A., Cuesta-López, S., López-Llano, J., Falo, F., and Sancho, J. (2005) A double-deletion method to quantifying incremental binding energies in proteins from experiment: example of a destabilizing hydrogen bonding pair. Biophys. J. 88, 1311-1321.
- 73) Swanwick, R.S., Daines, A.M., Tey, L.H., Flitsch, S.L. and Allemann, R.K. (2005) Increased thermal stability of site‐selectively glycosylated dihydrofolate reductase. ChemBioChem 6, 1338-1340.
- 74) Shental-Bechor, D. and Levy, Y. (2008) Effect of glycosylation on protein folding: a close look at thermodynamic stabilization. Proc. Natl. Acad. Sci. U.S.A. 105, 8256-8261.
- 75) Meldgaard, M. and Svendsen, L. (1994) Different effects of N-glycosylation on the thermostability of highly homologous bacterial (1,3-1,4)-β-glucanases secreted from yeast. Microbiol. 140, 159-166.
- 76) Fonseca-Maldonado, R., Vieira, D.S., Alponti, J.S., Bonneil, E., Thibault, P. and Ward, R.J. (2013) Engineering the pattern of protein glycosylation modulates the thermostability of a GH11 xylanase. J. Biol. Chem. 288, 25522-25534.
- 77) Nielsen, J. (2013) Production of biopharmaceutical proteins by yeast: advances through metabolic engineering. Bioengineered 4, 207-211.
- 78) Igarashi, K. (2013) Cooperative biomass breakdown. Nat. Chem. Biol. 9, 350-351.
- 79) Guo, B., Amano, Y. and Nozaki, K. (2016) Improvements in glucose sensitivity and stability of Trichoderma reesei β-glucosidase using site-directed mutagenesis. PLoS ONE 11, e0147301.
- 80) Lee, H.L., Chang, C.K., Jeng, W.Y., Wang, A.H.J. and Liang, P.H. (2012) Mutations in the substrate entrance region of β-glucosidase from Tricoderma reesei improve enzyme activity and thermostability. Protein Eng. Des. Sel. 25, 733-740.
Non-standard abbreviation list
Aa4MU-Glc
4-methylumbelliferyl-β-D-glucoside
AaBgl1
Aspergillus aculeatus Bgl1
AlBglA
Aspergillus luchuensis BglA
AnBgl1
Aspergillus niger Bgl1
AoBgl1
Aspergillus oryzae Bgl1
CBB
Coomassie Brilliant Blue R-250
CD
circular dichroism
DNJ
1-deoxynojirimycin
Ec
Escherichia coli
Endo-H
endo-β- N-acetylglucosaminidase H
GAPDH
glyceraldehyde 3-phosphate dehydrogenase
GH1
glycosyl hydrolase family 1
GH3
glycosyl hydrolase family 3
GS-PCS
ground and sieved-pretreated corn stover
Km
Kluyveromyces marxianus
M5
TaBglI mutant with M279V/T308S/K361R/D433N/N514C replacements
M5 Ec
M5 expressed in E. coli
M5 Km
M5 expressed in K. marxianus
MALDI-TOF MS
matrix-assisted laser desorption/ionization time of flight mass spectrometry
MS
mass spectrometry
pNP-Glc
4-nitrophenyl-β-D-glucoside
ReCel3a
Rasamsonia emersonii Cel3a
SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
t 1/2
thermal inactivation half-life
TaBglI
Thermoascus aurantiacus BglI
TLC
thin-layer chromatography
T m
midpoint of thermal unfolding transition
TrCel3B
Trichoderma reesei Cel3B
URA3
yeast gene encoding orotidine 5′-phosphatedecarboxylase
WT
wild type TaBglI
WT Ec
wild type TaBglI expressed in E. coli
WT Km
wild type TaBglI expressed in K. marxianus


