Highlights
The role of messenger (m) RNA vaccines in the outbreak of severe acute respiratory syndrome-related coronavirus
(SARS-CoV-2) has been very significant. It is necessary to consider whether there are any mRNA-specific aspects
to be taken into account regarding the vaccines’ efficacy and safety, as well as the design and quality control of
mRNA, with an eye toward the development of mRNA vaccines for infectious diseases other than SARS-CoV-2.
This review discusses regulatory requirements for assessing the quality, efficacy, and safety of developing mRNA
vaccines.
Introduction
The role of messenger (m) RNA vaccines in the 2020 outbreak of severe acute respiratory
syndrome-related coronavirus (SARS-CoV-2) has been significant. mRNA vaccines were the
earliest SARS-CoV-2 vaccines to be approved, and their efficacy in preventing the onset of
coronavirus disease (COVID)-19 has exceeded 90% [1,
2], with high efficacy in preventing severe disease
[2, 3]. The
mRNA vaccines have been commercialized to tackle the COVID-19 pandemic, although this
modality has not been used for other vaccines.
The high efficacy of mRNA vaccines is due not only to the high antibody titer induced by
the vaccines but also to the high-level induction of both humoral and cellular immunity
[4] by the expression of the antigen in cells.
Although this has not been established, it has been suggested that innate immune activation
provided by mRNA vaccines may also contribute to their high efficacy [5], as depicted in Fig. 1 The
application of mRNA vaccines to other infectious diseases, such as seasonal influenza, has
been attempted; however, the immune responses to these vaccines have not always been as
drastic as those to SARS-CoV-2, and the vaccines have not shown superiority over
conventional hemagglutinin split vaccines at this stage [6].

Evaluations of the quality, efficacy, and safety of mRNA vaccines have had to be conducted
in the absence of sufficient knowledge and accumulated experience; therefore, it is
necessary to consider the principles of these evaluations with a view toward the future
development of vaccines for other infectious diseases. In particular, it is necessary to
consider whether there are any mRNA-specific aspects to be taken into account regarding the
vaccines’ efficacy and safety, as well as the design and quality control of mRNA, with an
eye toward the development of mRNA vaccines for infectious diseases other than SARS-CoV-2.
mRNA products that have proliferative properties in administered cells have also been
developed [7, 8]. This review discusses regulatory requirements for assessing the quality,
efficacy, and safety of the development of mRNA vaccines. However, it is assumed that the
concept of non-clinical and clinical studies in vaccine development should be based on
relevant guidelines.
Development and Design of mRNA Vaccines
The design of mRNA vaccines for viral infections is based on the mechanism of infection of
the target virus within cells. In most designs, sequences encoding viral surface proteins
that bind to cell receptors are selected for viral infection. However, it is generally known
that (i) through viral infection, multiple cellular proteins are often
involved in viral binding to the cell for the introduction of the viral genome into the
cell, and (ii) multiple viral proteins are involved in the response to the
infection process and are thus involved in the expression of molecules involved in the
infection process other than viral proteins that bind to cell receptors. Therefore, in the
design of vaccines, it is very important to determine the types of proteins that are
expressed other than the viral proteins that bind to cell receptors and are involved in the
infection process [9, 10]. In addition, the form of the expressed target viral antigen, that is, as a
secretory protein, membrane protein, or particle formation, is closely related to the mRNA
vaccine development strategy. When the application for the vaccine’s approval by a
pharmaceutical regulatory is being processed, it will also be necessary to explain the
product design, including the reason (s) for selecting the protein to be expressed and its
validity from the viewpoint of preventing the target infectious disease.
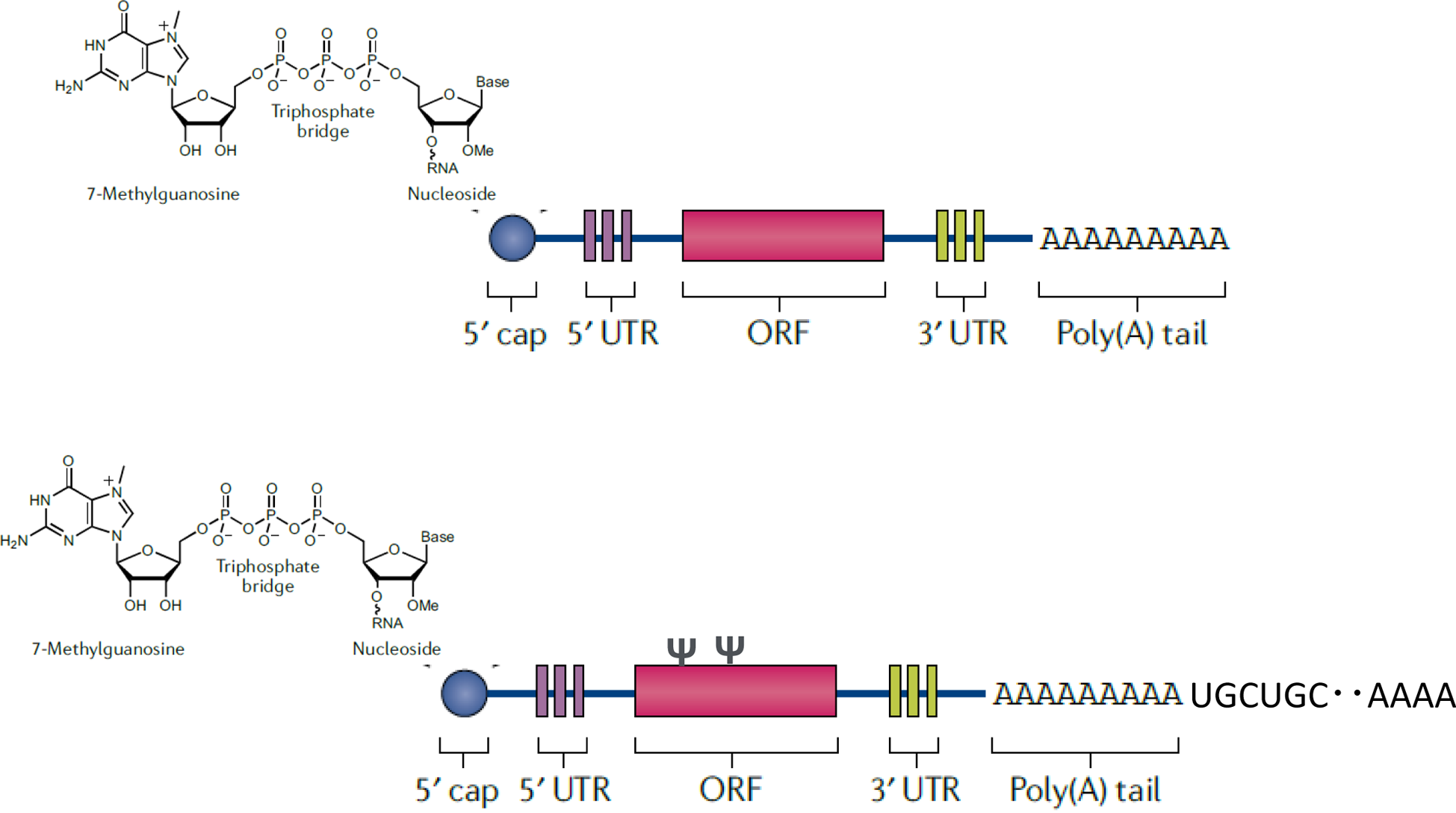
The basic structure of a non-proliferative mRNA vaccine consists of a 5′-Cap, a 5′
untranslated region upstream of the target gene, an open reading frame that encodes the
antigen, a 3′ untranslated region downstream of the target gene, and a poly (A) tail (Fig. 2). The nucleosides that make up mRNA activate
innate immunity [11,12,13]. Using modified nucleosides (such as
pseudouridine) instead of unmodified nucleosides to suppress the activation of innate
immunity is an important factor in mRNA designs for a better risk-benefit balance [14, 15]. In
addition, double-stranded (ds) RNA and the conformation of the product can activate innate
immunity. For example, dsRNA is known as a promoter of type I interferon production through
the TLR3, RIG-I, and MDA5 pathways [16].
Increasing G:C mRNA content has been reported to contribute to mRNA
stabilization during protein translation and expression both in vitro and
in vivo [17, 18]. Using more frequent codons in human cells may also lead to an
increased expression of target proteins. How mRNA sequences are designed may have a
significant impact on the persistence of antigen expression and induction of immunity.
Conversely, modifications of an mRNA sequence can lead to unexpected structural changes that
may significantly affect sequence translation [19].
The design used for constructing mRNA and its manufacturing process to achieve efficient
production also provides important information for the quality control of mRNA vaccines. For
example, it is important to develop appropriate control strategies for a drug substance and
product. This strategy includes the preparation of template DNA used in the in
vitro transcription process or the purification of mRNA from the raw materials
used in the transcription process. In addition, if an mRNA sequence or nucleoside is
modified during the development phase, evaluating how antigen expression is affected by such
changes is useful to justify the appropriateness of those modifications.
The design of mRNA constructs and the manufacturing process for efficient production of
mRNA are very important for understanding the properties and quality attributes of mRNA
vaccines used as pharmaceuticals.
A method for self-amplifying mRNA has been developed for the manufacture of
pharmaceuticals. This method amplifies the expression of target mRNA in human cells by
inserting an alpha virus RNA-dependent RNA polymerase genome into the region upstream of the
target antigen in the mRNA sequence [20]. When this
self-amplifying RNA system is used, the innate immune response is expected to be triggered
by the generated RNA, which may induce a strong adjuvant effect of the vaccine.
Development of mRNA Vaccine Manufacturing Processes
When producing mRNA vaccines, the in vitro transcription reaction and, in
some cases, the chemical addition reaction (i.e., adding a 5′-Cap structure or poly (A)) are
combined based on the design of the target mRNA. Since the composition of impurities differs
depending on how these reactions are used, it is important to evaluate the quality of mRNA
while considering the manufacturing process. mRNA production begins with in
vitro transcription. The raw materials used in the manufacturing process, such as
the template DNA, RNA polymerase, substrate nucleic acids, enzyme reaction solution,
5′-Capping enzymes and their substrate, and poly (A) polymerase, should be sufficiently
removed after the reaction. Thus, it is important to determine the residual amounts of these
impurities during the manufacturing of the final products.
The poly (A) sequence plays an important role in mRNA translation and intracellular
stability [21], and its chain length must be ≥100 mer
[10]. To add the optimal length of poly (A) [25] to mRNA, it is either transcribed directly from a DNA
template encoding the antigen and a poly (A) or added poly (A) sequence using poly (A)
polymerase after the transcription reaction to the end of the mRNA sequence. It is also
known that expressing a long poly (A) in a plasmid causes instability; therefore, UGC
sequences were inserted into the poly (A) chain to increase its stability (Fig. 2) [26,
27].
One of the most important processes in mRNA vaccine manufacturing is the production of the
template for the in vitro transcription, which is obtained using
Escherichia coli plasmids. Generally, purified plasmids produced in
E. coli are linearized and used as templates for in
vitro transcription. In some cases, one or more plasmids (which are also combined
with another plasmid) or target sequences amplified by polymerase chain reaction (PCR) are
used to manufacture templates. The mRNA is transcribed from these templates in an in
vitro transcription reaction using RNA polymerase. Because the plasmids are
amplified by E. coli, it is important to control impurities derived from
the host cells of E. coli (such as proteins and DNA) in the production of
templates during the extraction and purification processes. If the templates are
contaminated with fragmented DNA from the plasmid or the circular plasmid, incomplete mRNA
transcription may be contaminated in the final products. Thus, it is important to evaluate
incomplete mRNA transcription as potential product-related impurities.
The template used for the in vitro transcription reaction can be regarded
as the starting material for mRNA production. However, its quality attributes, including
purity, may affect the quality of the mRNA produced, and quality control is very important
for the production of mRNA vaccines of consistent quality. Thus, strict acceptance
specifications and appropriate process control for the plasmids, which are the raw materials
for templates, are required. It is also necessary to consider the possibility that the
amount of each nucleoside triphosphate to be added may be a critical process parameter in
the in vitro transcription reactions [29].
During transcription, mRNA is synthesized by a DNA-dependent RNA polymerase, such as T7,
SP6, or T3, using ATP, GTP, CTP, UTP, or their modified nucleoside triphosphates as
substrates. It is important for the poly (A) sequences of mRNA to have sufficient lengths,
but it is a challenge to control the strand length of poly (A) in the mRNA manufacturing
process; thus, it is important to evaluate the heterogeneity of poly (A) lengths for the
quality control of mRNA. In the transcription process, a 5′-Cap structure [21] is added to the 5′ end of the mRNA, and this addition
is important for the stability of mRNA, providing a shield to the 5′ end of the mRNA and
resistance to exonucleases. For this purpose, the 5′-Cap can be added by various methods,
such as the addition of a vaccinia virus-derived capping enzyme [22] during the transcription reaction, the addition of a synthetic Cap
after the transcription reaction, or the incorporation of an anti-reverse Cap analog [23, 24]. In the
process of these reactions, the products may contain incomplete mRNA to which Cap is not
added, and the efficiency of the reaction to the addition of a 5′-Cap is one of the key
issues to be checked when the quality of mRNA vaccines is evaluated.
After in vitro transcription, the target mRNA is purified. mRNA, as a drug
substance, is first treated by (i) degradation of the DNA used as the
template, (ii) removal of impurities using tangential flow filtration or
other purification processes, and (iii) concentration of the mRNA. Purified
drug substance (DS) must be prepared without RNases to ensure mRNA stability.
It is also expected that the DNA used as a template is degraded by DNase after the
transcription reaction and fully removed by the mRNA purification process. The World Health
Organization (WHO) has established the criteria for acceptable amounts of host cell-derived
DNA in conventional vaccines, which are classified into two groups: cancer cell-derived DNA
and non-cancer cell-derived DNA. This is based on the assumption that there is a certain
degree of cellular DNA uptake following local vaccine administration [28]. These criteria also mention the length of the cell-derived DNA. The
mRNA in mRNA vaccines is usually encapsulated in lipid nanoparticles (LNPs), and formulation
technology is used to ensure that the mRNA is actively taken up by the cells. Therefore,
when mRNA vaccines are administered, DNA contaminating the target mRNA as an impurity is
actively delivered into the cells together with the mRNA. Therefore, it is necessary to
determine whether the conventional WHO concept of DNA persistence is sufficient. It is
necessary to explain the reason (s) for the persistence of DNA, including not only the
amount of residual DNA but also its strand length.
Process control is important for the control of contamination and removal of impurities
that affect the quality of an mRNA DS, and appropriate process parameters should be
established in the purification process related to the removal of impurities. For impurities
derived from a manufacturing process that can be sufficiently removed in a specific process,
it may be more reasonable to ensure quality through in-process control testing instead of
controlling the final product.
During the formulation process, mRNA is encapsulated in lipid-nano-particles (LNPs) to
ensure mRNA stabilization, a drug delivery system for delivery to target cells, and
intracellular delivery. The LNP encapsulation process is the most important step in drug
formulation, and it is important to evaluate process-related impurities, such as the
encapsulation ratio after LNP encapsulation, particle size, content of intact mRNA, and
characteristics and content of degraded mRNA.
It is also necessary to evaluate the biological activity of a drug product, its ability to
express the target antigen using the formulation, and the characteristics of the expressed
antigen to explain the validity of the formulation process to a regulatory agency.
Characterization of the mRNA Products
To confirm that mRNA with the target quality attributes has been prepared, the physical and
chemical properties and biological activity of the DS must be determined. It is necessary to
identify the types of process-related and product-related impurities present and estimate
their amounts in the product. The basic sequence structure of the non-amplified mRNA is
shown in Fig. 2 The mRNA primary sequence is often
confirmed by oligonucleotide mapping analysis using liquid chromatography-tandem mass
spectrometry. Ultra-performance liquid chromatography/ultraviolet-mass spectrometry can also
be used to confirm the integrity of mRNA based on its strand length.
For example, capillary gel electrophoresis or reversed-phase-high-performance liquid
chromatography (RP-HPLC) can be used to clarify the ratio of complete to incomplete mRNA
contained in a DS. Quantitative PCR and digital PCR were used to estimate the amounts of
mRNA and incomplete mRNA. Digital PCR was used to estimate the ratio of complete to
incomplete mRNA. The heterogeneity of the added poly (A) chains may be difficult to fully
analyze using liquid chromatography-mass spectrometry, and the ratio of their presence
(heterogeneity) should be clarified using an analytical method such as RP-HPLC.
Heterogeneity may differ depending on whether the poly (A) addition reaction is performed
after the in vitro transcription reaction and/or whether the poly (T)
sequence is used as a template.
RP-HPLC is a useful tool for analyzing product-related impurities, such as mRNAs that have
no Cap structure added, incomplete transcripts, and mRNAs with insufficient poly (A) chain
length. Other product-related impurities may be required to analyze the dsRNA content.
Because dsRNA is considered a potent activator of innate immunity, it is often analyzed
using immunoblotting or other methods.
It is known that mRNAs have sequence-dependent secondary and higher-order structures,
information on which can be obtained using circular dichroism spectroscopy, nuclear magnetic
resonance spectroscopy, and analysis of thermodynamic parameters using differential
calorimetry. Even if these methods are applied, the precise extent to which the higher-order
structure of the target mRNA is involved in its biological characteristics, such as activity
and stability, remains unknown. These methods may be useful for stability tests and
assessments of product comparability before and after changes [29].
When confirming (i) the ability of mRNA to express proteins, including
target antigens, as the biological activity of the mRNA and (ii) the
characteristics of the expressed proteins, antigen proteins can be expressed by transfection
of mRNA into appropriate model cells in vitro, and the proteins can be
analyzed by western blotting. However, it is difficult to analyze the mode of existence of
proteins expressed from mRNA using western blotting alone. For example, if the expressed
protein is designed to be distributed on the cell membrane surface, characterization of the
protein may not be sufficient unless the distribution of the protein in the expressing cell
is analyzed, and the expressed protein is confirmed to be on the cell membrane surface. In
other cases, it may be appropriate to reveal biological activity by performing a
quantitative cellular immunological analysis of the expression of the target protein in
mRNA-treated cells. It is also possible to demonstrate the usefulness of a protein expressed
by mRNA as a target antigen by analyzing the reactivity of the target protein expressed in
cells using antibodies against the target viral antigen contained in the plasma of
convalescent patients [30, 31].
Potential impurities from the manufacturing process include template DNA used for
in vitro transcription reactions, RNA polymerase, DNase used to degrade
DNA after a transcription reaction, residual host cell proteins, cellular DNA, and
endotoxins in the templates derived from E. coli. It is important to
evaluate not only the amount of DNA present but also the strand length of the remaining DNA.
Measuring the amount of long-stranded DNA is particularly important.
For enzymatic elongation of the Cap structure or poly (A), the amount of enzyme used should
also be analyzed.
The sponsor may be encouraged to develop state-of-the-art technology methods, if necessary,
and conduct quality assessment and quality control using these methods. The development of
assay methods may include the preparation of new reagents and other materials to analyze
process-derived impurities.
Characterization of Drug Formulations
As mRNA is easily degraded by RNase and is not efficiently internalized into cells, it is
encapsulated into LNPs during the formulation process to ensure in vivo
stability, delivery to target cells, and sustained intracellular release. In the
encapsulation of purified mRNA DS into LNPs, multiple lipid molecules are used to anticipate
various changes in the behavior of mRNA in the human body, and a formulation using
microfluidic channels with baffle structures is used to efficiently encapsulate mRNA into
lipid molecules (Fig. 3).

When seeking the approval of a vaccine, vaccine developers must be prepared to
(i) explain the chemical characteristics of each lipid applied to the
design of the formulation process using LNPs, (ii) clarify whether the
desired properties have been obtained in terms of the physical parameters of the
mRNA-encapsulated nanoparticles, and (iii) demonstrate the purpose of the
formulation’s design and the justifications for its specifications. Therefore, it is
necessary to confirm the consistency and stability of the formulation quality of the final
product. In addition to evaluating the potential toxicity of the lipids that comprise the
LNPs, it may be useful to characterize the function of each lipid component with positive
and/or negative effects on the immune response and vaccine efficacy.
The role and safety of the lipid components used in vaccine formulations must be evaluated.
For example, if one of the lipid components has an adjuvant effect, the lipid content of the
LNP may be an important quality characteristic. Thus, the consistency of the lipid
composition and content in the formulated particles should also be evaluated.
Establishment of Analytical Procedures and Specifications for mRNA Vaccines
Analytical procedures and specifications for DSs
Because DNA templates used for in vitro transcription reactions are
important raw materials for the DS of mRNA, it is important to evaluate whether the
template DNA is sufficiently removed during the purification process from the standpoint
of quality and safety. To ensure the quality of the DS, it is reasonable to set acceptance
criteria for the templates. Product- and process-related impurities such as incomplete
sequences and endotoxins are examples of items that should be addressed in DS
specifications.
The specifications for a DS are expected to include the identity of the DS, its content,
physical and chemical properties, biological activity, and product- and process-related
impurities. In particular, it is necessary to consider (i) whether the
addition of a 5′-Cap or poly (A) is appropriate, (ii) the mRNA purity
(percentage of intact mRNA), and (iii) other impurities (e.g., the
percentage of dsRNA, RNA shorter or longer than the desired length, and template DNA)
(Table 1). However, if some quality
attributes can be adequately controlled using in-process control tests, it would be
acceptable not to conduct a release test. Vaccine developers must establish specifications
that guarantee sufficient safety margins for impurities. It is also necessary to determine
whether analytical procedures have sufficient sensitivity and accuracy.
Table 1.Specification testing of formulations
| WHO |
Comirnaty® intramuscular injection |
Moderna Spikevax intramuscular injection |
| Drug substance: |
| Identity |
Identity |
Identity |
| Purity and impurities |
RNA integrity, purity (double- strand RNA and
template DNA), 5′-Cap and poly (A) tail |
Purity (mRNA purity, product-related impurities,
residual DNA, 5′-Cap rate additional rate, poly (A) tail) |
| Quantification and physical state |
Contents |
Contents |
| Safety attributes |
Bioburden, endotoxins |
Bioburden, endotoxins |
| Additional quality attributes |
Clarity, pH |
Characteristics, pH |
| Drug product: |
| Identity |
Identity (RNA) |
Identity (RNA) |
| Purity and impurities |
RNA integrity |
Purity test (RNA, product-related impurities) |
| Quantification and physical state |
Content |
Content |
| Safety attributes |
Sterility test and endotoxin test |
Sterility test and endotoxin test |
| Additional quality attributes |
Appearance, pH, identity (lipid), lipid content,
particle size and polydispersity, RNA encapsulation, osmolality, insoluble
particle matter, vial content volume |
Characteristics, pH, identity (lipid), lipid
content, purity test (lipid impurity), particle size and polydispersity,
encapsulated RNA, osmolality, insoluble foreign matter, insoluble particulate
matter, vial content volume |
| Potency |
Biological activity |
In vitro translation |
This table was compiled from Ref. [35] and the
review report for Comirnaty® intramuscular injection and the Moderna
COVID-19 vaccine intramuscular injection with some modifications. Masked information
in the review report is not included in this table. WHO: World Health Organization;
RNA: ribonucleic acid; DNA: deoxyribonucleic acid.
Although tests for the drug products listed in Table
1 are envisioned, the table does not cover all the tests, and not all tests are
necessary. However, tests for identifying the contents of a drug product are essential for
dose setting and may be useful for demonstrating the accuracy and validity of the drug
product. It is important to consider how formulation processes affect the biological
activity and constancy of a drug product.
Reference materials
Reference materials are often prepared using highly purified mRNA DSs that have been
fully characterized from orthogonal points. For example, the formulation batches used in
clinical trials can be fully characterized by their chemical composition, purity,
biological activity, complete sequence analysis, and other parameters for use as reference
materials regarding their physical and/or biological properties.
Stability testing and validity periods
Since mRNA DSs and products have characteristics that are sensitive to storage
temperatures, a shelf-life that can basically guarantee that a certain level of quality is
maintained should be set; it should be considered to ensure the consistency of efficacy
and safety based on a thorough understanding of the stability profile from the results of
long-term storage studies evaluated under actual storage conditions and periods, as is the
case with biopharmaceuticals. Conducting stability studies at multiple temperatures based
on actual usage scenarios and discussing the results of studies conducted under
freeze-thaw conditions may provide useful information for understanding the stability of
drug substances and products and the control of the appropriate storage temperatures of
drug products in clinical settings.
Non-clinical Studies
In Japan, non-clinical studies on mRNA vaccines are conducted in accordance with the
country’s Guidelines for Non-clinical Studies of Vaccines for the Prevention of Infectious
Diseases [32]. They provide point-to-consider
principles for evaluating vaccines against COVID-19 [33]. Nevertheless, the safety and efficacy of mRNA vaccines should be evaluated by
considering their unique quality characteristics. For example, in the case of LNP-mRNA
vaccines, if the LNPs include chemical substances that have not been previously used in
humans, a non-clinical safety evaluation of these chemical substances, which is required for
a new additive in a drug product, would be necessary. In addition, if chemical modifications
are used for the mRNA (including 5′-Cap and poly (A)), a safety evaluation focusing on the
modifications should be considered.
In mRNA vaccines approved in Japan (as of May 2023), uridine in the mRNA is replaced with
pseudouridine to reduce the high innate immune activation potential of the mRNA, as
mentioned above. Because pseudouridine is a naturally occurring nucleic acid, there are no
genotoxicity concerns. There is concern that innate immune activation by mRNA may cause
adverse effects such as inflammatory reactions, including fever and pain, at the
administration site. However, there are limitations in extrapolating test results from
animals to humans; thus, careful evaluation of these reactions in clinical trials is
necessary.
In the development of a SARS-CoV-2 vaccine during a pandemic period, if the platform
technology used to manufacture the vaccine is already approved or its use in a clinical
trial is sufficiently well defined, non-clinical safety study results of other products
manufactured using the same platform technology can be used as data to determine whether to
initiate clinical trials [3]. The WHO has stated that
for mRNA vaccines other than the SARS-CoV-2 vaccine, by utilizing the same platform concept,
non-clinical safety studies can be omitted if agreed upon by the national regulatory
authority [34].
Clinical Trials
Clinical trials on mRNA products are required in accordance with Japan’s Guidelines for
Clinical Trials of Vaccines for the Prevention of Infectious Diseases [35]. However, as in non-clinical studies, if matters are to be considered
specific to mRNA vaccine products, they should be evaluated separately.
Funding
This study was supported by a grant from the Japan Agency for Medical
Research and Development (AMED), No. 22mk010194j002).
Conflict of Interest
None.
Acknowledgments
This paper is described as a review, with further discussion based on a Japanese article
(PMDRS 2023:54 (4): 312–321). Therefore, we thank Junichi Fukuchi, Takaaki Yoshida, Mihiro
Kawamura, and Kengo Kawachi (Office of Vaccines and Blood Products, Japan Pharmaceuticals
and Medical Devices Agency) for their assistance in writing this manuscript.
References
- 1. Polack, F.
P., Thomas,
S. J.,
Kitchin,
N., Absalon,
J.,
Gurtman, A.,
Lockhart,
S., Perez,
J. L., Pérez
Marc, G.,
Moreira, E.
D., Zerbini,
C., Bailey,
R.,
Swanson, K.
A., Roychoudhury,
S., Koury,
K., Li,
P., Kalina,
W. V.,
Cooper, D.,
Frenck, R. W.
Jr., Hammitt,
L. L.,
Türeci, Ö.,
Nell, H.,
Schaefer,
A., Ünal,
S.,
Tresnan, D.
B., Mather,
S.,
Dormitzer, P.
R., Şahin,
U., Jansen,
K. U.,
Gruber, W.
C., C4591001 Clinical Trial
Group
2020. Safety and efficacy of the BNT162b2 mRNA Covid-19
vaccine. N. Engl. J. Med.
383: 2603–2615.
- 2. Baden, L.
R., El Sahly,
H. M.,
Essink, B.,
Kotloff,
K., Frey,
S., Novak,
R.,
Diemert, D.,
Spector, S.
A., Rouphael,
N., Creech,
C. B.,
McGettigan,
J., Khetan,
S., Segall,
N., Solis,
J., Brosz,
A., Fierro,
C.,
Schwartz,
H., Neuzil,
K., Corey,
L.,
Gilbert, P.,
Janes, H.,
Follmann,
D., Marovich,
M.,
Mascola, J.,
Polakowski,
L.,
Ledgerwood,
J., Graham,
B. S.,
Bennett,
H., Pajon,
R.,
Knightly,
C., Leav,
B., Deng,
W., Zhou,
H., Han,
S.,
Ivarsson,
M., Miller,
J., Zaks,
T., COVE Study
Group
2021. Efficacy and safety of the mRNA-1273 SARS-CoV-2
vaccine. N. Engl. J. Med.
384: 403–416.
- 3. Barda,
N., Dagan,
N., Cohen,
C., Hernán,
M. A.,
Lipsitch,
M., Kohane,
I. S.,
Reis, B. Y.
and Balicer, R.
D.
2021. Effectiveness of a third dose of the BNT162b2 mRNA
COVID-19 vaccine for preventing severe outcomes in Israel: an observational
study. Lancet
398: 2093–2100.
- 4. Zhang,
Z., Mateus,
J., Coelho,
C. H., Dan,
J. M.,
Moderbacher, C.
R., Gálvez,
R. I.,
Cortes, F.
H., Grifoni,
A., Tarke,
A., Chang,
J.,
Escarrega, E.
A., Kim,
C.,
Goodwin, B.,
Bloom, N.
I., Frazier,
A.,
Weiskopf,
D., Sette,
A. and
Crotty,
S.
2022. Humoral and cellular immune memory to four COVID-19
vaccines. Cell
185: 2434–2451.e17.
- 5. Park, J.
W., Lagniton,
P. N. P.,
Liu, Y. and
Xu, R.
H.
2021. mRNA vaccines for COVID-19: what, why and
how. Int. J. Biol. Sci.
17: 1446–1460.
- 6. https://news.modernatx.com/news/news-details/2023/Moderna-Announces-Interim-Phase-3-Safety-and-Immunogenicity-Results-for-mRNA-1010-a-Seasonal-Influenza-Vaccine-Candidate/default.aspx.
[accessed September 18, 2023].
- 7. Bloom,
K., van den
Berg, F. and
Arbuthnot,
P.
2021. Self-amplifying RNA vaccines for infectious
diseases. Gene Ther.
28: 117–129.
- 8. https://www.science.org/content/article/mrna-vaccine-twist-it-copies-itself-protects-against-covid-19.
[accessed September 18, 2023].
- 9. WHO/RNA/DRAFT/22 December 2020
Evaluation of the quality, safety and efficacy of RNA-based prophylactic vaccines for
infectious diseases: Regulatory considerations. https://www.who.int/docs/default-source/biologicals/ecbs/reg-considerations-on-rna-vaccines_1st-draft_pc_tz_22122020.pdf?sfvrsn=c13e1e20_3.
[accessed September 18, 2023].
- 10. Chaudhary,
N.,
Weissman,
D. and
Whitehead, K.
A.
2021. mRNA vaccines for infectious diseases: principles,
delivery and clinical translation. Nat. Rev. Drug
Discov.
20: 817–838.
- 11. Alexopoulou,
L., Holt,
A. C.,
Medzhitov,
R. and
Flavell, R.
A.
2001. Recognition of double-stranded RNA and activation of
NF-kappaB by Toll-like receptor 3.
Nature
413: 732–738.
- 12. Diebold, S.
S., Kaisho,
T., Hemmi,
H., Akira,
S. and Reis e
Sousa, C.
2004. Innate antiviral responses by means of TLR7-mediated
recognition of single-stranded RNA.
Science
303: 1529–1531.
- 13. Heil,
F., Hemmi,
H.,
Hochrein,
H.,
Ampenberger,
F.,
Kirschning,
C., Akira,
S.,
Lipford, G.,
Wagner, H.
and Bauer,
S.
2004. Species-specific recognition of single-stranded RNA via
toll-like receptor 7 and 8. Science
303: 1526–1529.
- 14. Karikó,
K.,
Buckstein,
M., Ni,
H. and
Weissman,
D.
2005. Suppression of RNA recognition by Toll-like receptors:
the impact of nucleoside modification and the evolutionary origin of
RNA. Immunity
23: 165–175.
- 15. Karikó,
K.,
Muramatsu,
H., Welsh,
F. A.,
Ludwig, J.,
Kato, H.,
Akira, S.
and Weissman,
D.
2008. Incorporation of pseudouridine into mRNA yields superior
nonimmunogenic vector with increased translational capacity and biological
stability. Mol. Ther.
16: 1833–1840.
- 16. Karikó,
K.,
Muramatsu,
H., Ludwig,
J. and
Weissman,
D.
2011. Generating the optimal mRNA for therapy: HPLC
purification eliminates immune activation and improves translation of
nucleoside-modified, protein-encoding mRNA. Nucleic
Acids Res.
39: e142.
- 17. Thess,
A., Grund,
S., Mui,
B. L.,
Hope, M. J.,
Baumhof,
P.,
Fotin-Mleczek,
M. and
Schlake,
T.
2015. Sequence-engineered mRNA without chemical nucleoside
modifications enables an effective protein therapy in large animals.
Mol. Ther.
23: 1456–1464.
- 18. Kudla,
G., Lipinski,
L., Caffin,
F., Helwak,
A. and
Zylicz,
M.
2006. High guanine and cytosine content increases mRNA levels
in mammalian cells. PLoS Biol.
4: e180.
- 19. Kudla,
G., Murray,
A. W.,
Tollervey,
D. and
Plotkin, J.
B.
2009. Coding-sequence determinants of gene expression in
Escherichia coli. Science
324: 255–258.
- 20. Blakney, A.
K., McKay,
P. F., Yus,
B. I.,
Aldon, Y.
and Shattock, R.
J.
2019. Inside out: optimization of lipid nanoparticle
formulations for exterior complexation and in vivo delivery of saRNA.
Gene Ther.
26: 363–372.
- 21. Gallie, D.
R.
1991. The cap and poly(A) tail function synergistically to
regulate mRNA translational efficiency. Genes
Dev.
5: 2108–2116.
- 22. Martin, S.
A., Paoletti,
E. and
Moss,
B.
1975. Purification of mRNA guanylyltransferase and mRNA
(guanine-7-) methyltransferase from vaccinia virions. J.
Biol. Chem.
250: 9322–9329.
- 23. Stepinski,
J.,
Waddell, C.,
Stolarski,
R.,
Darzynkiewicz,
E. and
Rhoads, R.
E.
2001. Synthesis and properties of mRNAs containing the novel
“anti-reverse” cap analogs 7-methyl(3′-O-methyl)GpppG and 7-methyl
(3′-deoxy)GpppG. RNA
7: 1486–1495.
- 24. Malone, R.
W., Felgner,
P. L. and
Verma, I.
M.
1989. Cationic liposome-mediated RNA
transfection. Proc. Natl. Acad. Sci.
USA
86: 6077–6081.
- 25. Holtkamp,
S.,
Kreiter, S.,
Selmi, A.,
Simon, P.,
Koslowski,
M., Huber,
C., Türeci,
O. and
Sahin,
U.
2006. Modification of antigen-encoding RNA increases
stability, translational efficacy, and T-cell stimulatory capacity of dendritic
cells. Blood
108: 4009–4017.
- 26. Stadler, C.
R., Bähr-Mahmud,
H., Celik,
L., Hebich,
B., Roth,
A. S.,
Roth, R. P.,
Karikó, K.,
Türeci, Ö.
and Sahin,
U.
2017. Elimination of large tumors in mice by mRNA-encoded
bispecific antibodies. Nat. Med.
23: 815–817.
- 27. Eberie,
F., Sahin,
U., Huhn,
A., et al.
Stabilization of poly(a) sequence encoding DNA sequences. Patent number: US 2017/0166905
A1 ( 2017).
- 28. Lebron, J.
A., Troilol,
P. J.,
Pacchione,
S.,
Griffiths, T.
G., Harper,
L. B.,
Mixson, L.
A., Jackson,
B. E.,
Michna, L.,
Barnum, A.
B., Denisova,
L.,
Johnson, C.
N., Maurer,
K. L.,
Morgan-Hoffman,
S., Niu,
Z., Roden,
D. F.,
Wang, Z.,
Wolf, J.
J., Hamilton,
T. R.,
Laux, K. M.,
Soper, K.
A. and Ledwith,
B. J.
2006. Adaptation of the WHO guideline for residual DNA in
parenteral vaccines produced on continuous cell lines to a limit for oral
vaccines. Dev. Biol. (Basel)
123: 35–44, discussion 55–73.
- 29. European Medicines Agency
Science Medicines Health Assessment report. 19 February 2021. https://www.ema.europa.eu/en/documents/assessment-report/comirnaty-epar-public-assessment-report_en.pdf.
[accessed September 18, 2023].
- 30. Sahin,
U., Muik,
A.,
Derhovanessian,
E., Vogler,
I., Kranz,
L. M.,
Vormehr,
M., Baum,
A., Pascal,
K., Quandt,
J., Maurus,
D.,
Brachtendorf,
S., Lörks,
V.,
Sikorski,
J., Hilker,
R., Becker,
D., Eller,
A. K.,
Grützner,
J., Boesler,
C.,
Rosenbaum,
C., Kühnle,
M. C.,
Luxemburger,
U.,
Kemmer-Brück,
A., Langer,
D., Bexon,
M., Bolte,
S., Karikó,
K.,
Palanche,
T., Fischer,
B.,
Schultz, A.,
Shi, P. Y.,
Fontes-Garfias,
C., Perez,
J. L.,
Swanson, K.
A., Loschko,
J., Scully,
I. L.,
Cutler, M.,
Kalina, W.,
Kyratsous, C.
A., Cooper,
D.,
Dormitzer, P.
R., Jansen,
K. U. and
Türeci,
Ö.
2020. COVID-19 vaccine BNT162b1 elicits human antibody and
TH1 T cell responses.
Nature
586: 594–599.
- 31. Mulligan, M.
J., Lyke,
K. E.,
Kitchin,
N., Absalon,
J.,
Gurtman, A.,
Lockhart,
S., Neuzil,
K., Raabe,
V., Bailey,
R.,
Swanson, K.
A., Li,
P., Koury,
K., Kalina,
W., Cooper,
D.,
Fontes-Garfias,
C., Shi,
P. Y.,
Türeci, Ö.,
Tompkins, K.
R., Walsh, E.
E., Frenck,
R., Falsey,
A. R.,
Dormitzer, P.
R., Gruber,
W. C.,
Şahin, U.
and Jansen, K.
U.
2020. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in
adults. Nature
586: 589–593.
- 32. Guidelines for Non-Clinical Studies of
Vaccines for the Prevention of Infectious Diseases. ( 2010).
Ministry of Health, Labour and Welfare, Japan. PSEHB/PED Notification No. 0527–1.
[accessed September 18, 2023].
- 33. Point-to-Consider. Principles for the Evaluation of
Vaccines Against the Novel Coronavirus SARS-CoV-2. Office of Vaccines and Blood Products,
Pharmaceuticals and Medical Devices Agency, Japan. September 2020. https://www.pmda.go.jp/files/000237021.pdf. Accessed Sept. 18, 2023. And the
supplement 1 to 4 have been also published.
- 34. Guideline, W. H.
O. Evaluation of the quality, safety and
efficacy of messenger RNA vaccines for the prevention of infectious diseases: Regulatory
considerations. Annex 3, Technical Report Series No. 1039. ( 2022). Available
at: https://www.who.int/publications/m/item/annex-3-mRNA-vaccines-trs-no-1039.
[accessed September 18, 2023].
- 35. Guidelines for Clinical Studies of
Vaccines for the Prevention of Infectious Diseases ( 2010).
Ministry of Health, Labour and Welfare, Japan. PSEHB/PED Notification No.
0527.
