Introduction
DNA base-pair mismatches are routinely generated by intrinsic factors such as DNA replication errors and oxygen species and extrinsic factors such as ultraviolet and ionizing radiation. It has been well-documented that these mismatches are corrected by the mismatch repair (MMR) pathway both in prokaryotes and eukaryotes [1,2]. In Escherichia coli (E. Coli), a base-pair mismatch is searched and recognized by a MutS protein [3]. The crystal structure of MutS that recognizes a mismatch contains an adenosine diphosphate (ADP) molecule in one of the two nucleotide-binding sites [4–6]. After the mismatch recognition, the bound ADP molecule is exchanged with an adenosine triphosphate (ATP) molecule, and the other nucleotide-binding site is also occupied by ATP. The ATP binding induces a MutS conformational change, binding of MutL and MutH proteins to MutS, and release of MutS from the mismatch [7–11]. The released MutS/MutL/MutH complex diffuses along DNA to search and recognize a GATC sequence around the mismatch [12], and MutH generates a nick on the sequence [7]. Then, an exonuclease digests one strand of double-stranded DNA from the nick to the site beyond the mismatch [13]. Finally, the new strand is re-synthesized using the undigested strand as a template [13].
The bacterial mismatch recognition protein MutS takes a homodimer consisting of 95 kDa monomers [14]. Each monomer contains the mismatch-binding, connector, lever, clamp, ATPase, and tetramerization domains from N to C terminus (Figure 1A). In the crystal structure of ADP-bound MutS that recognizes a mismatch, the mismatch binding domain in one of the dimer and the clamp domains of the dimer wrap around and attach to DNA (Figure 1B) [4,5]. The deuterium exchange mass spectrometry study suggested that similar protein/DNA contacts form on both mismatched and homo-duplex DNA, indicating that the search conformation is almost the same as the recognition conformation [15].

Previous single-molecule fluorescence microscopy [12,16–18] and fluorescence resonance energy transfer (FRET) [19,20] studies have suggested that MutS temporarily binds to DNA and one-dimensionally diffuses along it to search a base-pair mismatch. The diffusion coefficient does not depend on ion concentration, suggesting that the one-dimensional diffusion is not accompanied by microscopic dissociation and reassociation [18]. Notably, the narrow distribution of polarization of fluorescent dyes on the diffusing MutS indicated that the diffusion is coupled with the protein rotation around DNA [19]. Also, ATP binding to MutS makes the polarization distribution wider, suggesting that the diffusion after the MutS conformational change is less coupled with the protein rotation [19]. Recently, the molecular dynamics (MD) simulations of human MutS homolog, Msh2-Msh6, were performed using coarse-grained (CG) models [21]. However, structural dynamics details of bacterial MutS sliding along DNA before and after ATP binding have not been addressed yet.
The MutS structures in various conformations have been published [4–6,10,11,22]. They differ in their nucleotide state and DNA binding. We were interested in MutS sliding along DNA in three conformations. The first one is the ADP bound conformation, which recognizes a base-pair mismatch (MutSADP) and was proposed to be relevant to the mismatch search as described above [18] (Figure 1B). We put special focus on this conformation because the previous experiment indicated that MutS in this conformation rotates around DNA long axis while diffusing along it, though the structural dynamics have not been directly observed. The second one is the conformation obtained in the presence of ATP and homo-duplex DNA in the recent cryo-electron microscopy (EM) study [22]. In this conformation, the two clamp domains from each MutS monomer are distal to each other, and hence we name it as MutSOpenATP (Figure 1C). We were interested in the searchability of this conformation because it was also proposed to be relevant to the mismatch search. The third one is the ATP-bound sliding clamp conformation, which binds to MutL in the previously published crystal structure [11]. In this conformation, the two clamp domains from each MutS monomer are proximal to each other, and hence we name it as MutSClosedATP (Figure 1D). In this study, we performed CGMD simulations of MutSADP, MutSOpenATP, and MutSClosedATP (Special focus on MutSADP, and MutSOpenATP and MutSClosedATP for comparison). In the simulations, we successfully observed MutSADP, MutSOpenATP, and MutSClosedATP diffusing along DNA. The simulation results suggested that MutSADP and MutSClosedATP diffuses along DNA in a rotation-coupled manner with rare and frequent groove-crossing events, respectively, in the presence of a physiological concentration of ions. In the groove-crossing events, the protein overcame an edge of a groove and temporarily diffused in a rotation-uncoupled manner. The simulation results also suggested that DNA rotations around its long axis significantly facilitate the MutS diffusion. It was also indicated that mismatch searches by MutSOpenATP is inefficient in terms of mismatch checking even though MutSOpenATP diffuses along DNA and reaches unchecked regions more rapidly than MutSADP. Together, our simulations elucidated unprecedented structural dynamics details of bacterial MutS sliding along DNA.
Material and Methods
Simulation Model
The E. coli MutS protein is 853 residues long. Even if we omit the tetramerization domain, which is known to be dispensable based on the finding that the tetramer-disrupting D835R and D840E mutations only modestly affected the MutS function in vivo [23], it is still 800 residues long. It is a daunting task to observe the MutS sliding along DNA in MD simulations with all atoms treated explicitly. Therefore, we used a CG model in which multiple atoms are represented by a single mass point (bead) [24].
For the MutS protein, we used the AICG2+ model (Please refer to the original work [25] for details). In this model, each amino acid was represented by one bead located on the Cα atom position. Consecutive amino acids were connected by elastic bonds. Sequence-based statistical potentials were used to model bond angles and dihedral angles [26]. Excluded volume interactions prevented two beads from overlapping each other. Native-structure-based contact potentials (Gō potential) restrained the distance between amino acid pairs that contact each other in native structures. Parameters in the AICG2+ model were decided so that the fluctuation of each amino acid in reference proteins reproduced that of all-atom simulations.
For the DNA molecule, we used the 3SPN.2C model (Please refer to the original work [27] for details). In this model, each nucleotide was represented by three beads located at the positions of base, sugar, and phosphate units. Neighboring sugar-phosphate and sugar-base were connected by virtual bonds. Bond angles and dihedral angles were restrained to their values in reference B-type DNA. Excluded volume interactions prevented any two beads from overlapping each other. Orientation-dependent attractive potentials were applied to base-pairs, cross-stacking pairs, and intra-chain-stacking pairs. The parameters were decided so that the model reproduced several types of experimental data.
For interactions between MutS and DNA, electrostatic and excluded volume interactions were applied unless otherwise stated. Previous studies have shown that electrostatic interactions dominate sequence-nonspecific protein/DNA interactions [28]. It was also demonstrated that CGMD simulations that consider only electrostatic and excluded volume interactions as interactions between proteins and DNA could predict protein/DNA complex structures with a certain degree of accuracy [29–31]. The electrostatic interactions were modeled with the Debye-Hückel potential, which can represent the ion concentration dependency of ion screening effects. For MutS, charges were distributed on surface residue beads according to the RESPAC algorithm [29] so that the resulting charge arrangement reproduces electrostatic potential around MutS theoretically calculated using all charged atom positions. For intra-DNA interactions, constant –0.6 charges were placed on the phosphate beads. For protein/DNA interactions, we added –0.4 extra charges to them to model releases of counter ions upon protein/DNA bindings. This simple treatment of protein/DNA interactions has been successfully applied to various biological systems in our and other groups [21,32–37].
Initial Structures
We performed CGMD simulations using three MutS conformations: ADP-bound (MutSADP), ATP-bound open clamp (MutSOpenATP), and ATP-bound closed clamp (MutSClosedATP) conformations. We used the crystal structure [PDB ID: 1e3m] [4], the cryo-EM structure [PDB ID: 7ai5] [22], and the crystal structure [PDB ID: 5akb] [11] as the reference structures for MutSADP, MutSOpenATP, and MutSClosedATP simulations, respectively. For double-stranded DNA, we used two homogeneous sequences of 75 base-pairs (bp): GC repeats and AT repeats. The reference B-type DNA structure was prepared using 3DNA software [38]. For MutSADP and MutSOpenATP, we replaced the DNA molecule included in the crystal structure with 75 bp coarse-grained DNA without steric clashes. For MutSClosedATP, we omitted the MutL included in the crystal structure and manually threaded 75 bp coarse-grained DNA through the middle hole without steric clashes. The initial positions of MutS were 32 and 45 bp away from the end for MutSADP and MutSClosedATP simulations and MutSOpenATP simulations, respectively.
Simulation Time Evolution
The coordinates of CG beads were updated for 5×107 steps according to the Langevin equation of motion with a step size of 0.3 in the CafeMol time unit and were recorded every 1×103 steps [39]. We did not observe the MutS dissociation from the middle of DNA. We stopped the simulations when MutS dissociates from DNA ends. Temperature and the friction constant were set to 300 K and 0.843, respectively. The dielectric constants for solution and solutes were set to 78.0 and 1.0, respectively. We performed all the simulations using CafeMol 3.2 (https://www.cafemol.org) [40]. The molecular structures were illustrated using PyMOL (https://pymol.org) and VMD (https://www.ks.uiuc.edu/Research/vmd).
Results
Simulations of MutS with an ADP-Bound Conformation on DNA
The crystal structure of bacterial MutS recognizing a DNA base-pair mismatch was previously solved. In that structure, ADP binds to one of the nucleotide-binding sites in ATPase domains. We sought to observe the sliding dynamics of this conformation (MutSADP) along DNA, assuming that the searching and recognition states share the common conformation [15]. Therefore, we performed CGMD simulations of MutSADP on 75 bp DNA with GC and AT repeat sequences at 100 mM monovalent ion. The simulation movies and trajectories showed bidirectional movements of MutS along DNA, which are indicative of diffusion (Figure 2AB, Supplementary Figure S1 to S2, and Supplementary Movie S1). To confirm that these bidirectional movements are simple diffusion dynamics, we calculated mean square displacements (MSD) from the trajectories and plotted them against step intervals (Figure 2C). MSD was defined as ⟨(x(t1)–x(t0))2⟩∆t where x(t) is a DNA base-pair index closest to MutS at simulation step t, and ⟨·⟩∆t represents an ensemble average of snapshots separated by a step interval ∆t=t1–t0. The plot showed linear lines regardless of sequences, supporting that the MutS movements along DNA are diffusion and the dynamics do not significantly depend on sequences. We also calculated the slopes of the linear lines (diffusion coefficients) and confirmed that the difference caused by the sequences is within an error (Figure 2D). Together, we successfully observed MutS sliding along DNA using our CGMD simulation technique.

Next, we sought to investigate the ion concentration dependency of the sliding dynamics. Therefore, we performed additional simulations of MutSADP on 75 bp DNA with a GC repeat sequence at 50, 150, 500 mM ion. We also performed simulations in which no electrostatic interactions were applied for interactions between MutS and DNA. Interestingly, the simulation trajectories showed that MutS can hold DNA even without electrostatic interactions since it wraps around it with mismatch binding and clamp domains. In all the setups, we observed bidirectional movements of MutS along DNA, which are indicative of diffusion (Figure 3A and Supplementary Figure S3 to S6). We plotted MSDs against step intervals, and the plot shows linear lines regardless of ion concentrations, confirming that the MutS movements along DNA are diffusion irrespective of the electrostatic interaction strength (Figure 3B). We calculated diffusion coefficients and the fold difference between 50 mM and 500 mM cases was only 1.5 (Figure 3C). The weak ion concentration dependency of the diffusion coefficients was consistent with the previous single-molecule experiment [18].

The previous study suggested that ADP-bound MutS diffuses along DNA in a rotation-coupled manner [18]. To clarify if the diffusion we observed in the simulations is coupled to MutS rotations around the DNA long axis, we plotted the time trajectories of MutS positions against cumulative angle changes (Figure 4AB and Supplementary Figure S7 to S11). We defined the angle formed by two lines connecting the center of masses (COMs) of MutS and the base-pairs most proximal to the MutS COM, and the angle change was calculated by summing the displacement of the angle between two consecutive simulation frames up to a certain time point. Thus, the cumulative angle is not limited to the range of 2π. In the 100 mM ion case, the representative plot showed one linear line with occasional fluctuations (Figure 4A and Supplementary Figure S7). This result clearly showed that MutS movement along DNA is tightly coupled to its rotation around the DNA long axis. Also, the pitch was consistent with that of the DNA groove. Therefore, MutS diffuses along the groove and hence moves along DNA in a rotation-coupled manner. In the 4/20 simulation trajectories, we observed more than one linear line, suggesting the groove-crossing events. In the groove-crossing events, MutS originally diffusing along a major groove overcomes the two edges of the adjacent minor groove and translocates in the direction of the DNA long axis to the major groove ~5 bp away from the original position. After groove-crossing, MutS started rotation-coupled diffusion again on the new groove. Together, the simulation results suggested that MutS diffuses along DNA groove with rare groove-crossing events in the presence of 100 mM ions.

Next, we sought to investigate the ion concentration dependency of the degree of coupling between diffusion along and rotation around DNA. Therefore, we plotted the time trajectories of MutS positions against cumulative angle changes for the 50, 150, 500 mM ion case and the case without electrostatic interactions between MutS and DNA (Figure 4B and Supplementary Figure S8 to S11). In the 50 mM case, we observed one linear line in most of the trajectories, suggesting that the MutS movement along DNA is tightly coupled to its rotation in this ion concentration range. Also, we observed no groove-crossing events in the 50 mM case and few (6/20) in the 150 mM case. Together, the simulation results suggested that MutS diffuses along the DNA groove with no or rare groove-crossing events in a wide ion concentration range. In the 500 mM ion case (18/20) or the case without electrostatic interactions between MutS and DNA (19/20), although the frequency of groove-crossing events significantly increased, MutS still slid along the DNA groove. These results suggested that the excluded volume interactions between MutS and DNA mainly dictate the sliding path, and the electrostatic interactions prevent groove-crossing events.
To quantitatively visualize groove-crossing events and their frequency, we plotted time trajectories of MutS positions on the axis orthogonal to linear lines with the slope of 10 bp/2π in the position vs cumulative angle change plots (Figure 4C and Supplementary Figure S12 to S16). This plot would show a horizontal line if MutS slid along DNA in a rotation-coupled manner with the groove pitch, and the position would change if a groove-crossing event took place. As expected, we observed one linear horizontal line in each trajectory in the presence of 50 mM ion, suggesting the rotation-coupled diffusion with the groove pitch and no groove-crossing events. We observed a few groove-crossing events in the 100 mM and 150 mM cases and a lot more in the 500 mM case and the case without electrostatic interactions between MutS and DNA (Supplementary Figure S12 to S16). To quantify the frequency, we calculated and plotted probabilities of diffusing without groove-crossing against duration. In the 50 mM, 100 mM, and 150 mM ion cases, the survival probabilities decreased sharply as the duration increased from 0 to around 8×105 steps (Figure 4D). Then the probabilities became nearly flat in the range from around 8×105 to 5×106 steps. We attributed the initial sharp decrease to the rapid back and forth groove-crossing observed in the time trajectories, the frequency of which is thought to be highly susceptible to the definition of protein position. The second flat phase confirmed that few groove-crossing events took place in this ion concentration range. In the 500 mM ion case and the case without electrostatic interaction between MutS and DNA, on the other hand, the survival probabilities decreased linearly as the duration increased from 8×105 to 5×106 steps. The linear decrease indicated that the “one-way” groove-crossing events take place as a Poisson process of a constant rate. To quantitatively compare the frequency of the one-way groove-crossing events, we calculated the rate constants from the second phases and plotted them in Figure 4E. We confirmed that the groove-crossing events are rare in the 50 mM, 100 mM, and 150 mM ion cases, but more frequent in the 500 mM case and the case without electrostatic interaction between MutS and DNA. Together, the simulation results suggested that the frequency of the groove-crossing events is dictated by the strength of electrostatic interactions between MutS and DNA.
The ADP-Bound MutS Domain-Wise Interactions with DNA
Next, we sought to investigate the interaction of the clamp domain when MutS diffuses along DNA. Therefore, we plotted (Figure 5AC and Supplementary Figure S17 to S21) the base-pair indexes most proximal to the clamp domain (COM of residues 467 to 470 and 495 to 498; purple in Fig. 5D). In these plots, each point was colored according to which groove the clamp domains are in: Blue if in the major groove and red if in the minor groove. We judged the domain is in the major (minor) groove if it is closer to the contour line drawn by connecting the position of phosphate beads located at both edges of the major (minor) groove than that of the minor (major) groove. The clamp domain was likely to stay in the major groove in the presence of 100 mM ions (Figure 4A left), while it frequently traveled to a minor groove in the presence of 500 mM ions (Figure 5A right). The statistics clearly showed that the domain is more likely to travels to the minor groove as the ion concentration increases (Figure 5B). This result was consistent with the notion above that MutSADP diffuses along DNA in a rotation-coupled manner. In the rare groove-crossing events, we observed that the clamp domain initially contacting with a base-pair in the major groove travels to and temporarily stays in the minor groove and again travels to and contacts with the base-pair in the major groove ~10 bp away from the original one (Figure 5CD). These results suggested that the groove-crossing event may be rare due to the high free energy barrier for the clamp domain to travel to the minor groove and travel again to the distal major groove base-pairs instead of coming back to the original ones.

We also analyzed the location of one of the mismatch recognition domains (COM of residues 36 to 38, purple in Figure 6AB) when MutS diffuses along DNA (Figure 6D). Interestingly, the domain almost always stays in the major groove in the presence of 100 mM ions in the simulations, though it is in the minor groove in the crystal structure [PDB ID: 1e3m]. From the simulation trajectories, we identified the DNA contacting residues (the minimum distance to DNA is less than 10 Å in >90% of the simulation frames): Residues 11 to 13, 35, 71, 72, 99, 100, 103 to 105, and 469 to 470. The residues contacting DNA in the crystal structure are associated with DNA in the simulation (Figure 6C). As is in the case of the clamp domain, the statistics clearly showed that the mismatch recognition domain is more likely to travel to the minor groove as the ion concentration increases. In the crystal structure, MutS widens the minor groove around the mismatch binding domain to ~26 Å and bends DNA by ~55° (Figure 6A). Therefore, we analyzed the snapshots of the simulations in the presence of 100 mM ions to observe the DNA deformation around MutS. Here, the minor groove width was defined as the distance between (i-3)-th and (i+3)-th phosphate particles from each DNA strand where the i-th base-pair is most proximal to MutS. The DNA bending angle was defined as the average of the angles formed by (i–j)-th, i-th, and (i+j)-th base-pairs (j=1, 2, 3). First, we plotted the probability distributions of the minor groove width in the presence and absence of MutS, finding that MutS widened the minor groove from 15.6±1.7 Å to 17.7±1.8 Å (Figure 6E). Interestingly, the minor groove width around MutS was smaller than that in the crystal structure (~26 Å). Second, we plotted the probability distributions of the DNA bending angle in the presence and absence of MutS, finding that MutS bent DNA from 29±5° to 35±6° (Figure 6F). Again, the DNA bending angle around MutS was also smaller than that in the crystal structure (~55°), qualitatively consistent with the previous experiment [41,42]. The DNA in the crystal structure contains a mismatched base-pair, which was not included in the current simulations. Therefore, it is attractive to assume that the mismatched base-pair alters DNA deformability and facilitates the minor groove binding of the mismatch recognition domain. The natural extension of the simulation technique in the current work, especially precise modeling of the DNA deformation by mismatch base-pairs, may allow us to directly show it in the future.

In the simulations above, DNA was free to move. Hence, DNA rotated around its long axis and bent. To investigate the effects of the DNA rotation and bending on MutS diffusion, we performed three additional sets of simulations in which some ends of DNA strands were anchored in space: i) all (four) strand ends, ii) two strand ends at one duplex DNA end, and iii) two ends of one strand. The GC repeat sequence was used, and the ion concentration was set to 100 mM. Interestingly, when all strand ends were anchored in space, MutS diffusion along DNA was almost completely suppressed (Figure 7A right and Supplementary Figure S22 to S24). We confirmed this by calculating the diffusion coefficient (Figure 7BC) from the MSD curve as above. This result suggested that the DNA rotation and (or) bending have significant effects on MutS diffusion. The marginally same result was obtained when two strand ends at one duplex DNA end were anchored (Figure 7A center). Since DNA bending was hardly suppressed in this setup (Figure 7D; The DNA curvature was calculated as the previous study [34]), this result suggested that DNA rotations have more significant effects on MutS diffusion than the bending. Consistent with this suggestion, the MutS diffusion along DNA was moderately suppressed when two ends of one strand were anchored (Figure 7A). Together, these simulation results suggested that DNA rotations around its long axis significantly affect MutS diffusion dynamics. As suggested above, MutS sliding along DNA requires MutS rotation relative to DNA or DNA rotation relative to MutS. Since the diffusion rate in the rotation direction is inversely proportional to the molecular radius from the rotation axis according to the Stokes-Einstein equation, it is reasonable that the latter is more likely to take place.

The cryo-EM structure of bacterial MutS on DNA without mismatch was recently published [22]. In that structure, ATP binds to the two nucleotide-binding sites. Also, two clamp domains from each monomer are distal to each other. We sought to observe the sliding dynamics of this conformation (MutSOpenATP) along DNA and compare its searchability with MutSADP because both were proposed to be relevant to the mismatch search. Therefore, we performed CGMD simulations of MutSOpenATP on 75 bp DNA with a GC repeat sequence in the presence of 100 mM ions. The simulation movies and trajectories showed bidirectional movements of MutS along DNA, which are indicative of diffusion (Figure 8AB, Supplementary Figure S25, and Supplementary Movie S2). To confirm that these bidirectional movements are simple diffusion dynamics, we calculated MSDs from the trajectories and plotted them against MD step intervals (Figure 8C). The plot showed linear lines, supporting that the MutSOpenATP movements along DNA are diffusion. The ~10 bp2 shift at Δstep of 2×105 is thought to be caused by the swivel motion of MutS with the DNA binding portion as the fulcrum and is the potential artifact due to our choice of the mass center coordinate for the large molecule to calculate the MSDs. We calculated the slopes of the linear lines (diffusion coefficients) using data with Δstep from 1×105 to 5×105 and compared them with those of MutSADP. The result suggested that the MutSOpenATP diffusion (4.8×10–6 bp2/step) is moderately faster than MutSADP (2.8×10–6 bp2/step) (Figure 8D).

In the cryo-EM structure, ATP-bound MutS binds to homo-duplex DNA [22]. Therefore, this conformation has been proposed to be the searching mode. For mismatch recognition, the 36th phenylalanine residue in the mismatch recognition domain must contact the mismatched base-pair. To investigate how efficiently MutS can check the mismatches, we calculated the total number of base-pairs contacted by the phenylalanine bead within a certain period in our simulations (Figure 8E). Here, we considered the phenylalanine bead contacted to a base-pair when the nearest distance was less than 10 Å. For comparison, we performed the same analysis for the MutSADP simulations. In the plot, the total number of contacted base-pairs increased more rapidly for MutSADP than for MutSOpenATP. The total number of checked base-pairs are 28±8 and 4±2 for MutSADP and MutSOpenATP, respectively. These results indicated that the mismatch search by MutSOpenATP is inefficient in terms of the mismatch checking though MutSOpenATP diffuses along DNA and reaches unchecked regions more rapidly than MutSADP. The simulations indicated that, as has been suggested for other DNA binding proteins [43], the searching MutS may repeatedly change its conformation between the search (MutSOpenATP) and the recognition (MutSADP) mode.
Simulations of MutS with an ATP-Bound Closed Clamp Conformation on DNA
The crystal structure of ATP bound MutS not associating with DNA but with MutL was previously solved [11]. In that structure, two clamp domains from each monomer are proximal to each other (MutSClosedATP). Previously, the difference in the sliding dynamics between MutSADP and MutSClosedATP was suggested based on the single-molecule experimental results. It was proposed that diffusion of MutSClosedATP along DNA is not tightly coupled to rotation around it [18]. Therefore, we performed CGMD simulations of MutSOpenATP on 75 bp DNA with a GC repeat sequence in the presence of 100 mM ions. To make the comparison with other setups easy, we omitted MutL from the system. The simulation movies and trajectories showed bidirectional movements of MutSClosedATP along DNA, which are indicative of diffusion (Figure 9AB, Supplementary Figure S26, and Supplementary Movie S3). To confirm that these bidirectional movements are simple diffusion, we calculated MSDs from the trajectories and plotted them against MD step intervals (Figure 9C). The plot showed linear lines, supporting that the MutS movements along DNA are diffusion. We also calculated the slopes of the linear lines (diffusion coefficients) and compared them with those of MutSADP and MutSClosedATP (Figure 9D). The result suggested that the MutSClosedATP diffusion (6.6×10–6 bp2/step) is significantly and moderately faster than MutSADP (2.8×10–6 bp2/step) and MutSOpenATP (4.8×10–6 bp2/step), respectively. To investigate the main factors that cause the difference, we calculated the average numbers of residues that contact with DNA (minimum distance is less than 10Å) regardless of the residue types, and they are 84±14, 70±14, and 59±11 for MutSADP, MutSOpenATP, and MutSClosedATP, respectively. Therefore, as the number of DNA contacting residues (so as the excluded volume effect) increases, the diffusion rate decreases. On the other hand, we also calculated the average numbers of basic residues that contact with DNA, and they are 14±3, 14±2, and 14±2 for MutSADP, MutSOpenATP, and MutSClosedATP, respectively. In sum, we attributed the diffusion rate difference mainly to the excluded volume difference.

Next, we sought to investigate the ion concentration dependency of the sliding dynamics of MutSClosedATP. Therefore, we performed additional CGMD simulations of MutSClosedATP on 75 bp DNA with a GC repeat sequence in the presence of 50, 150, 500 mM ions. We also performed simulations in which only excluded volume interactions (no electrostatic interactions) were applied for interactions between MutS and DNA. The simulation trajectories showed that MutSClosedATP can hold DNA even without electrostatic interactions since MutS wraps around DNA with the lever and clamp domains. In all the setups, we observed bidirectional movements of MutSClosedATP along DNA, which are indicative of diffusion (Supplementary Figure S27 to S30). We plotted MSDs against MD step intervals, and the plot showed linear lines regardless of ion concentrations (Figure 9C), confirming that the MutSClosedATP movements along DNA are diffusion irrespective of the strength of the electrostatic interactions. We calculated diffusion coefficients and found that those do not significantly depends on ion concentration in the investigated (50 to 500 mM) concentration range contrary to the previous experimental result (Figure 9E) [18]. In the experiment, the protein conformation may change spontaneously (to MutSOpenATP) or upon ATP hydrolysis the rate of which may depend on ion concentration. In each ion concentration, the diffusion coefficient of MutSClosedATP is higher than that of MutSADP, consistent with the experimental result [18]. When there were no electrostatic interactions between MutS and DNA, the diffusion coefficient was significantly increased. Therefore, the MutSClosedATP diffusion was modulated by electrostatic interactions.
To clarify if the diffusion we observed in the simulations is coupled to MutSCloseATP rotations, we plotted the time trajectories of MutS positions against cumulative angle changes around the DNA long axis (Figure 10A and Supplementary Figure S31). The angle change was calculated as above. In the 100 mM ion case, we observed a few linear lines with occasional transitions to neighboring lines. The pitch was consistent with that of the DNA grooves. The simulation results suggested that MutSClosedATP diffuses along a DNA groove with frequent groove-crossing events in the presence of 100 mM ions. This is contrary to the simulation results of MutSADP in which MutS diffuses with rare groove-crossing events in the presence of 100 mM ions. The different frequencies of groove-crossing events can explain the altered diffusion rates between MutSClosedATP and MutSADP. From the simulation trajectories, we identified the DNA contacting residues (the minimum distance to DNA is less than 10Å in >90% of the simulation frames): Residues 350, 354, 359 to 364, and 416 to 418 in the lever domain. The R354 in MutS corresponds to K393 in human Msh2, mutation of which causes Hereditary Non-Polyposis Colorectal Cancer [5]. The positively charged residue at this position conserves from E. coli to humans and most likely is functionally important.

Next, we sought to investigate ion concentration dependency of the degree of coupling between diffusion along and rotation around DNA. Therefore, we plotted the time trajectories of MutSCloseATP positions against cumulative angle changes for the 50, 150, and 500 mM ion cases and the case without electrostatic interactions between MutS and DNA (Figure 10B and Supplementary Figure S32 to S35). In all the setups, we observed a few linear lines with frequent fluctuations, suggesting that MutSClosedATP diffuses along a groove with frequent groove-crossing events regardless of the ion concentration. The simulation results suggested that the MutSOpenATP diffuses along a groove with frequent groove-crossing events in a broad range of ion concentrations. When there are no electrostatic interactions between MutS and DNA, the linear lines were hardly observed, suggesting that the diffusion was uncoupled to the protein rotation around DNA. Therefore, the simulation results suggested that the rotation-coupled diffusion is caused by electrostatic interactions.
To quantitatively visualize groove-crossing events and their frequency, we plotted time trajectories of MutS positions on the axis orthogonal to linear lines with the slope of 10 bp/2π in the position vs cumulative angle change plots as above (Figure 10C and Supplementary Figure S36 to S40). We observed linear horizontal lines with frequent height changes in a broad range of the ion concentration (50 to 500 mM), suggesting the rotation-coupled diffusion with the pitch of 2π/10 bp with frequent groove-crossing events. The horizontal line was much shorter in the case of no electrostatic interactions between MutS and DNA, suggesting again that the coupling is caused by electrostatic interactions. To quantify the frequency, we calculated and plotted probabilities of diffusing along a groove without groove-crossing against duration (Figure 10D). In the 50 mM, 100 mM, and 150 mM ion cases, the survival probabilities decreased sharply as the duration increased from 0 to around 8×105 steps. The slope was significantly lower in the range from around 8×105 to 5×106 steps. We attributed the initial sharp decrease to the rapid back and forth groove-crossing observed in the time trajectories (Figure 10AB and Supplementary Figure S31 to S35). The second low slope phase confirmed that groove-crossing events frequently took place in this ion concentration range. In the 500 mM ion case, the survival probabilities decreased more rapidly as the duration increased from 8×105 to 5×106 steps. In the no electrostatics case, on the other hand, the survival probabilities decreased to ~0.01 from 8×105 to 4×106 steps, suggesting that most of the groove-crossing events took place in this time range. The linear decrease indicated that the “one-way” groove-crossing events took place as a Poisson process of a constant rate. To quantitatively compare the frequency of the one-way groove-crossing events, we calculated the rate constants from the second phases and plotted them in Figure 10E. In the 50 mM, 100 mM, and 150 mM ion case, we found that the groove-crossing events of MutSClosedATP were more frequent than MutSADP. In the 500 mM ion case, the frequency difference between MutSClosedATP and MutSADP was within error. When there were no electrostatic interactions between MutS and DNA, the frequency of MutSClosedATP groove-crossing was significantly higher than that of MutSADP. Together, the simulation results suggested that MutSClosedATP is more likely to cross a groove edge while diffusing in a rotation-coupled manner and the frequency of the groove-crossing events is dictated by the strength of electrostatic interactions between MutS and DNA.
Discussion and Conclusion
The single-molecule experiment has shown that MutS searches a base-pair mismatch while diffusing along DNA in a rotation-coupled manner [18]. Due to the low resolution of the experiments, however, the structural dynamics details of the search process remain unknown. In this study, we performed CGMD simulations of MutS moving along DNA using previously solved conformations (MutSADP, MutSOpenATP, and MutSClosedATP) as initial structures. In the simulations, MutSADP, MutSOpenATP, and MutSClosedATP diffuses along DNA irrespective of the ion concentration. MutSADP and MutSClosedATP diffuse along DNA in a rotation-coupled manner and experience rare and frequent groove-crossing events, respectively. The groove-crossing events have never been indicated in any of the previous experiments. Therefore, the simulations revealed the unprecedented structural dynamics details of the MutS searching along DNA. It was also indicated that the mismatch search by MutSOpenATP is inefficient in terms of mismatch checking even though MutSOpenATP diffuses along DNA and reaches unchecked regions more rapidly than MutSADP.
In the experiment [18], it was suggested that the protein diffuses without microscopic DNA dissociation and reassociation because the diffusion coefficients do not depend on ion concentration. Also, the rotation-coupled diffusion was suggested from the distribution of fluorescent polarization. On the other hand, the simulations here suggested that the search mechanism depends on ion concentration: The frequency of the groove-crossing events is high and low at high and low ion concentration, respectively. The ion concentration-dependent mechanism alteration and the rare groove-crossing events have never been observed in the experiment due to its resolution and averaging. In the future, high-resolution experiments would validate these simulation results.
As for MutSClosedATP, the simulations showed that the diffusion coefficients do not significantly depend on ion concentration at least in the low ion concentration range (50 to 150 mM). The single-molecule experiment [18], on the other hand, has suggested that the diffusion coefficients increase ~1.5 fold as the ion concentration increases from 50 to 150 mM, supporting that MutS repeats the microscopic DNA dissociation and reassociation while diffusing. This discrepancy may be attributed to two reasons. First, the timescale of the simulations may be too short for MutS to dissociate from DNA. The coarse-graining for a simulation timescale boost is not enough for the experimental minute timescale though the simulations can provide fine details of micro-second timescale events. Second, the MutS conformation was restrained to ATP-bound closed clamp one in the simulations. In the experiments, conformational changes associated with ATP hydrolysis cycles can take place. The conformational changes may alter the diffusion mechanism and so the ion concentration dependency of the diffusion coefficients. Future studies should address the nucleotide state dependent MutS conformations while it diffuses along DNA in the presence of ATP.
When the rotation of DNA around the DNA long axis was prevented in the simulations, the diffusion coefficient of ADP-bound MutS was dramatically decreased. This result is reasonable because ADP-bound MutS should relatively rotate around DNA while diffusing, though the DNA molecule more easily rotates around its long axis than MutS due to the less hydrodynamic friction. This reasoning can be applied to any proteins that diffuse along DNA in a rotation-coupled manner including human MutS homolog, Msh2-Msh6. It was reported that base-pair mismatches on a lagging strand are more efficiently corrected than those on a leading strand during human genome replication. The lagging strand is replicated in a form of 100 to 300 bp Okazaki fragments, and the short length may facilitate the fragment rotation around its long axis and hence the search by Msh2-Msh6. Further experiments are required to prove the causal relationship. Notably, the previous CGMD simulation study of the eukaryotic MutS homolog protein, Msh2-Msh6, suggested that the sliding along DNA in the mismatch recognition conformation is slow and conformational change must be assumed to facilitate diffusion [21]. In that work, however, DNA rotation along its long axis was not modeled. The DNA rotation may facilitate Msh2-Msh6 diffusion.
In this study, we used the CG model to perform simulations of MutS sliding along DNA. The coarse-graining increased the calculation speed with some compromise on accuracy compared with all-atom models. Our CG model neglected the protein-DNA hydrophobic interactions, based on the assumption that the electrostatic interaction was dominant in the protein-DNA interaction. However, the simulations could be more accurate if we introduced hydrophobic interactions into the CG model. The fine detail of the MutS sliding mechanism might depend on excluded volume interactions between amino acid residues and nucleotides. Currently, all the CG beads representing every amino acid residue share the same radius, which can be refined in the future. Also, the simulations of MutSClosedATP did not include MutL proteins. The effects of MutL on the sliding dynamics should be addressed in the future. The simulations of MutSOpenATP in various ion concentrations are one of the future directions. Aside from these limitations, the current study elucidated unprecedented structural dynamics details of bacterial MutS sliding along DNA.


 ), ATP-bound open clamp (
), ATP-bound open clamp ( ), and ATP-bound closed clamp (
), and ATP-bound closed clamp (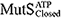 ) conformations. In the simulations, we observed conformation-dependent diffusion of MutS along DNA.
) conformations. In the simulations, we observed conformation-dependent diffusion of MutS along DNA.