
Volume 19
Displaying 1-48 of 48 articles from this issue
- |<
- <
- 1
- >
- >|
Review Article (Invited)
-
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190001
Published: 2022
Released on J-STAGE: February 22, 2022
Advance online publication: February 08, 2022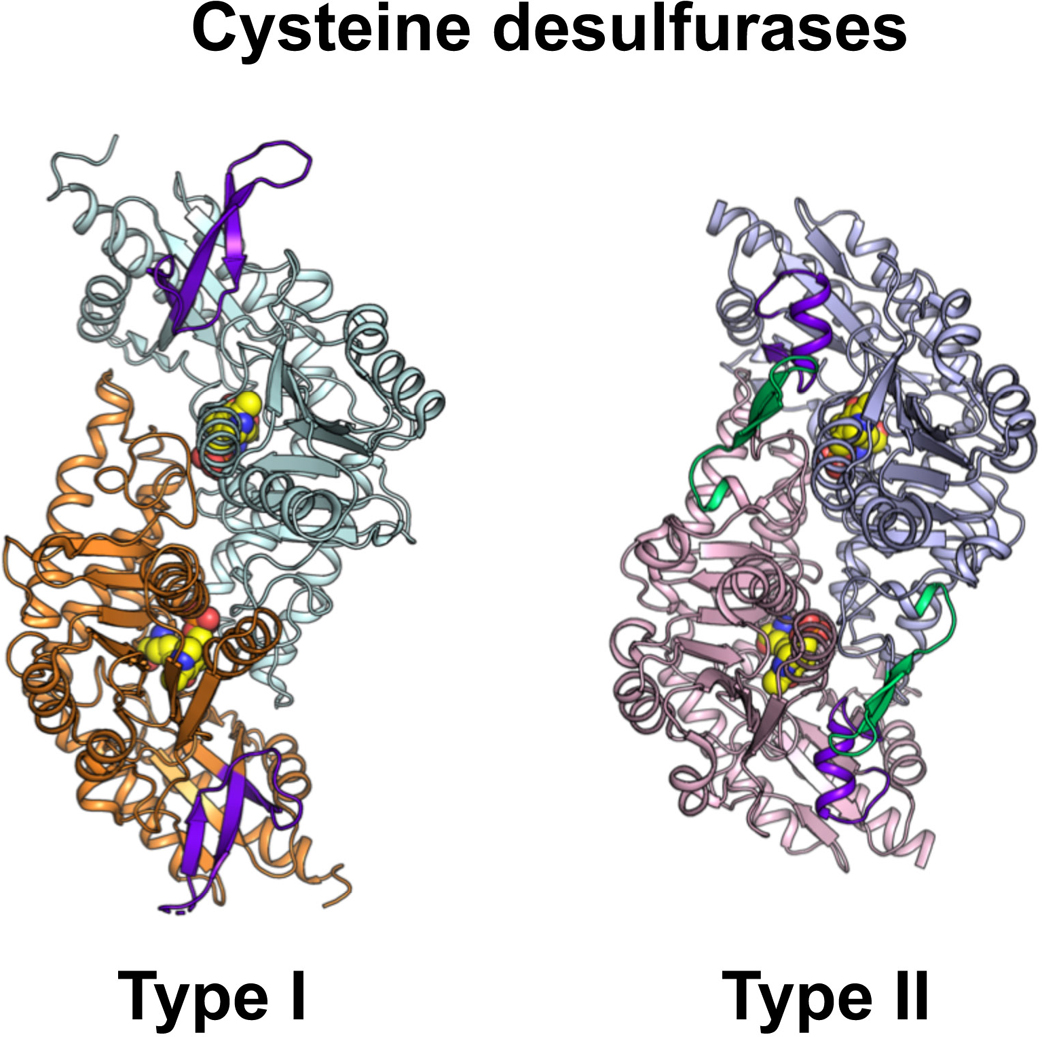 Download PDF (2450K) Full view HTML
Download PDF (2450K) Full view HTML
Regular Article
-
Article type: Regular Article
2022Volume 19 Article ID: e190002
Published: 2022
Released on J-STAGE: February 22, 2022
Advance online publication: February 08, 2022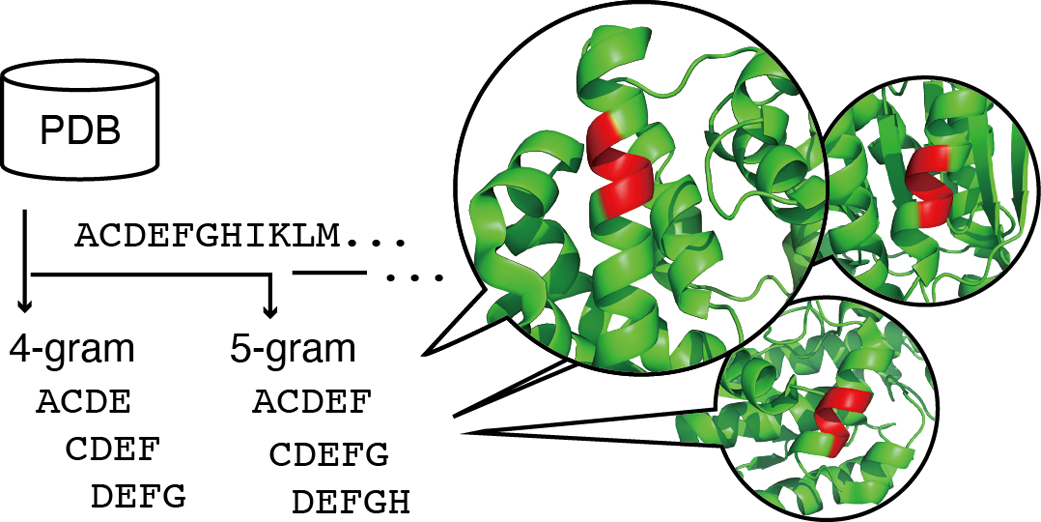 Download PDF (1846K) Full view HTML
Download PDF (1846K) Full view HTML -
Article type: Regular Article
2022Volume 19 Article ID: e190003
Published: 2022
Released on J-STAGE: February 26, 2022
Advance online publication: February 09, 2022 Download PDF (1081K) Full view HTML
Download PDF (1081K) Full view HTML
Commentary and Perspective
-
Article type: Commentary and Perspective
2022Volume 19 Article ID: e190004
Published: 2022
Released on J-STAGE: February 26, 2022
Advance online publication: February 09, 2022Download PDF (508K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190005
Published: 2022
Released on J-STAGE: February 26, 2022
Advance online publication: February 15, 2022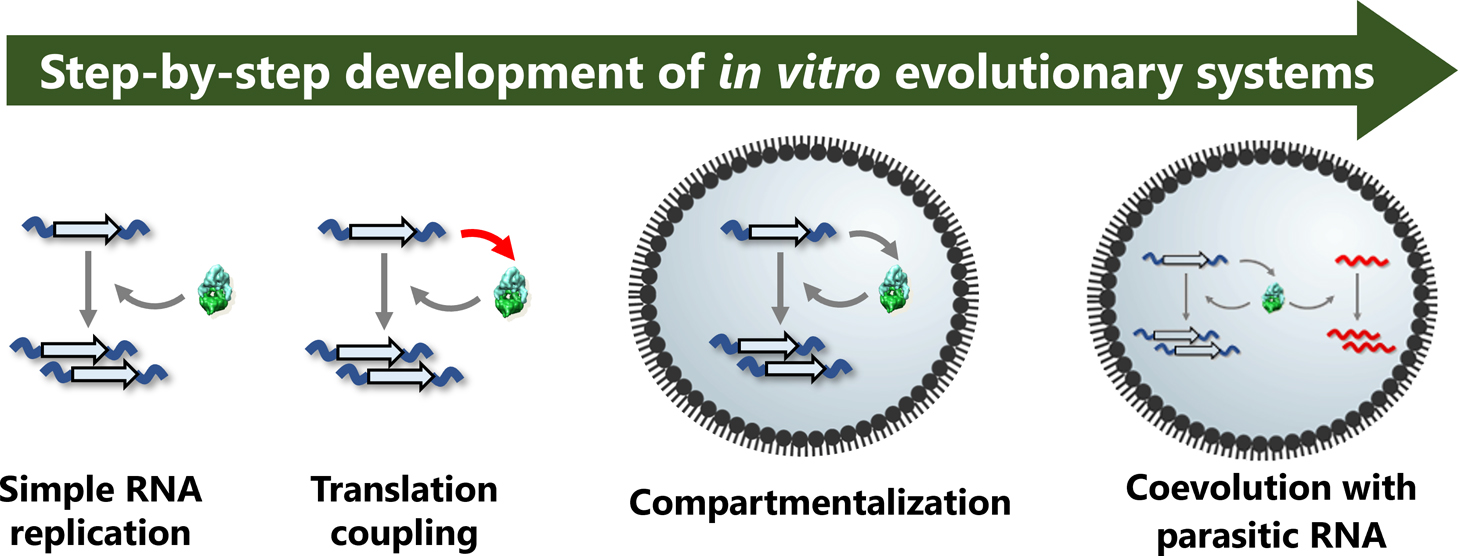 Download PDF (1923K) Full view HTML
Download PDF (1923K) Full view HTML -
Quantitative and kinetic single-molecule analysis of DNA unwinding by Escherichia coli UvrD helicaseArticle type: Review Article (Invited)
2022Volume 19 Article ID: e190006
Published: 2022
Released on J-STAGE: March 25, 2022
Advance online publication: March 10, 2022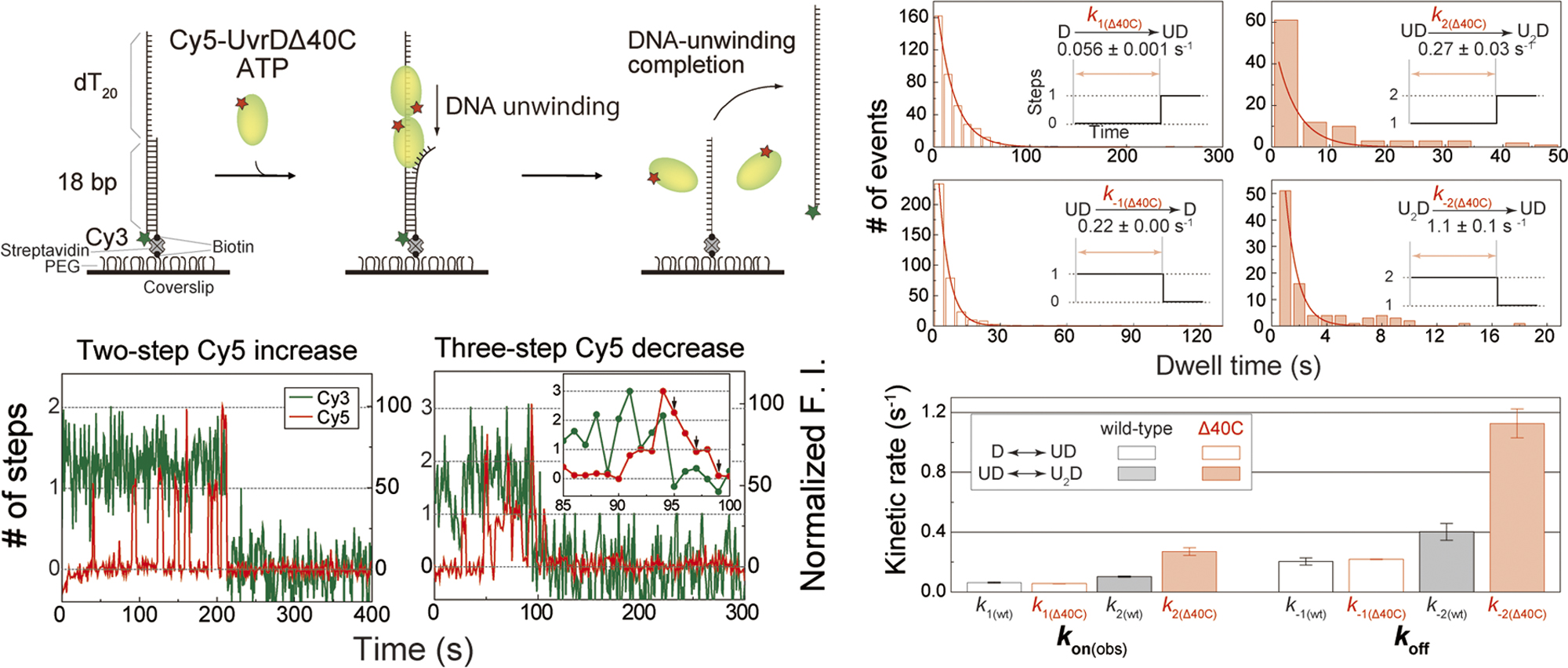 Download PDF (2589K) Full view HTML
Download PDF (2589K) Full view HTML -
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190007
Published: 2022
Released on J-STAGE: March 26, 2022
Advance online publication: March 11, 2022 Download PDF (1524K) Full view HTML
Download PDF (1524K) Full view HTML
Regular Article
-
Article type: Regular Article
2022Volume 19 Article ID: e190008
Published: 2022
Released on J-STAGE: April 14, 2022
Advance online publication: March 30, 2022 Download PDF (2704K) Full view HTML
Download PDF (2704K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190009
Published: 2022
Released on J-STAGE: April 16, 2022
Advance online publication: April 01, 2022 Download PDF (1324K) Full view HTML
Download PDF (1324K) Full view HTML -
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190010
Published: 2022
Released on J-STAGE: April 20, 2022
Advance online publication: April 02, 2022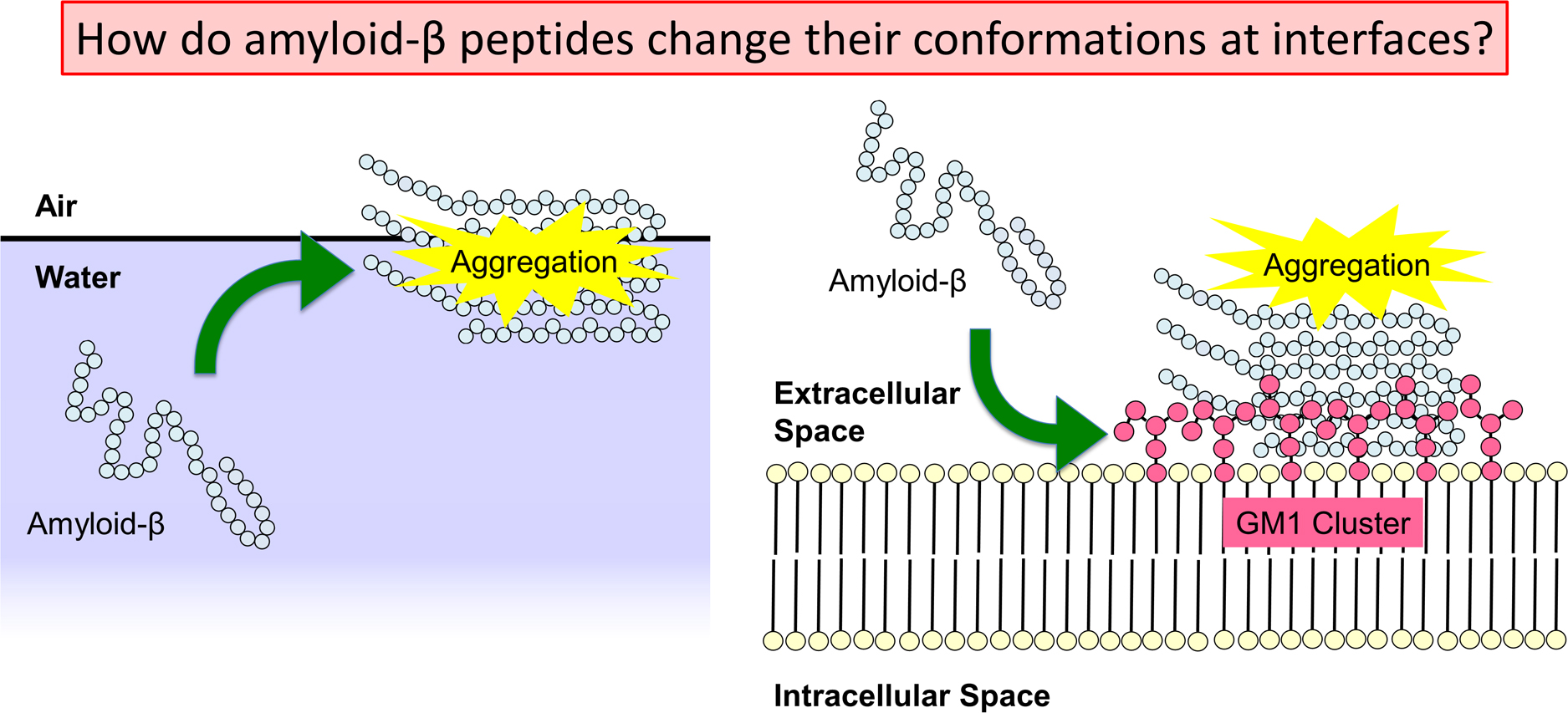 Download PDF (4393K) Full view HTML
Download PDF (4393K) Full view HTML -
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190011
Published: 2022
Released on J-STAGE: April 20, 2022
Advance online publication: April 05, 2022 Download PDF (3086K) Full view HTML
Download PDF (3086K) Full view HTML
Regular Article
-
Article type: Regular Article
2022Volume 19 Article ID: e190012
Published: 2022
Released on J-STAGE: April 20, 2022
Advance online publication: April 05, 2022 Download PDF (2411K) Full view HTML
Download PDF (2411K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190013
Published: 2022
Released on J-STAGE: April 28, 2022
Advance online publication: April 08, 2022 Download PDF (4110K) Full view HTML
Download PDF (4110K) Full view HTML
Review Article
-
Article type: Review Article
2022Volume 19 Article ID: e190014
Published: 2022
Released on J-STAGE: April 28, 2022
Advance online publication: April 08, 2022 Download PDF (860K) Full view HTML
Download PDF (860K) Full view HTML
Regular Article
-
Article type: Regular Article
2022Volume 19 Article ID: e190015
Published: 2022
Released on J-STAGE: May 11, 2022
Advance online publication: April 14, 2022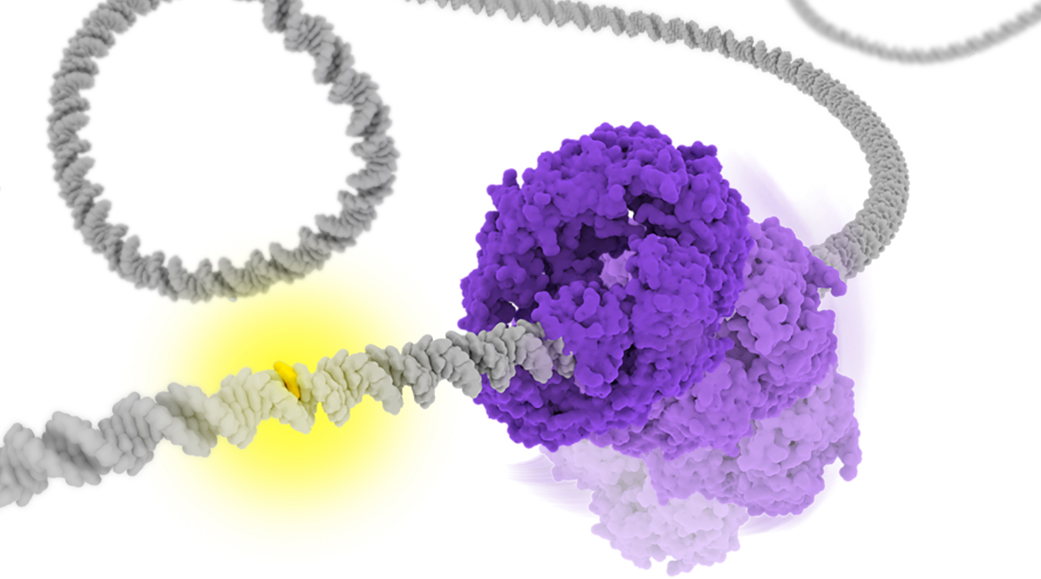 Download PDF (3107K) Full view HTML
Download PDF (3107K) Full view HTML -
Article type: Regular Article
2022Volume 19 Article ID: e190016
Published: 2022
Released on J-STAGE: May 18, 2022
Advance online publication: April 15, 2022 Download PDF (1861K) Full view HTML
Download PDF (1861K) Full view HTML
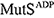 ), ATP-bound open clamp (
), ATP-bound open clamp (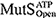 ), and ATP-bound closed clamp (
), and ATP-bound closed clamp (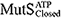 ) conformations. In the simulations, we observed conformation-dependent diffusion of MutS along DNA.
) conformations. In the simulations, we observed conformation-dependent diffusion of MutS along DNA. Review Article (Invited)
-
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190017
Published: 2022
Released on J-STAGE: June 01, 2022
Advance online publication: May 10, 2022 Download PDF (2106K) Full view HTML
Download PDF (2106K) Full view HTML
Regular Article
-
Article type: Regular Article
2022Volume 19 Article ID: e190018
Published: 2022
Released on J-STAGE: June 01, 2022
Advance online publication: May 10, 2022 Download PDF (1697K) Full view HTML
Download PDF (1697K) Full view HTML -
Article type: Regular Article
2022Volume 19 Article ID: e190019
Published: 2022
Released on J-STAGE: June 01, 2022
Advance online publication: May 12, 2022 Download PDF (3384K) Full view HTML
Download PDF (3384K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190020
Published: 2022
Released on J-STAGE: June 03, 2022
Advance online publication: May 12, 2022 Download PDF (1803K) Full view HTML
Download PDF (1803K) Full view HTML -
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190021
Published: 2022
Released on J-STAGE: June 21, 2022
Advance online publication: June 01, 2022 Download PDF (6291K) Full view HTML
Download PDF (6291K) Full view HTML -
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190022
Published: 2022
Released on J-STAGE: June 30, 2022
Advance online publication: June 07, 2022 Download PDF (3477K) Full view HTML
Download PDF (3477K) Full view HTML
Commentary and Perspective
-
Article type: Commentary and Perspective
2022Volume 19 Article ID: e190023
Published: 2022
Released on J-STAGE: July 29, 2022
Advance online publication: July 14, 2022Download PDF (2224K) Full view HTML -
Article type: Commentary and Perspective
2022Volume 19 Article ID: e190024
Published: 2022
Released on J-STAGE: August 09, 2022
Advance online publication: July 27, 2022Download PDF (384K) Full view HTML
Note
-
Article type: Note
2022Volume 19 Article ID: e190025
Published: 2022
Released on J-STAGE: August 20, 2022
Advance online publication: July 28, 2022 Download PDF (1511K) Full view HTML
Download PDF (1511K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190026
Published: 2022
Released on J-STAGE: August 26, 2022
Advance online publication: August 09, 2022 Download PDF (1285K) Full view HTML
Download PDF (1285K) Full view HTML -
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190027
Published: 2022
Released on J-STAGE: September 08, 2022
Advance online publication: August 23, 2022 Download PDF (8158K) Full view HTML
Download PDF (8158K) Full view HTML
Commentary and Perspective
-
Article type: Commentary and Perspective
2022Volume 19 Article ID: e190028
Published: 2022
Released on J-STAGE: September 08, 2022
Advance online publication: August 24, 2022Download PDF (465K) Full view HTML -
Article type: Commentary and Perspective
2022Volume 19 Article ID: e190029
Published: 2022
Released on J-STAGE: September 08, 2022
Advance online publication: August 27, 2022Download PDF (613K) Full view HTML -
Article type: Commentary and Perspective
2022Volume 19 Article ID: e190030
Published: 2022
Released on J-STAGE: September 13, 2022
Advance online publication: August 27, 2022Download PDF (767K) Full view HTML -
Article type: Commentary and Perspective
2022Volume 19 Article ID: e190031
Published: 2022
Released on J-STAGE: September 13, 2022
Advance online publication: August 27, 2022Download PDF (554K) Full view HTML
Review Article
-
Article type: Review Article
2022Volume 19 Article ID: e190032
Published: 2022
Released on J-STAGE: September 14, 2022
Advance online publication: August 30, 2022 Download PDF (1552K) Full view HTML
Download PDF (1552K) Full view HTML
Commentary and Perspective
-
Article type: Commentary and Perspective
2022Volume 19 Article ID: e190033
Published: 2022
Released on J-STAGE: September 14, 2022
Advance online publication: August 30, 2022Download PDF (391K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190034
Published: 2022
Released on J-STAGE: September 29, 2022
Advance online publication: September 08, 2022 Download PDF (3088K) Full view HTML
Download PDF (3088K) Full view HTML
Commentary and Perspective
-
Article type: Commentary and Perspective
2022Volume 19 Article ID: e190035
Published: 2022
Released on J-STAGE: September 29, 2022
Advance online publication: September 08, 2022Download PDF (400K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190036
Published: 2022
Released on J-STAGE: October 05, 2022
Advance online publication: September 13, 2022 Download PDF (1729K) Full view HTML
Download PDF (1729K) Full view HTML
Commentary and Perspective
-
Article type: Commentary and Perspective
2022Volume 19 Article ID: e190037
Published: 2022
Released on J-STAGE: October 05, 2022
Advance online publication: September 17, 2022Download PDF (392K) Full view HTML
Editorial
-
Article type: Editorial
2022Volume 19 Article ID: e190038
Published: 2022
Released on J-STAGE: October 05, 2022
Advance online publication: September 21, 2022Download PDF (385K) Full view HTML
Commentary and Perspective
-
Article type: Commentary and Perspective
2022Volume 19 Article ID: e190039
Published: 2022
Released on J-STAGE: October 05, 2022
Advance online publication: September 22, 2022Download PDF (399K) Full view HTML
Regular Article
-
Article type: Regular Article
2022Volume 19 Article ID: e190040
Published: 2022
Released on J-STAGE: October 07, 2022
Advance online publication: September 22, 2022 Download PDF (1676K) Full view HTML
Download PDF (1676K) Full view HTML -
Article type: Regular Article
2022Volume 19 Article ID: e190041
Published: 2022
Released on J-STAGE: October 07, 2022
Advance online publication: September 22, 2022 Download PDF (3223K) Full view HTML
Download PDF (3223K) Full view HTML
Editorial
-
Article type: Editorial
2022Volume 19 Article ID: e190042
Published: 2022
Released on J-STAGE: October 19, 2022
Advance online publication: October 13, 2022Download PDF (319K) Full view HTML
Commentary and Perspective
-
Article type: Commentary and Perspective
2022Volume 19 Article ID: e190043
Published: 2022
Released on J-STAGE: November 02, 2022
Advance online publication: October 20, 2022Download PDF (706K) Full view HTML -
Article type: Commentary and Perspective
2022Volume 19 Article ID: e190044
Published: 2022
Released on J-STAGE: November 29, 2022
Advance online publication: November 16, 2022Download PDF (385K) Full view HTML
Regular Article
-
Article type: Regular Article
2022Volume 19 Article ID: e190045
Published: 2022
Released on J-STAGE: December 07, 2022
Advance online publication: November 19, 2022 Download PDF (2651K) Full view HTML
Download PDF (2651K) Full view HTML
Review Article (Invited)
-
Article type: Review Article (Invited)
2022Volume 19 Article ID: e190046
Published: 2022
Released on J-STAGE: December 07, 2022
Advance online publication: November 19, 2022 Download PDF (2473K) Full view HTML
Download PDF (2473K) Full view HTML
Commentary and Perspective
-
Article type: Commentary and Perspective
2022Volume 19 Article ID: e190047
Published: 2022
Released on J-STAGE: December 14, 2022
Advance online publication: November 25, 2022Download PDF (483K) Full view HTML
Review Article (Invited)
-
Intestinal and optic-cup organoids as tools for unveiling mechanics of self-organizing morphogenesisArticle type: Review Article (Invited)
2022Volume 19 Article ID: e190048
Published: 2022
Released on J-STAGE: January 12, 2023
Advance online publication: December 21, 2022 Download PDF (1058K) Full view HTML
Download PDF (1058K) Full view HTML
- |<
- <
- 1
- >
- >|