Abstract
The anaplastic lymphoma kinase gene (ALK) is located at chromosome 2p23 and encodes a transmembrane receptor tyrosine kinase. As a result of chromosomal translocation/inversion, the 3′ sequence of ALK fuses to the 5′ sequence of a partner gene, producing an oncogenic chimeric protein that activates downstream signal transduction pathways. ALK fusion genes are found in diverse tumor types of different lineages, including hematological neoplasms. ALK-positive (ALK+) anaplastic large cell lymphoma (ALCL) is a distinct subtype of peripheral T-cell lymphoma that is characterized by the expression of CD30 and ALK by immunohistochemistry (IHC). Approximately 70 to 80% of ALK+ ALCL cases have t(2;5)(p23;q35)/NPM1::ALK associated with the nuclear/cytoplasmic ALK IHC staining pattern. In the remaining cases, ALK fuses to various partner genes and ALK staining pattens vary with these partners. ALK+ large B-cell lymphoma (LBCL), accounting for <1% of all diffuse large B-cell lymphoma cases, is characterized by an immunoblast/plasmablast-like cytomorphology. ALK+ LBCL cases with t(2;17)(p23;q23)/CLTC::ALK show the cytoplasmic granular pattern of ALK IHC. Acute myeloid leukemia (AML) harboring inv(2)(p23q13)/RANBP2::ALK defines a small subset of AML characterized by monocytic differentiation and monosomy 7. Extramedullary plasmacytoma/multiple myeloma with the ALK fusion gene shares cytomorphological and immunophenotypic features with ALK+ LBCL. ALK+ histiocytosis is an emerging rare histiocytic entity and most cases carry KIF5B::ALK. The antibody-drug conjugate, brentuximab vedotin, which targets cell-surface CD30, is approved for the treatment of ALK+ ALCL as a single agent or in combination with other cytotoxic agents in both the front-line and relapsed/refractory (R/R) settings. R/R ALK+ hematological neoplasms respond well to first- to third-generation ALK tyrosine kinase inhibitors and an optimal treatment strategy including these novel agents is being tested.
Translated Abstract
ALK (anaplastic lymphoma kinase)遺伝子は染色体2p23に位置し,膜貫通型受容体チロシンキナーゼをコードしている.染色体転座・逆位によってALKの3′側の配列がパートナー遺伝子の5'側の配列に結合し,腫瘍原性キメラ蛋白質をコードする.その結果,下流のシグナル伝達経路が活性化し腫瘍発生に至る.ALK融合遺伝子は,造血器腫瘍を含む異なる系統の多様な腫瘍に認められる.ALK陽性未分化大細胞型リンパ腫(ALCL)は,末梢性T細胞リンパ腫のサブタイプで,免疫組織化学染色(IHC)によるCD30とALKの発現を特徴とする.ALK陽性ALCLの約70–80%の症例ではt(2;5)(p23;q35)/NPM1::ALKを認め,ALK IHCで核・細胞質染色パターンを示す.残りの症例では,ALKは多様なパートナー遺伝子と融合し,パートナーによって様々なALK染色パターンを示す.ALK陽性大細胞型B細胞リンパ腫(LBCL)は,びまん性大細胞型B細胞性リンパ腫全体の1%未満を占めるに過ぎないが,免疫芽球・形質芽球様の細胞形態を特徴とする.t(2;17)(p23;q23)/CLTC::ALKを伴うALK陽性LBCLは,ALK IHCで細胞質顆粒状染色パターンを示す.inv(2)(p23q13)/RANBP2::ALK を伴う急性骨髄性白血病(AML)はAMLのなかの稀なサブセットで,単球分化とモノソミー7を特徴とする.ALK融合遺伝子が認められた髄外形質細胞腫・多発性骨髄腫は,ALK陽性LBCLと細胞形態や免疫形質が共通する.ALK陽性組織球症は,近年見出された稀な組織球症で,大半の症例でKIF5B::ALKが認められる.細胞表面CD30分子を標的とする抗体薬物複合体であるブレンツキシマブ・ベドチンは,ALK陽性ALCLの一次治療または再発・難治症例に対する二次治療として承認を受け,単剤または他の抗腫瘍剤と組み合わせて投与される.第1世代から第3世代のALKチロシンキナーゼ阻害剤は,再発・難治性のALK陽性造血器腫瘍に対して顕著な効果を示し,これらの新規薬剤を含む治療戦略が試みられている.
INTRODUCTION
Since the discovery of a fusion gene between the 5' sequence of BCR and 3' sequence of ABL1 resulting from t(9;22)(q34;q11.2) in chronic myeloid leukemia (CML), multiple fusion genes have been found not only in leukemias and lymphomas, but also in solid tumors.1 The fusion gene is invariably composed of the 5' end of one gene and 3' end of the other. Since the 5' and 3' genes are fused in-frame, the fusion gene generates a chimeric protein, consisting of the amino (N)-terminal structure of the 5' gene, encoding domains affecting the properties and activity of the chimera, and the carboxy (C)-terminal structure of the 3' gene, typically encoding a transcriptional factor or tyrosine kinase.1 The chimera acts as a single unit affecting various cellular processes, thereby playing central roles in the development of tumors. On the other hand, some fusion genes are closely associated with specific tumor subtypes, e.g., BCR::ABL1 in CML, while others are found in many tumor subtypes, e.g., ETV6::NTRK3 in Philadelphia chromosome-like acute lymphoblastic leukemia, mesoblastic nephroma of the kidney, soft tissue fibrosarcoma, and adenocarcinoma of the breast.1 Another characteristic of fusion genes is their promiscuity; 3' genes are rearranged with a diverse range of 5' partner genes in one tumor type, while the 5' genes, in turn, are rearranged with other 3' genes in another type of tumor.1
This review describes hematological neoplasms associated with anaplastic lymphoma kinase (ALK) fusion genes, along with the clinical characteristics and outcomes of relevant patients treated in our institution. Since effective targeting therapies have been developed for these hematological tumors,2-4 a more detailed understanding of their clinicopathological features and treatments is becoming increasingly important for not only physicians in the field of hematology, but also clinical laboratory technologists involved in their diagnosis and the monitoring of treatment courses.
ALK FUSION GENES WITH DIVERSE PARTNERS IN VARIOUS TUMORS
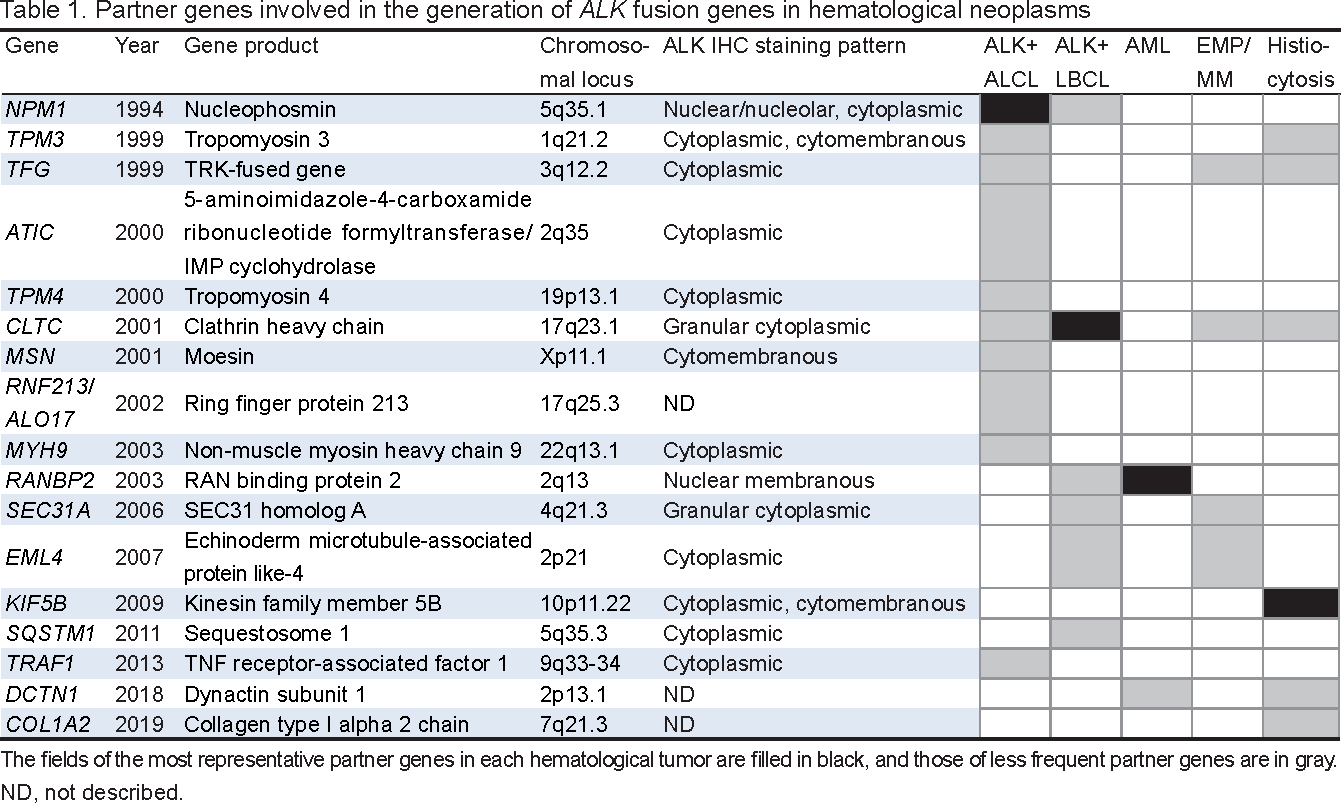
ALK was discovered from the breakpoint of the t(2;5)(p23;q35) translocation, which was initially identified in the Ki-1 (CD30)-positive anaplastic large cell lymphoma (ALCL) cell line, Karpas 299 (Figure 1A).5-7ALK is located at chromosome 2p23 and encodes a transmembrane receptor tyrosine kinase with a molecular weight of 177 kDa, belonging to the insulin receptor subfamily with the closest homology to the leukocyte tyrosine kinase.8, 9

The ALK protein is, in its orientation from the N terminus towards the C terminus, composed of a unique extracellular ligand-binding domain, comprising two MAM domains and a low-density lipoprotein class A (LDL-A) domain, transmembrane domain, and intracellular tyrosine kinase domain.8 The expression of ALK is restricted to the nervous system, and it plays a role in neuronal development in mice.4, 9 The activation of the catalytic kinase function of ALK kinase is induced by the ligand-mediated dimerization of the molecules, resulting in the intramolecular transphosphorylation of the kinase domain and the subsequent activation of downstream signaling, which is important for cellular growth and proliferation.8, 9
In tumors, ALK is aberrantly expressed due to chromosomal translocation or inversion that fuses the 3′ sequence of ALK with the 5′ sequence of a partner gene, which provides promoter activity and leads to the constitutive expression of chimeric protein products.3, 9The prototypical fusion partner is NPM1 encoding for nucleophosmin (NPM). NPM1 is located at 5q35 and, as a result of t(2;5)(p23;q35), fuses to ALK to generate the NPM1::ALK fusion gene, thereby leading to the aberrant production of the p80 NPM::ALK chimeric protein (Table 1, Figure 1B).7, 10 NPM is a 23-kDa nucleolar phosphoprotein composed of distinct domains that account for its multiple biochemical functions.11The N terminus contains the regions responsible for the self-oligomerization and chaperone activities of the molecule. On the other hand, the C terminus contains regions that are crucial for binding to the nucleolus (the nucleolar localization signal). NPM is involved in the transport of pre-ribosomal particles and ribosome biogenesis, the regulation of cell division, DNA repair, transcription, and genomic stability.3
Another predominant fusion gene is EML4::ALK, which was initially described in 5 (6.7%) out of 75 Japanese patients with non–small-cell lung cancer (NSCLC).12
Since EML4 is located at 2p21, EML4::ALK is generated by the paracentric inversion of a small chromosomal segment within the short arm of chromosome 2, i.e. inv(2)(p21p23).
13, 14 EML4 belongs to the family of echinoderm microtubule-associated protein (EMAP)-like proteins and is composed of an N-terminal basic region, hydrophobic EMAP-like protein domain, and tryptophan-aspartic acid (WD) repeat region.13 EML4 retains an important function for the formation of an intact microtubule network and plays an essential role in cell survival and proliferation. Several variants of the EML4::ALK fusion have been reported based on the different breakpoints on EML4;15the most common is variant 1 (33%), in which exon 13 of EML4 is fused to exon 20 of ALK (E13; A20), while variant 2 (10%) and variant 3a/b (29%) involve exon 20 and exon 6a or 6b of EML4, respectively, fused to exon 20 of ALK.13, 15
Multiple partner genes were found to be rearranged with ALK to produce each chimeric protein.3, 4, 7-9, 16, 17 These partner genes share the properties of their protein products being ubiquitously expressed and having the domain for self-dimerization.9 Similar to the ligand-mediated dimerization of wild-type ALK, ALK chimeric proteins are homodimerized through the self-dimerization domain, resulting in the constitutive activation/phosphorylation of the ALK tyrosine kinase. Downstream molecules activated by dimeric ALK chimeric proteins include the phosphoinositide-3 kinase pathway, leading to AKT activation (PI3K/AKT), phospholipase-γ, RAS/ERK/MAPK, mTOR, and the Janus kinase/signal transducer and activator of transcription pathways, which promote growth, survival, and proliferation.2-4, 7-9ALK fusion genes are found not only in ALCL and NSCLC, but also in diverse tumor types of different lineages, including renal cell carcinoma, colorectal cancer, thyroid cancer, breast cancer, inflammatory myofibroblastic tumors, and Spitz melanocytic tumors.4, 7, 8, 17 Hematological tumors other than ALCL include diffuse large B-cell lymphoma (DLBCL), acute myeloid leukemia (AML), extramedullary plasmacytoma/multiple myeloma (EMP/MM), and histiocytosis. A preferential, but not specific, relationship appears to exist between partner genes and disease subtypes (Table 1).
DETECTION OF ALK FUSION GENES
G-banding and fluorescence in situ hybridization
Metaphase spreads are prepared by short-term cultures of neoplastic cells followed by standard cytogenetic processing, and chromosomes are banded by trypsin-Giemsa; however, metaphase cells that are adequate for karyotyping are not always obtained. The Vysis ALK Break Apart FISH Probe Kit (Abbott Laboratories, Abbott Park, IL, USA) consists of a green-labeled centromere probe corresponding to the 5′ sequence of ALK and a red-labeled telomere probe corresponding to the 3′ sequence of the gene (Figures 1B and C). The probe kit is applied to not only cytogenetic preparations (Figure 1D), but also tissue sections prepared from formalin-fixed, paraffin-embedded (FFPE) specimens. When ALK is rearranged with a partner gene, one pair of the green and red signals is separated. Copy number increases may occur. FISH using the ALK break apart probe kit cannot identify partner genes.
Polymerase chain reaction
mRNA and genomic DNA are extracted from tumor materials. Although fresh tumor biopsies or frozen tissues in OTC compound are preferable, mRNA/DNA may be extracted from FFPE specimens. The reverse transcriptase (RT)-mediated polymerase chain reaction (PCR) targets cDNA complementary to mRNA transcribed from the fusion gene. Forward primers are designed for the partner genes and a reverse primer is for ALK. RT-PCR products are common among cases carrying each partner::ALK fusion gene. The rapid amplification of cDNA ends (RACE) has revealed previously unknown fusion partners. The genomic sequences of the fusion gene are obtained by PCR using DNA as the template. Since the breakpoints of both the partner gene and ALK occur within specific introns, primers are designed for exons immediately adjacent to each intron. DNA-PCR products are specific to individual cases.
Immunohistochemistry
Since ALK expression is normally confined to the central nervous system, positive ALK immunohistochemistry (IHC) is a surrogate for the presence of a rearrangement of the ALK gene. Depending on the partner gene, ALK staining may be observed in various subcellular locations (Figures 2 and 3). Mouse (clone 5A4) and rabbit (clone D5F3) monoclonal antibodies as well as polyclonal antibodies for ALK IHC are provided from manufacturers.
Next-generation sequencing
Next-generation sequencing (NGS) assays have rapidly emerged as a preferred option over FISH and IHC for the molecular testing of NSCLC.18Comprehensive genomic profiling has also been applied to hematological neoplasms,19 and, in the upcoming NGS panel for hematological neoplasms from the Japan Society of Hematology, ALK is listed at evidence level 1 for the diagnosis of ALCL. The advantage of the NGS platform is that it allows for the detection of not only known ALK fusion genes, but also unknown partners, along with other gene mutations, fusions, and amplifications.19, 20
ALK-POSITIVE ALCL
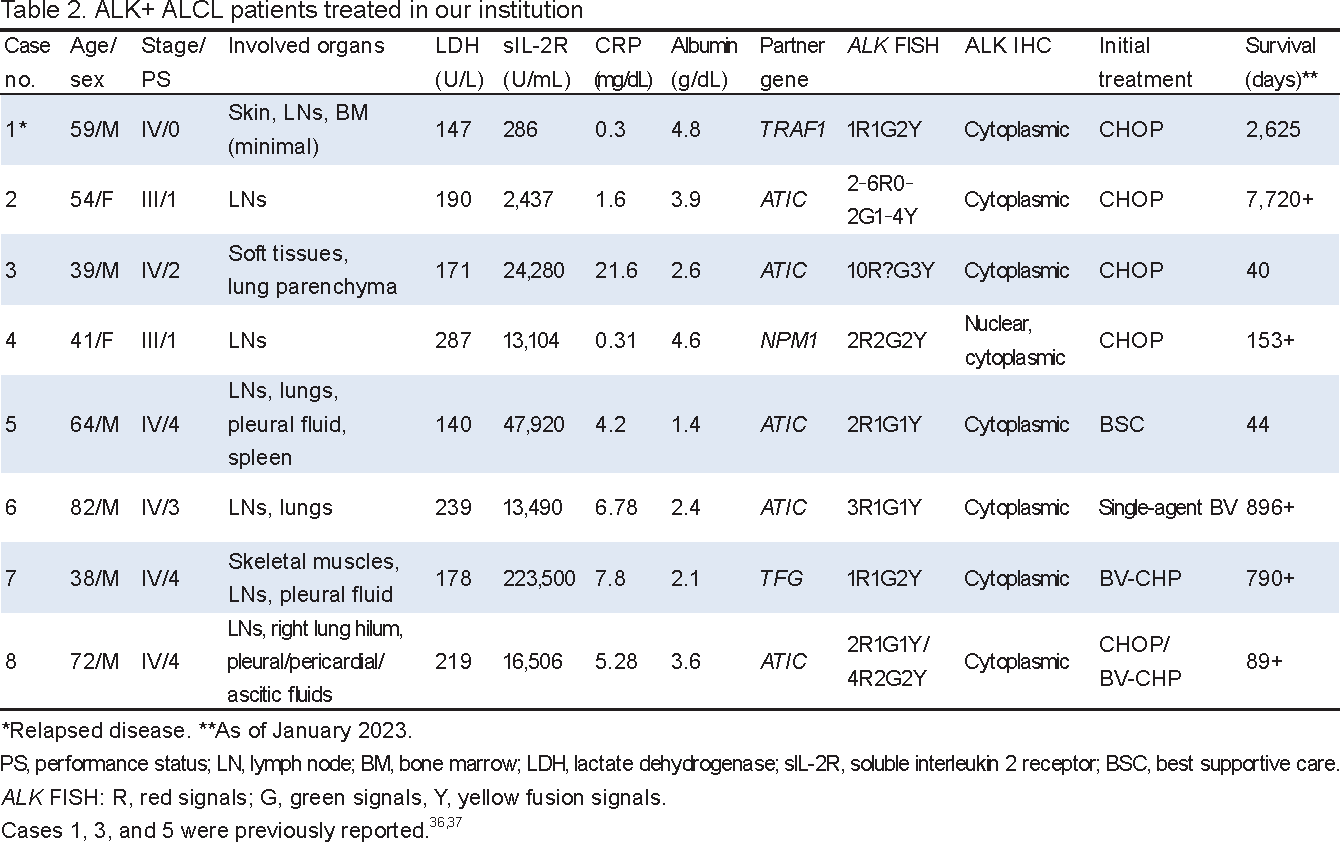
ALCL is the third most common subtype of peripheral T-cell lymphoma (PTCL). A significant fraction of ALCL carries the rearrangement of ALK, leading to ALK protein expression, and clinically has a distinctly favorable treatment outcome. Therefore, the WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues classifies ALK-positive (ALK+) ALCL as a distinct clinicopathologic entity.16 The disease is most frequently detected in the first 3 decades of life, accounting for between 10 and 20% of childhood lymphoma cases, but only between 2 and 3% of all non-Hodgkin lymphomas in adults.9, 16 Patients present with rapidly progressive lymphadenopathy associated with systemic symptoms, such as fever, night sweats, and weight loss. Approximately two-thirds of patients present with stage III or IV disease, and extranodal involvement is common, including the skin, bone, soft tissue, lungs, and liver. Bone marrow (BM) involvement is detected in 10% of cases and a rare leukemic manifestation has been described.21 The central nervous system is rarely affected.22
The lymph node and tissue involvement of ALK+ ALCL comprises a sheet-like cohesive pattern of the proliferation of large tumor cells (Figures 2A and 3). When the lymph node architecture is only partially effaced, tumor cells grow within the sinusoids (Figure 2B), potentially being misdiagnosed as metastatic carcinoma.16 Although the majority of cases show the common histopathology pattern, several morphological variants have been described, including the lymphohistiocytic pattern, small cell pattern, Hodgkin-like pattern, and a composite of two or more patterns within a single lymph node. Nevertheless, variable percentages of hallmark cells characterized by eccentric, horseshoe-shaped, or kidney-shaped nuclei with an eosinophilic region near the nucleus are present in all morphological variants (Figure 3).16 Tumor cells are universally positive for CD30 on the cell membrane and in the Golgi region (Figures 2A and 3). CD4 and CD5 are positive, whereas CD3 is negative in >75% of cases. B-cell surface markers are negative. The so-called null-cell type represents the absence of both T- and B-cell markers. Epithelial membrane antigen (EMA) is positive in most cases (Figure 3, g). Tumor cells may express TIA1, perforin, and granzyme B cytotoxic molecules (Figure 2A, e). There is no evidence for Epstein-Barr virus infection. Clonal rearrangements of T-cell receptor genes (TCRs) are detected in most cases.
Approximately 70 to 80% of ALK+ ALCL cases have t(2;5)(p23;q35)/NPM1::ALK (Figure 1).2, 7, 9, 16,17, 23, 24Initial transgenic mouse models of the NPM1::ALK fusion gene driven by different promoters developed B-cell lymphoma with rearrangements of the immunoglobulin genes or T-cell lymphoma with an immature thymocyte phenotype.25, 26 Direct evidence for the involvement of NPM1::ALK in the development of ALCL was obtained by an experiment, in which NPM1::ALK was lentivirally transduced into human CD4+ T lymphocytes.27 Cells became immortalized and displayed the morphology and immunophenotype characteristic of ALCL. Most importantly, the implantation of NPM1::ALK-transformed CD4+ T lymphocytes into immunodeficient mice resulted in the formation of tumors with similar features to those of human ALK+ ALCL, i.e., the anaplastic large cell morphology, the expression of NPM::ALK and CD30, the variable loss of T-cell antigens, and a high proliferative rate. Nevertheless, since a TCR rearrangement analysis indicated that the tumor cells were of a monoclonal to oligoclonal nature, additional genetic changes may be required to achieve a complete malignant phenotype.27
ALK IHC of tumor tissues with t(2;5)(p23;q35)/NPM1::ALK showed cytoplasmic and nuclear/nucleolar positivity (Figure 2A, c), the latter of which was due to the transport of NPM::ALK into the nucleus/nucleolus through the formation of heterodimers of NPM::ALK with wild-type NPM that shuttled between the cytoplasm and nucleus.16 In the remaining 15% of cases, ALK fused to various partners (Table 1). The ALK IHC staining patterns of non-NPM1::ALK cases were attributed to the normal subcellar distribution of each partner protein, i.e., diffuse cytoplasmic, diffuse cytoplasmic with peripheral intensification, granular cytoplasmic, or membranous staining, whereas nuclear/nucleolar staining was uniformly absent (Table 1, Figures 2B and 3).7, 9, 16, 17 In one experiment, Ba/F3 cells carrying the ATIC::ALK fusion gene generated by CRISPR/Cas9 genome editing showed a significantly lower transformation potential and slower cell growth than cells carrying NPM1::ALK.28 Nevertheless, it currently remains unclear whether the clinical behavior of ALK+ ALCLs with the non-NPM1::ALK rearrangement is identical to or different from that of NPM1::ALK ALCL.
The nuclei of ALK+ ALCL expressed high levels of BCL3 (Figure 2A, f) and gene copies increased in Karpas 299 and SU-DHL-1 ALCL cell lines.29BCL3 was initially discovered on chromosome 19 adjacent to the breakpoints of t(14;19)(q32;q13) observed in some patients with B-cell chronic lymphocytic leukemia.30The BCL3 protein includes 7 tandem copies of the ankyrin repeat element in the central domain, representing the characteristic structure of the IκB family of inhibitors of NF-κB transcription factors. BCL3 is unique in that the protein localizes within the nucleus and is predominantly associated with p50 or p52 homodimers. Upon a stimulation with CD30, BCL3 was found to be phosphorylated and associated with the p52 homodimer by immunoprecipitation and Western blotting analyses.31 We found that BCL3 and p52 colocalized within ALK+ ALCL cell nuclei by IHC of biopsy specimens.
The standard treatment for patients with PTCL for decades has been 6 to 8 cycles of cyclophosphamide, doxorubicin, vincristine, and prednisolone (CHOP), with the addition of etoposide (CHOEP) for younger patients.22, 32 ALK+ ALCL responds very well to these treatments among PTCLs; the estimated 5-year overall survival (OS) rate for ALK+ ALCL patients treated with an anthracycline-based regimen ranged between 70 and 93%,32-34 and the 3-year event-free survival rate for CHOEP-treated ALK+ ALCL patients was as high as 91.2%.32 However, these favorable survival rates may be attributed to a younger age distribution in ALK+ ALCL among PTCL subtypes; an increased age was associated with poor clinical outcomes in most studies.32-34In one study, OS was worse in patients with CD56 positivity by IHC than in those lacking its expression.35 The international prognostic index (IPI; age >60 years, LDH >normal range, ECOG performance status [PS] ≥2, Ann Arbor stage III/IV, and number of extranodal sites >1) had a better predictive value for OS than the prognostic index for PTCL, not otherwise specified (age >60 years, ECOG PS ≥2, LDH >normal range, and the presence of BM involvement).22 The International PTCL project reported estimated 5-year OS rates for ALK+ ALCL patients of 90, 68, 23, and 33% for those with IPI of 0/1, 2, 3, and 4/5, respectively.33
Table 2 summarizes the characteristics and laboratory data of 8 patients with ALK+ ALCL treated in our institution in the past 20 years.36, 37 Ages ranged between 38 and 82 years with a median of 56.5 years. Six patients were male. Four patients presented with disseminated disease involving extranodal sites and poor PS. Three patients had pleural/ascitic fluid retention and anasarca, likely representing capillary leak syndrome.37 Abnormal laboratory data included markedly high levels of soluble interleukin-2 receptor in 7 patients. C-reactive protein was positive in all patients. In contrast, lactate dehydrogenase levels were not elevated. Hypoalbuminemia was detected in 6 patients. Rearrangements of 2p23/ALK were identified by G-banding and/or FISH and RT- and DNA-PCR. In our series, ATIC was the most common partner of ALK, which was located at 2q35 and fused with ALK by pericentric inversion inv(2)(p23q35) (Figure 4). FISH uniformly demonstrated an increase of red signals representing the 5′ ATIC::3′ ALK fusion gene that encodes the ATIC::ALK chimeric protein (Table 2, Figure 4).28 ALK IHC confirmed the restrictive cytoplasmic expression of the protein in 7 non-NPM1::ALK cases. Five patients were initially treated with CHOP. Case 1 showed multiple relapses after a short-term remission with 6 cycles of CHOP,36 while case 2 relapsed after 18 relapse-free years with the treatment (Table 2).

ALK+ LARGE B-CELL LYMPHOMA
ALK+ large B-cell lymphoma (ALK+ LBCL) was initially described by Delsol et al. in 1997 as a subtype of DLBCL characterized by the expression of the ALK protein, but lacking t(2;5)(p23;q35).38, 39Subsequent studies reported that lymphoma was very rare, accounting for <1% of all DLBCL cases.40 ALK+ LBCL showed a male predominance with a male to female ratio of 3.5 and occurred in younger individuals with an average age of 38.4 years.41 Patients present with generalized lymphadenopathy, a mediastinal mass, or extranodal diseases.40-42 BM involvement is detected in 30% of cases,40, 41 and 60% of cases present with stage III/IV disease.39, 41, 42
Involved lymph nodes comprise marked infiltrates of lymphoma cells often with a sinusoidal growth pattern.38, 41-43 These cells exhibit a monomorphic immunoblast/plasmablast-like morphology with an abundant amphophilic cytoplasm and round pale nuclei that contain large central nucleoli.38-42, 44 These cells are negative for pan–B-cell markers (i.e., CD20 and CD79a), but are positive for plasma cell markers, such as CD138, and express monotypic immunoglobulins in the cytoplasm; the heavy chain is typically IgA.38, 40, 41, 45PAX5 is negative or weakly positive, while MUM1 is positive.40-42, 45 CD30 is negative. EMA is positive. EBV is negative by EBER-ISH. Lymphoma cells in the majority of cases express ALK with a restricted cytoplasmic granular staining pattern by IHC (Table 1; Figure 2B, c),38, 40-42 which is associated with the generation of the CLTC::ALK chimeric protein, resulting from t(2;17)(p23;q23) that fuses CLTC at 17q23 to ALK at 2p23.44, 46, 47 In a few cases, ALK fuses to NPM1, SEC31A, SQSTM1, RANBP2, IGL, or EML4,40-42, 48, 49 leading to variable ALK staining patterns.41 A differential diagnosis includes B-cell neoplasms with an immunoblastic/plasmablastic cytomorphology and CD20 negativity, such as plasmablastic lymphoma, plasmablastic plasmacytoma, primary effusion lymphoma, and HHV8+ DLBCL.41
CLTC, alias clathrin heavy chain, is a major protein constituent of the coat that surrounds the cytoplasmic face of intracellular organelles. These coats are involved in receptor-mediated endocytosis and intracellular trafficking as well as the recycling of receptors, which accounts for its characteristic punctate cytoplasmic and perinuclear cellular distribution.50 Structurally, CLTC forms a triskelion-shaped protein complex that is composed of a trimer of CLTC, each of which is bound to a single light chain. CLTC is a 1,675 amino-acid residue protein comprising an N-terminal globular domain that interacts with adaptor proteins, a light-chain–binding region, and a trimerization domain near the C terminus. Since the CLTC::ALK fusion point in CLTC is the C terminus to the trimerization domain, CLTC is considered to provide the CLTC::ALK chimera with deregulated expression driven by its constitutively activated promoter and constitutive oligomerization through the trimerization domain, which is typically used for CLTC coat assembly.50
ALK+ LBCL exhibits an aggressive clinical behavior. Patients with the disease have been treated with CHOP, CHOEP, or more intensive chemotherapies, excluding rituximab due to the lack of CD20 expression, with or without consolidated radiotherapy or high-dose chemotherapy/auto-HSCT.39, 41, 42 The 2- and 5-year OS rates of patients with stage I/II ALK+ LBCL treated with CHOP or CHOP-like regimens were reported to be 76 and 66%, respectively, while those of stage III/IV disease were as low as 27 and 8%, respectively.
41A review of case series of ALK+ LBCL showed that the granular ALK IHC staining pattern, which is the surrogate of the CLTC::ALK fusion gene, was associated with a higher response rate to CHOP-like treatment and better survival than the non-granular staining pattern.49
ALK+ AML
Although myeloproliferative diseases with ALK fusion genes were initially described in the pediatric literature, older patients with identical cytogenetic/molecular abnormalities have since been reported.51-56 The disease presents as juvenile myelomonocytic leukemia (JMML) or AML with monocytic differentiation and a myelodysplastic morphology (Table 3). The white cell count in peripheral blood ranged between 12 and 159 × 103/μL (median, 84 × 103/μL) with monocytes between 1.6 and 55.4 × 103/μL (median, 27 × 103/μL). Leukemia cells carry inv(2)(p23q13) or its translocation equivalent, t(2;2)(p23;q13), which fuses RANBP2 at 2q13 to ALK at 2p23, generating the RANBP2::ALK fusion gene (Table 1). A single case with the DCTN1::ALK fusion gene was found in a large series of JMML using the RNA-seq–based study.54 Leukemia cells mostly carry monosomy 7 identified by G-banding and/or FISH (Table 3).
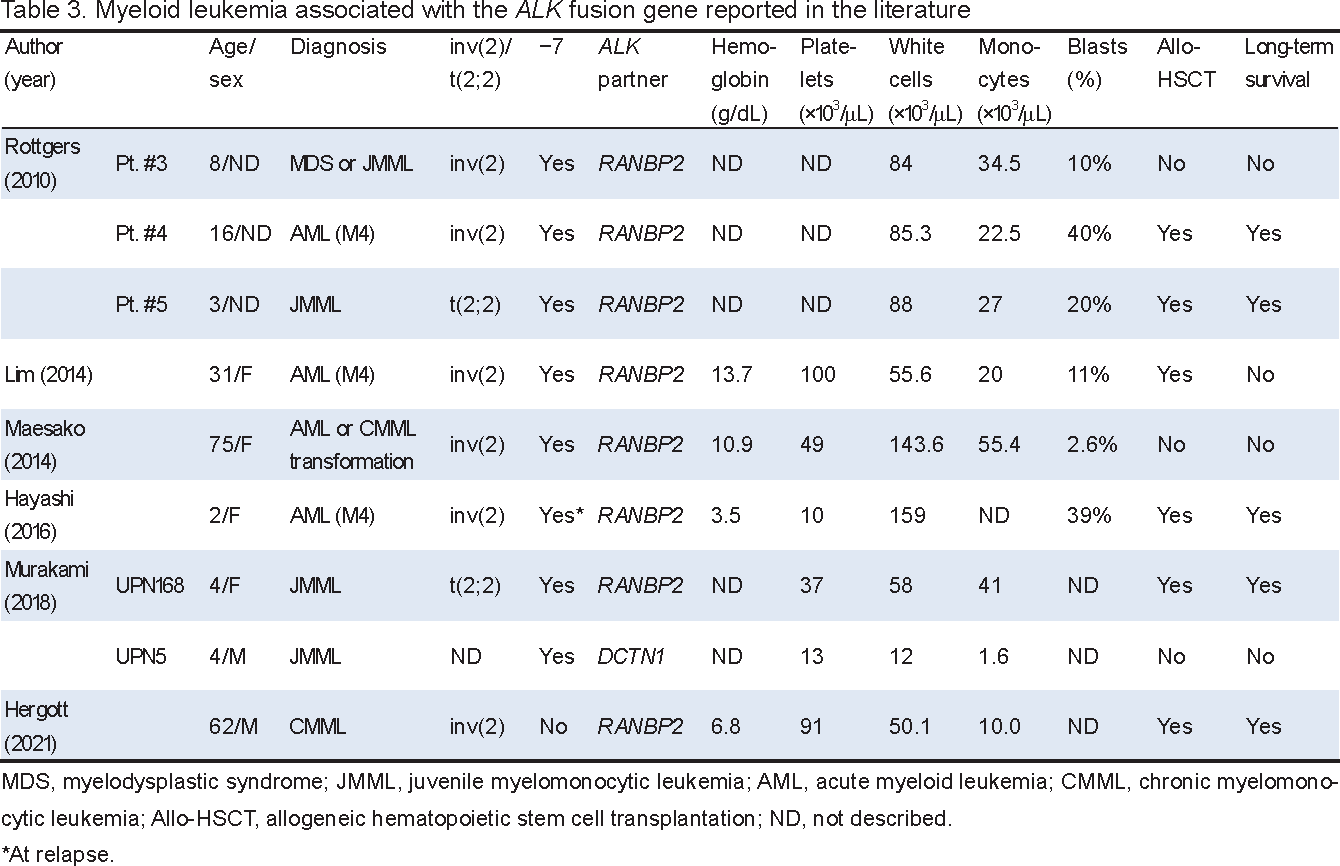
RANBP2, also known as nucleoporin NUP358, is a component of the cytoplasmic face of the nuclear pore complex (NPC) and is a major component of filaments exposed to the cytoplasm.57 RANBP2/NUP358 is a multidomain protein consisting of a large leucine-rich domain, including a leucine zipper motif, in the N terminus, four RAN-binding domains, each of which may interact with RAN-GTP, eight zinc finger motifs, and a C-terminal domain with homology to cyclophilin A.58 The primary structure of the RANBP2::ALK chimeric protein resulting from inv(2)(p23q13) is composed of the leucin-rich domain of RANBP2, which is encoded by the first 18 exons of RANBP2, and the intracellular tyrosine kinase domain of ALK encoded by exon 20 and the downstream exons of ALK.51, 59 Since the leucine-rich domain may be involved in protein-protein interactions and the site of attachment to the NPC core,58 this domain may mediate the heterodimerization of wild-type RANBP2 and chimeric RANBP2::ALK, accounting for the nuclear membrane localization of ALK IHC (Table 1, Figure 5).

We previously described a 75-year-old woman with ALK+ AML.52 The white cell count at presentation was 143.6 × 103/μL with 38.6% monocytes and 13.6% immature granulocytes, including blasts. BM showed >90% cellularity with hyperplasia of myeloid lineage cells, 14.6% monocytes, and 32.1% blasts (Figure 5A, a). The granulocyte series showed a range of dysplastic morphologies. The rate of peroxidase positivity was 51.5% (Figure 5A, b). CD36+ cells with monocytic differentiation comprised 64.6% mononuclear cells. G-banding revealed −7 and a submetacentric marker chromosome derived from chromosome 2, which was identified as inv(2)(p23q13) by FISH using the ALK break apart probe (Figure 5B), and FISH applied to a BM smear slide showed that myeloid precursor cell nuclei carried ALK break apart signals (Figure 5A, c). RANBP2::ALK fusion mRNA was confirmed by RT-PCR and nucleotide sequencing of the products.52 ALK IHC of a BM biopsy specimen demonstrated the nuclear membrane staining of leukemia cells (Figure 5A, d). Therefore, inv(2)(p23q13)/RANBP2::ALK appears to define a small subset of myeloid leukemia characterized by monocytic differentiation and sharing the features of myelodysplastic syndrome/myeloproliferative neoplasm.52
EMP/MM
EMP/MM carrying the ALK fusion gene has been reported in the literature.20, 60-62 Partner genes included CLTC, EML4, TFG, and SEC31A and the generation of the ALK fusion gene with the latter 3 partners was identified by the NGS platform (Table 1). One case harbored t(14;16)(q32;q23) and EML4::ALK, suggesting the secondary occurrence of the latter fusion gene.61A 49-year-old male patient reported by Masood et al. presented with κ-restricted EMP that initially developed in the small intestine, but subsequently disseminated to systemic sites, including the central nervous system (CNS).20 No paraproteins were found. The disease responded to drugs for MM, but relapsed shortly thereafter. Since the cytomorphology and immunophenotype identified by IHC of the reported cases overlapped with those of ALK+ LBCL, it currently remains unclear whether ALK+ EMP/MM is distinct from ALK+ LBCL.20
ALK+ HISTIOCYTOSIS
ALK+ histiocytosis is an emerging rare histiocytic entity harboring the ALK fusion gene,63 and has been listed in the histiocytic and dendritic cell neoplasms category of both the 5th edition of the WHO Classification of Haematolymphoid Tumours focusing on myeloid and histiocytic/dendritic neoplasms and the International Consensus Classification of Mature Lymphoid Neoplasms.64, 65 Involved tissues contain proliferating histiocytes with irregularly folded, deeply clefted, or lobulated nuclei, fine chromatin, and small nucleoli, with some cells being multinucleated.66 Cells express histiocytic (CD68 and CD163) and dendritic cell-associated markers (fascin and factor XIIIa), but lack Langerhans cell markers (CD1a and langerin [CD207]).66, 67 Cells are negative for the clone VE1 IHC against V600E-mutated BRAF,66, 67 whereas in other histiocytic entities, Langerhans histiocytosis and Erdheim-Chester disease, approximately 50% of cases react with the antibody, indicating the presence of a BRAF V600E mutation.68 The expression of phosphorylated ERK suggests the activation of the MAPK pathway.63 Staining for the S-100 protein is variable, being negative in most cases, but patchy to diffuse in a few cases.67
KIF5B is the most frequent partner of the ALK fusion gene, showing the cytoplasmic and cytomembranous staining pattern of ALK IHC;66 however, all staining patterns, except for nuclear staining, were detected in KIF5B::ALK cases in another study.63 Other ALK partners are listed in Table 1. Although the disease was originally described in infants who presented with hepatosplenomegaly and hematopoietic involvement,66subsequent studies showed that ALK+ histiocytosis occurred not only in pediatric patients, but also in adults, and exhibited a spectrum of clinical features, ranging from multisystem disease to single-system disease and the CNS was often involved.63, 66, 67
TARGET THERAPIES FOR ALK+ HEMATOLOGICAL NEOPLASMS
Brentuximab vedotin for ALK+ ALCL
Brentuximab vedotin (BV) is an antibody-drug conjugate consisting of the chimeric monoclonal antibody brentuximab targeting cell-surface CD30 and the antimitotic agent monomethyl auristatin E (MMAE) through a cathepsin-cleavable linker.17, 24 Once BV binds to cell-surface CD30, the conjugate is internalized by endocytosis and MMAE is released within lysosomes, thereby exerting anti-tumor effects.17, 24 BV was approved in 2019 as a first-line treatment for Hodgkin lymphoma and CD30+ PTCL, including ALK+ ALCL, in combination with cytotoxic drugs.69
Six cycles of 1.8 mg/kg BV in the combination of cyclophosphamide, doxorubicin, and prednisolone (CHP) every 3 weeks are currently recommended as the first-line treatment for ALK+ ALCL. This is based on the superior progression-free survival (PFS) and OS without added toxicity among 452 patients with CD30+ lymphomas in the multicenter, double-blind, ECHELON-2 trial, which included 98 (22%) patients with ALK+ ALCL who were randomly assigned to BV-CHP (49 patients) or CHOP (49 patients).70, 71 Although this trial was not powered to compare efficacy between individual histological subtypes, PFS (hazard ratio [HR], 0.29; 95% confidential interval [CI], 0.11–0.79) and OS (HR, 0.38; 95% CI, 0.12–1.22) advantages for BV-CHP over CHOP were observed for ALK+ ALCL.70 In the 5-year findings of the trial, the PFS HR for BV-CHP over CHOP was 0.40 (CI, 0.17–0.98) and the OS HR was 0.48 (CI, 0.16–1.40) in the ALK+ ALCL subset.72
On the other hand, in a phase II multicenter trial, 58 patients with relapsed/refractory (R/R) systemic ALCL, including 16 with ALK+ ALCL, were treated with single-agent BV (1.8 mg/kg every 3 weeks for up to 16 doses).73 The overall response rate was 86%, including 57% (33 patients) with a complete response (CR), and the median response duration was 12.6 months with a median PFS of 13.3 months and 1-year OS rate of 70%.73 After a more than 5-year follow-up of 38 patients who achieved a best response of CR, 16, including 5 with ALK+ ALCL, remained in remission.74 Single-agent BV was also effective for relapsed cases after BV-CHP.72In a Japanese post-marketing surveillance study, 101 R/R systemic ALCL patients were registered between April and September, 2014, 69 (68.3%) of whom had been treated with CHOP and 16 (15.8%) had undergone autologous and/or allogeneic hematopoietic stem cell transplantation (allo-HSCT).75 These patients were treated with between one and 17 cycles of BV and 49 (62.0%) out of 79 evaluable patients showed an objective response. The most common adverse events were peripheral sensory neuropathy (any grade, 36.6%; grade ≥3, 3.0%) and neutropenia (any grade, 33.7%; grade ≥3, 20.8%). Grade ≥3 pulmonary toxicities occurred in 3 patients.75
In our series of ALK+ ALCL, one patient was initially treated with BV-CHP and one with single-agent BV (Table 2). Case 7 (a 38-year-old male) presented with marked neoplastic involvement of the rectus abdominis, iliopsoas, quadriceps, and abductor muscles of the right hemi-trunk and skin (Figure 3). After the first dose of BV-CHP, the hemodynamic and respiratory status deteriorated. Since the second dose of BV-CHP was considered infeasible, single-agent BV every 3 weeks was substituted for BV-CHP. When the patient recovered sufficiently to receive multiagent chemotherapy, we resumed BV-CHP and completed 5 additional cycles. 18F-fluorodeoxyglucose positron-emission tomography combined with computed tomography (18F-FDG-PET/CT) performed after the completion of treatment confirmed a complete metabolic response and the patient is currently free from relapse for 26 months. Figure 6 shows the course of case 6 (an 82-year-old male patient). Since he had long-lasting cytopenia due to BM failure syndrome and cytotoxic drugs were considered to be unfeasible, we treated the patient with single-agent BV. After the first dose of BV, fever and hypoxemia improved, and pulmonary infiltrates resolved accordingly. The patient was moved to the outpatient clinic and received 12 doses of BV every 3 to 4 weeks. He has remained free from relapse for 30 months, even though he has grade 3 neurotoxicity. Based on the courses of these two patients, we suggest that since BV is very active against ALK+ ALCL, single-agent BV or prophase use of the drug to reduce the tumor burden needs to be considered for elderly patients or those who are initially not candidates for multiagent chemotherapy because of significant comorbidities and poor PS.76

Although ALK tyrosine kinase inhibitors (TKIs) have transformed the therapeutic landscape of NSCLC harboring ALK rearrangements,13, 14 the antitumor activity of the prototype small-molecule ALK TKI, crizotinib, was initially demonstrated by an experimental model of ALK+ ALCL.77 This study showed that crizotinib markedly inhibited the proliferation of Karpas 299 and SU-DHL-1 ALK+ ALCL cells with t(2;5)(p23;q35) in vitro and induced the complete regression of Karpas 299 xenotransplant tumors in vivo. These antitumor effects were associated with the potent inhibition of the downstream signal transduction of NPM::ALK and induction of apoptosis. Similar findings were obtained in a model of the ALK+ LBCL cell line, LM1, carrying the CLTC::ALK fusion gene,78 indicating that ALK inhibitors are effective for tumors with ALK fusion genes irrespective of the type of tumor or partner gene.
The clinical activity of crizotinib was initially demonstrated in two young adult patients with relapsed ALK+ ALCL (Table 4).79 After the initiation of crizotinib therapy, their constitutional symptoms readily resolved and both patients achieved CR. The effects of crizotinib for ALK+ hematological tumors was confirmed in a phase 1 trial by the Children’s Oncology Group and a compassionate use study by an Italian group, in which 8 out of 9 pediatric patients with R/R ALK+ ALCL and 10 out of 11 adult patients with ALK+ lymphoma, respectively, showed an objective tumor response (Table 4).80, 81 We previously described an elderly patient with RANBP2::ALK AML who relapsed shortly after daunorubicin/cytarabine, was refractory to azacytidine, and was treated with crizotinib.52, 82 After the administration of crizotinib, circulating immature myelomonocytic cells and blasts readily disappeared and, after 3 months of crizotinib therapy, BM was composed of normal erythroid/myeloid precursors and megakaryocytes with <5% blasts, achieving the level of hematological CR.82 On the other hand, two Japanese children with RANBP2::ALK AML achieved second CR by crizotinib and underwent allo-HSCT,53, 54 suggesting that the crizotinib-containing treatment provides a potential bridge to allo-HSCT. A cavernous sinus tumor of ALK+ histiocytosis that developed in a 15-year-old male was reported to be resolved by crizotinib.66

Treatment with crizotinib is limited by the development of drug resistance due to secondary mutations within the ALK protein kinase domain, re-inducing kinase activation and signaling despite the presence of crizotinib.4, 83 These resistance mutations may directly hinder crizotinib binding to the ALK kinase, alter the conformation of the kinase, and/or alter the ATP-binding affinity of the kinase. The most frequent post-crizotinib mutation is the L1196M mutation that alters the gatekeeper residue at the bottom of the ATP-binding pocket and impairs crizotinib binding.4 The second most frequent G1269A mutation, which was identified in a case of crizotinib-treated AML (Figure 5C),83 also lies in the ATP-binding pocket, thereby hindering crizotinib binding.4
Following the first-generation ALK TKI, crizotinib, second-generation alectinib, ceritinib, and brigatinib, and third-generation lorlatinib have been approved for the treatment of NSCLC, and alectinib has been approved for ALK+ ALCL in the R/R setting in Japan.84 These second/third-generation agents bind to ALK kinase with higher affinities than crizotinib and may show efficacy in crizotinib-resistant tumors. The effective treatment of ALK+ hematological tumors with second/third-generation ALK TKIs has been reported in the literature, albeit in a small number of cases (Table 4).63 In the study that led to the approval of alectinib for R/R ALK+ ALCL, 10 patients were enrolled, and objective responses were documented in 8 (80%) out of 10 patients, including 6 CR, with favorable toxicity profiles, and 1-year PFS and OS rates were 58.3 and 70.0%, respectively.85 R/R ALK+ ALCL with CNS involvement was effectively treated with second/third-generation ALK TKIs, which were designed to cross the blood-brain barrier more effectively than crizotinib, thereby achieving a higher concentration in the cerebrospinal fluid (Table 4).86 Nevertheless, second/third-generation ALK TKIs may induce a distinct spectrum of resistance mutations that are rare in post-crizotinib tumors.4
Off-target resistance mechanisms, such as the activation of bypass signaling pathways, are found in ALK TKI-treated NSCLC. Therefore, the aim of targeting therapy is not only to effectively treat the relapsed disease by overcoming the dominant resistance mechanism, but also to enhance the depth and duration of the tumor response up front by preventing the emergence of resistance.4 To achieve this, different therapeutic strategies of sequential or combined ALK TKIs or the combination of ALK TKIs with chemotherapy, immunotherapy, or other target agents are being tested.13
ACKNOWLEDGMENTS
This study was supported by the Tenri Foundation.
ETHICAL APPROVAL
The present study was performed according to the regulations of the Institutional Review Board (Approval No. 1324).
Reference List
- 1. Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 2007;7:233-245.
- 2. Werner MT, Zhao C, Zhang Q, et al. Nucleophosmin-anaplastic lymphoma kinase: The ultimate oncogene and therapeutic target. Blood 2017;129:823-831.
- 3. Boi M, Zucca E, Inghirami G, et al. Advances in understanding the pathogenesis of systemic anaplastic large cell lymphomas. Br J Haematol 2015;168:771-783
- 4. Lin JJ, Riely GJ, Shaw AT. Targeting ALK: Precision medicine takes on drug resistance. Cancer Discov 2017;7:137-155
- 5. Fischer P, Nacheva E, Mason DY, et al. A Ki-1 (CD30)-positive human cell line (Karpas 299) established from a high-grade non-Hodgkin's lymphoma, showing a 2;5 translocation and rearrangement of the T-cell receptor β-chain gene. Blood 1988;72:234-240
- 6. Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 1994;263:1281-1284
- 7. Chiarle R, Voena C, Ambrogio C, et al. The anaplastic lymphoma kinase in the pathogenesis of cancer.Nat Rev Cancer 2008;8:11-23
- 8. Barreca A, Lasorsa E, Riera L, et al. Anaplastic lymphoma kinase in human cancer. J Mol Endocrinol 2011;47:R11-23
- 9. Leventaki V, Bhattacharyya S, Lim MS. Pathology and genetics of anaplastic large cell lymphoma. Semin Diagn Pathol 2020;37:57-71
- 10. Shiota M, Fujimoto J, Takenaga M, et al. Diagnosis of t(2;5)(p23;q35)-associated Ki-1 lymphoma with immunohistochemistry. Blood 1994;84:3648-3652
- 11. Falini B, Nicoletti I, Bolli N, et al. Translocations and mutations involving the nucleophosmin (NPM1) gene in lymphomas and leukemias.Haematologica 2007;92:519-532
- 12. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561-566
- 13. Cognigni V, Pecci F, Lupi A, et al. The landscape of ALK-rearranged non-small cell lung cancer: A comprehensive review of clinicopathologic, genomic characteristics, and therapeutic perspectives. Cancers (Basel) 2022;14:4765
- 14. Shaw AT, Solomon B. Targeting anaplastic lymphoma kinase in lung cancer. Clin Cancer Res 2011;17:2081-2086
- 15. Du X, Shao Y, Qin HF, et al. ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac Cancer 2018;9:423-430.
- 16. Falini B, Stein H, Lamant-Rochaix L, et al. Anaplastic large cell lymphoma, ALK-positive. In: Swerdlow SH, Campo E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th edition). Lyon: IARC; 2017:413-418
- 17. Tsuyama N, Sakamoto K, Sakata S, et al. Anaplastic large cell lymphoma: Pathology, genetics, and clinical aspects. J Clin Exp Hematop 2017;57:120-142
- 18. Lin C, Shi X, Yang S, et al. Comparison of ALK detection by FISH, IHC and NGS to predict benefit from crizotinib in advanced non-small-cell lung cancer. Lung Cancer 2019;131:62-68
- 19. Fukuhara S, Oshikawa-Kumade Y, Kogure Y, et al. Feasibility and clinical utility of comprehensive genomic profiling of hematological malignancies. Cancer Sci 2022;113:2763-2777
- 20. Masood A, Christ T, Asif S, et al. Non-secretory multiple myeloma with unusual TFG-ALK fusion showed dramatic response to ALK inhibition. NPJ Genom Med 2021;6:23
- 21. Grewal JS, Smith LB, Winegarden JD, 3rd, et al. Highly aggressive ALK-positive anaplastic large cell lymphoma with a leukemic phase and multi-organ involvement: a report of three cases and a review of the literature. Ann Hematol 2007;86:499-508
- 22. Sibon D, Nguyen DP, Schmitz N, et al. ALK-positive anaplastic large-cell lymphoma in adults: an individual patient data pooled analysis of 263 patients. Haematologica2019;104:e562-e565
- 23. Freedman AS, Aster JC. Clinical manifestations, pathologic features, and diagosis of systemic anaplastic large cell lymphoma. In: Lister A, ed. UpToDate (accessed on October 29, 2021).
- 24. Hapgood G, Savage KJ. The biology and management of systemic anaplastic large cell lymphoma. Blood 2015;126:17-25
- 25. Kuefer MU, Look AT, Pulford K, et al. Retrovirus-mediated gene transfer of NPM-ALK causes lymphoid malignancy in mice. Blood 1997;90:2901-2910
- 26. Chiarle R, Gong JZ, Guasparri I, et al. NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood 2003;101:1919-1927
- 27. Zhang Q, Wei F, Wang HY, et al. The potent oncogene NPM-ALK mediates malignant transformation of normal human CD4+ T lymphocytes. Am J Pathol 2013;183:1971-1980
- 28. van der Krogt JA, Bempt MV, Ferreiro JF, et al. Anaplastic lymphoma kinase-positive anaplastic large cell lymphoma with the variant RNF213-, ATIC- and TPM3-ALK fusions is characterized by copy number gain of the rearranged ALK gene. Haematologica 2017;102:1605-1616.
- 29. Nishikori M, Maesako Y, Ueda C, et al. High-level expression of BCL3 differentiates t(2;5)(p23;q35)-positive anaplastic large cell lymphoma from Hodgkin disease. Blood 2003;101:2789-2796
- 30. Ohno H, Takimoto G, McKeithan TW. The candidate proto-oncogene bcl-3 is related to genes implicated in cell lineage determination and cell cycle control. Cell 1990;60:991-997
- 31. Nishikori M, Ohno H, Haga H, et al. Stimulation of CD30 in anaplastic large cell lymphoma leads to production of nuclear factor-kB p52, which is associated with hyperphosphorylated Bcl-3. Cancer Sci 2005;96:487-497.
- 32. Schmitz N, Trümper L, Ziepert M, et al. Treatment and prognosis of mature T-cell and NK-cell lymphoma: an analysis of patients with T-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin Lymphoma Study Group. Blood 2010;116:3418-3425.
- 33. Savage KJ, Harris NL, Vose JM, et al. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood 2008;111:5496-5504.
- 34. Sibon D, Fournier M, Briere J, et al. Long-term outcome of adults with systemic anaplastic large-cell lymphoma treated within the Groupe d'Etude des Lymphomes de l'Adulte trials. J Clin Oncol 2012;30:3939-3946
- 35. Suzuki R, Kagami Y, Takeuchi K, et al. Prognostic significance of CD56 expression for ALK-positive and ALK-negative anaplastic large-cell lymphoma of T/null cell phenotype. Blood 2000;96:2993-3000
- 36. Takeoka K, Okumura A, Honjo G, et al. Variant translocation partners of the anaplastic lymphoma kinase (ALK) gene in two cases of anaplastic large cell lymphoma, identified by inverse cDNA polymerase chain reaction. J Clin Exp Hematop 2014;54:225-235
- 37. Okamori S, Maesako Y, Ehara J, et al. An elderly man with anaplastic large cell lymphoma carrying inv(2)(p23q35) who presented with cardiopulmonary failure: Clinical, pathological, and cytogenetic features and postmortem findings. Tenri Med Bull 2013;16:89-100
- 38. Delsol G, Lamant L, Mariame B, et al. A new subtype of large B-cell lymphoma expressing the ALK kinase and lacking the 2;5 translocation. Blood 1997;89:1483-1490
- 39. Laurent C, Do C, Gascoyne RD, et al. Anaplastic lymphoma kinase-positive diffuse large B-cell lymphoma: A rare clinicopathologic entity with poor prognosis. J Clin Oncol 2009;27:4211-4216
- 40. Campo E, Gascoyne RD. ALK-positive large B-cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th edition). Lyon: IARC; 2017:319-320
- 41.
Pan Z, Hu S, Li M, et al. ALK-positive large B-cell lymphoma: A clinicopathologic study of 26 cases with review of additional 108 cases in the literature. Am J Surg Pathol 2017;41:25-38
- 42. Jiang XN, Yu BH, Wang WG, et al. Anaplastic lymphoma kinase-positive large B-cell lymphoma: Clinico-pathological study of 17 cases with review of literature. PLoS One 2017;12:e0178416
- 43. Delsol G, Jaffe ES, Falini B, et al. Anaplastic large cell lymphoma (ALCL), ALK-positive. In: Swerdlow SH, Campo E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008:312-316
- 44. Ohno H, Takeoka K, Kishimori C, et al. ALK-positive large B-cell lymphoma showing long-term response to conventional chemoradiotherapy. Tenri Medical Bulletin 2021;24:100-107
- 45. Valera A, Colomo L, Martinez A, et al. ALK-positive large B-cell lymphomas express a terminal B-cell differentiation program and activated STAT3 but lack MYC rearrangements. Mod Pathol 2013;26:1329-1337
- 46. Gascoyne RD, Lamant L, Martin-Subero JI, et al. ALK-positive diffuse large B-cell lymphoma is associated with Clathrin-ALK rearrangements: report of 6 cases. Blood 2003;102:2568-2573.
- 47. De Paepe P, Baens M, van Krieken H, et al. ALK activation by the CLTC-ALK fusion is a recurrent event in large B-cell lymphoma. Blood 2003;102:2638-2641
- 48. Takeuchi K, Soda M, Togashi Y, et al. Identification of a novel fusion, SQSTM1-ALK, in ALK-positive large B-cell lymphoma. Haematologica 2011;96:464-467
- 49. Sakamoto K, Nakasone H, Togashi Y, et al. ALK-positive large B-cell lymphoma: identification of EML4-ALK and a review of the literature focusing on the ALK immunohistochemical staining pattern. Int J Hematol 2016;103:399-408
- 50. Argani P, Lui MY, Couturier J, et al. A novel CLTC-TFE3 gene fusion in pediatric renal adenocarcinoma with t(X;17)(p11.2;q23). Oncogene 2003;22:5374-5378
- 51.
Röttgers S, Gombert M, Teigler-Schlegel A, et al. ALK fusion genes in children with atypical myeloproliferative leukemia. Leukemia 2010;24:1197-1200
- 52.
Maesako Y, Izumi K, Okamori S, et al. inv(2)(p23q13)/RAN-binding protein 2 (RANBP2)-ALK fusion gene in myeloid leukemia that developed in an elderly woman. Int J Hematol 2014;99:202-207
- 53.
Hayashi A,
Tanoshima R, Tsujimoto SI, et al. Crizotinib treatment for refractory pediatric acute myeloid leukemia with RAN-binding protein 2-anaplastic lymphoma kinase fusion gene. Blood Cancer J 2016;6:e456
- 54. Murakami N, Okuno Y, Yoshida K, et al. Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 2018;131:1576-1586
- 55. Lim JH, Jang S, Park CJ, et al.RANBP2-ALK fusion combined with monosomy 7 in acute myelomonocytic leukemia. Cancer Genet 2014;207:40-45
- 56. Hergott CB, Dal Cin P, Hornick JL, et al. Characteristic nuclear membrane ALK reactivity in chronic myelomonocytic leukemia with RANBP2-ALK fusion. Am J Hematol 2021.
- 57. Bernad R, van der Velde H, Fornerod M, et al. Nup358/RanBP2 attaches to the nuclear pore complex via association with Nup88 and Nup214/CAN and plays a supporting role in CRM1-mediated nuclear protein export. Mol Cell Biol 2004;24:2373-2384
- 58. Avis JM, Clarke PR. Ran, a GTPase involved in nuclear processes: its regulators and effectors. J Cell Sci 1996;109:2423-2427
- 59. Ma Z, Hill DA, Collins MH, et al. Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosomes Cancer 2003;37:98-105
- 60. Wang WY, Gu L, Liu WP, et al. ALK-positive extramedullary plasmacytoma with expression of the CLTC-ALK fusion transcript. Pathol Res Pract 2011;207:587-591
- 61. Morgan GJ, He J, Tytarenko R, et al. Kinase domain activation through gene rearrangement in multiple myeloma. Leukemia
2018;32:2435-2444
- 62. Subbiah V, Kuravi S, Ganguly S, et al. Precision therapy with anaplastic lymphoma kinase inhibitor ceritinib in ALK-rearranged anaplastic large cell lymphoma. ESMO Open 2021;6:100172
- 63. Kemps PG, Picarsic J, Durham BH, et al. ALK-positive histiocytosis: A new clinicopathologic spectrum highlighting neurologic involvement and responses to ALK inhibition. Blood 2022;139:256-280
- 64. Campo E, Jaffe ES, Cook JR, et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee. Blood 2022;140:1229-1253.
- 65. Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022;36:1703-1719.
- 66. Chang KTE, Tay AZE, Kuick CH, et al. ALK-positive histiocytosis: An expanded clinicopathologic spectrum and frequent presence of KIF5B-ALK fusion. Mod Pathol 2019;32:598-608.
- 67. Qiu L, Weitzman SP, Nastoupil LJ, et al. Disseminated ALK-positive histiocytosis with KIF5B-ALK fusion in an adult. Leuk Lymphoma 2021;62:1234-1238.
- 68. Rech KL, He R. Challenges in the histopathologic diagnosis of histiocytic neoplasms. J Natl Compr Canc Netw 2021;19:1305-1311.
- 69. 武田薬品工業. アドセトリス点滴静注用50mg. (https://www.info.pmda.go.jp/go/pack/4291425D1021_1_12/). Accessed 2022 December 9.
- 70. Horwitz S, O'Connor OA, Pro B, et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): A global, double-blind, randomised, phase 3 trial. Lancet 2019;393:229-240
- 71. Jacobsen E. Treatment of systemic anaplastic large cell lymphoma. In: Freedman AS, ed. UpToDate (acccessed on October 29, 2021).
- 72. Horwitz S, O'Connor OA, Pro B, et al. The ECHELON-2 Trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann Oncol 2022;33:288-298
- 73. Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J Clin Oncol 2012;30:2190-2196.
- 74. Pro B, Advani R, Brice P, et al. Five-year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood 2017;130:2709-2717
- 75. Izutsu K, Ogura M, Tobinai K, et al. Safety profile of brentuximab vedotin in Japanese patients with relapsed/refractory Hodgkin lymphoma or systemic anaplastic large cell lymphoma: a post-marketing surveillance study. Int J Hematol 2021;113:404-412
- 76. Fanale MA, Horwitz SM, Forero-Torres A, et al. Brentuximab vedotin in the front-line treatment of patients with CD30+ peripheral T-cell lymphomas: Results of a phase I study. J Clin Oncol 2014;32:3137-3143
- 77. Christensen JG, Zou HY, Arango ME, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther 2007;6:3314-3322
- 78. Cerchietti L, Damm-Welk C, Vater I, et al. Inhibition of anaplastic lymphoma kinase (ALK) activity provides a therapeutic approach for CLTC-ALK-positive human diffuse large B cell lymphomas. PLoS One 2011;6:e18436
- 79. Gambacorti-Passerini C, Messa C, Pogliani EM. Crizotinib in anaplastic large-cell lymphoma. N Engl J Med 2011;364:775-776
- 80.
Mosse YP, Lim MS, Voss SD, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol 2013;14:472-480
- 81. Gambacorti Passerini C, Farina F, Stasia A, et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J Natl Cancer Inst 2014;106:djt378
- 82.
Maesako Y, Okumura A, Takeoka K, et al. Reduction of leukemia cell burden and restoration of normal hematopoiesis at 3 months of crizotinib treatment in RAN-binding protein 2 (RANBP2)-anaplastic lymphoma kinase (ALK) acute myeloid leukemia. Leukemia 2014;28:1935-1937
- 83.
Takeoka K, Okumura A, Maesako Y, et al. Crizotinib resistance in acute myeloid leukemia with inv(2)(p23q13)/RAN binding protein 2 (RANBP2) anaplastic lymphoma kinase (ALK) fusion and monosomy 7. Cancer Genet 2015;208:85-90
- 84.
中外製薬. アレセンサカプセル150mg. (https://www.info.pmda.go.jp/go/pack/4291032M3021_1_06/). Accessed 2022 December 9.
- 85.
Fukano R, Mori T, Sekimizu M, et al. Alectinib for relapsed or refractory anaplastic lymphoma kinase-positive anaplastic large cell lymphoma: An open-label phase II trial. Cancer Sci 2020;111:4540-4547
- 86.
Rigaud C, Abbou S, Ducassou S, et al. Profound and sustained response with next-generation ALK inhibitors in patients with relapsed or progressive ALK-positive anaplastic large cell lymphoma with central nervous system involvement. Haematologica 2022;107:2255-2260

