Abstract
trans-Fatty acids (TFAs) are food-derived fatty acids that possess one or more trans double bonds between carbon atoms. Compelling epidemiological and clinical evidence has demonstrated the association of TFA consumption with various diseases, such as cardiovascular diseases, and neurodegenerative diseases. However, the underlying etiology is poorly understood since the mechanisms of action of TFAs remain to be clarified. Previous studies have shown that single treatment with TFAs induce inflammation and cell death, but to a much lesser extent than saturated fatty acids (SFAs) that are well established as a risk factor for diseases linked with inflammation and cell death, which cannot explain the particularly higher association of TFAs with atherosclerosis than SFAs. In our series of studies, we have established the role of TFAs as an enhancer of inflammation and cell death. We found that pretreatment with TFAs strongly promoted apoptosis induced by either extracellular ATP, one of the damage-associated molecular patterns (DAMPs) leaked from damaged cells, or DNA damaging-agents, including doxorubicin and cisplatin, thorough enhancing activation of the stress-responsive mitogen-activated protein (MAP) kinase p38/c-jun N-terminal kinase (JNK) pathways; pretreatment with SFAs or cis isomers of TFAs had only minor or no effect, suggesting the uniqueness of the pro-apoptotic role of TFAs among fatty acids. Our findings will provide an insight into understanding of the pathogenesis mechanisms, and open up a new avenue for developing prevention strategies and therapies for TFA-related diseases.
1. INTRODUCTION
Fatty acids are carboxylic acids with a variety of lengths of hydrocarbon chains, which are categorized into saturated fatty acids (SFAs) or unsaturated fatty acids (UFAs), containing no or at least one double bond between carbon atoms, respectively. Among UFAs, those that harbor one or more carbon-carbon double bonds in trans configuration are called trans-fatty acids (TFAs) (Fig. 1). In the human body, only cis double bonds are enzymatically produced during UFA synthesis, and the resulting products, henceforth referred to as cis-fatty acids (CFAs), play key roles as a component of cellular membranes, an energy source, and a regulator of various biological functions.1) TFAs are therefore not produced in humans, and taken into the body via food consumption, which are classified into two types according to their origin: the industrial TFAs (iTFAs) and the ruminant TFAs (rTFAs)2) (Fig. 1). iTFAs, such as elaidic acid (EA, C18 : 1 t9) and linoelaidic acid (LEA, C18 : 2 t9, t12), are produced in the process of food manufacturing, mainly through partial hydrogenation and deodorization of edible oils that contain CFAs, and are present in a variety of processed foods including bakery products, snacks and fast foods; on the other hand, rTFAs, such as trans-vaccenic acid (TVA, C18 : 1 t11) and rumenic acid (TVA, C18 : 2 c9, t11), are produced by microbial biohydrogenation of CFAs in the rumen of cows and sheep. and contained in dairy products and meat.1,2) Accumulating epidemiological studies have shown that consumption of TFAs, particularly iTFAs, is associated with various diseases, such as metabolic syndrome, neurodegenerative diseases (NDs), and cardiovascular diseases (CVDs).3–6) However, the molecular mechanisms underlying TFA-related diseases remain unknown. In this review, I summarize the recent progress in dissecting the mechanisms of actions of TFAs that can account for their etiology.
2. DELETERIOUS EFFECTS OF TFAS ON HUMAN HEALTH
Potentially harmful effects of TFAs were first proposed by Kummerow and colleagues in 1950s, who demonstrated that TFAs were contained between 2 to 15% in various tissues from 24 human subjects who died of heart disease.7) Another group showed an evidence that the concentration of TFAs (particularly C18 : 1 and C16 : 1) was significantly higher in the adipose tissue of people who died of ischemic heart disease than in those who died of other causes.8,9) In early 1990s, much attention was focused on TFAs, after large cohort studies and intervention trials suggested that TFA intake increases the risk of CVDs accompanied with increased low-density lipoprotein cholesterol (LDL-C) levels and decreased high-density lipoprotein cholesterol (HDL-C) levels in blood.10,11) In 2003, the joint expert committee of WHO and Food and Agriculture Organization of the United Nations (FAO) recommended that the daily intake of TFAs should be less than 1% of total energy.12) Based on this recommendation, various laws and regulations have been introduced to limit TFA consumption in many countries. Notably, in New York State counties, after restrictions for the use of iTFAs in eateries from 2007, the incidence of myocardial infarction and the mortality rate from CVDs were reduced by 7.8 and 4.5%, respectively.13,14)
It has been widely believed that TFAs are dispensable for humans as they are not synthesized in the body, and that’s one of the reasons why strong efforts have been made to regulate and reduce the content of TFAs in foods. Another major reason is that prospective cohort studies and intervention trials have shown that the intake of TFAs increases the risk of CVDs more than that of SFAs, which have been also established as a risk factor for CVDs15); while SFA intake upregulated plasma LDL-C levels without affecting HDL-C levels, TFA intake caused both upregulation of LDL-C levels and downregulation of HDL-C levels, resulting in a greater increase in the LDL/HDL ratio and the risk of atherosclerosis.4) Supporting this notion, evidence from mouse models of atherosclerosis also showed that TFA intake caused more severe atherosclerosis than SFA intake.16–18) Several mechanisms have been proposed which can account for these adverse effects of TFAs on plasma lipoprotein regulation. Previous clinical studies demonstrated that TFA-rich diet intake upregulated apolipoprotein B-100 (a major constituent protein of LDL) and downregulated apolipoprotein A-I (a major component of HDL),19) and that it promoted activation of cholesterol ester transfer protein transferring cholesteryl ester from HDL to very LDL (VLDL) and LDL,20,21) which may in turn contributed to the increase in LDL/HDL ratio. Interestingly, it was previously reported that treatment of EA with human hepatoma cell lines, but not that of OA, induced expression of sterol regulatory element-binding protein (SREBP) target genes that encode cholesterogenic enzymes, such as 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase, squalene epoxidase and mevalonate kinase, and promoted de novo synthesis of fatty acid and cholesterol.22,23) However, although TFA intake consistently exacerbated atherosclerosis across human and mouse model studies, plasma lipoprotein levels were not always altered by TFA intake,18,24) which implicates that unknown TFA-specific biological effects other than dysregulation of plasma lipoprotein levels also contribute to the exacerbation of atherosclerosis.
There is also epidemiological and clinical evidence linking TFA consumption to a wide variety of disorders other than CVDs, including systemic inflammation, cognitive disorders, Alzheimer’s disease, diabetes, obesity, non-alcoholic steatohepatitis (NASH) and cancer, which were supported by experimental evidence from animal model studies.6,25) However, these relationships have not been determined yet, except for that of TFA intake with CVDs, due to insufficient evidence and inconsistency of the results between studies.
3. PREVIOUS REPORTS ON STRESS RESPONSES, INFLAMMATION AND CELL DEATH, INDUCED BY TFAS
Cells are constantly exposed to a wide variety of exogenous and endogenous stresses, including UV, oxidants, chemicals, cytokines and pathogens. When exposed to such stresses, cells sense their types, strength and duration, and then induce appropriate cellular responses, such as inflammation and cell death, to maintain homeostasis.26,27) Excessive activation of inflammation and cell death, due to either their dysregulation or cumulative stress, plays a deleterious role in numerous diseases such as CVDs, NDs, and metabolic syndrome,26,27) which are all associated with TFA consumption. Indeed, mounting evidence has shown a close link of TFA with inflammation and cell death. Epidemiological and mouse model studies have demonstrated a positive correlation between TFA intake and plasma levels of a systemic inflammation marker C-reactive protein, and inflammatory cytokines including interleukin-6 (IL-6) and tumor-necrosis factor-α (TNF-α).24,28) It has been shown that in human vascular endothelial cell lines and 3T3-L1 adipocytes, TFA treatment upregulates expression of IL-6 and TNF-α possibly through nuclear factor-κB (NF-κB) activation, although the underlying molecular mechanism has not been elucidated yet. However, pro-inflammatory function of TFAs is controversial since there are reports that show TFAs do not induce inflammatory cytokines.29,30) Moreover, TFAs promote inflammation much less efficiently than SFAs.3)
Reactive oxygen species (ROS) are produced in various stress conditions, promote oxidation of lipids, DNA and proteins, and thereby induce inflammation and cell death, leading to progression of diseases including CVDs and NDs.31–33) Meanwhile, ROS also plays a key role as a second messenger in diverse signaling pathways related to inflammation and cell death by regulating molecular modifications and functions,34–36) and thus dysregulation of ROS-dependent signaling contributes to development of various diseases as well.37) A number of in vivo and in vitro studies showed that TFA treatment induces ROS generation.24) In atherosclerotic lesions in LDL receptor deficient mice fed with EA, ROS accumulated more abundantly than in those fed with oleic acid (OA, C18 : 1 c9) as a cis isomer of EA, accompanied with the increase in inflammation and areas of atherosclerotic plaque.18) TFA-induced ROS production was observed in several types of cells, such as rat vascular smooth muscle cells, human aortic endothelial cells and a human neuroblastoma cell line SH-SY5Y.28,38) Nevertheless, it is still unknown by what mechanism TFA treatment induces ROS generation in a direct manner.
Endoplasmic reticulum (ER) stress is triggered by protein unfolding, and notably, SFA accumulation, which initially induces an adaptive response, called as the unfolded protein responses (UPRs), in order to eliminate unfolded proteins and restore ER homeostasis; excessive accumulation of ER stress eventually induces inflammatory responses and cell death.39) A previous report showed that EA treatment causes cell death in SH-SY5Y cells, along with ROS production and ER stress response.38) However, there are controversial reports showing only a minor to no effect of EA on ER stress, and it remains totally unclear whether and how TFAs induce ER stress,40,41) while SFAs have a well-established role as a strong ER stress inducer.3)
4. SYNERGETIC EFFECT OF TFAS ON STRESS RESPONSES
To verify the previous findings, we first examined whether single treatment with EA induces cell death and inflammation using various types of cell lines. Unexpectedly, however, even though the cells were treated with high concentrations of EA (100–500 µM), no significant cell death or inflammatory cytokine induction was observed. We therefore assumed that EA itself does not serve as a potent pro-inflammatory factor or a cell death inducer, but might rather function as an enhancer of stress responses only when cell death and inflammation are initiated. To address this possibility, we conducted a comprehensive screen to identify any synergetic effect of EA on stress responses; a variety types of cells were pretreated with or without EA or OA, and then treated with various drugs that induce cell death and inflammation, in order to examine whether EA, but not OA, has any significant effect on cell viability and inflammatory cytokine induction. As a result, we found that EA dramatically promoted cell death induced by extracellular ATP (eATP), one of the damage-associated molecular patterns (DAMPs) leaked from damaged cells or tissues, and DNA damage. In the following sessions, I describe in detail and discuss about the molecular mechanisms underlying synergetic effects of TFAs on stress responses, particularly inflammation and cell death.
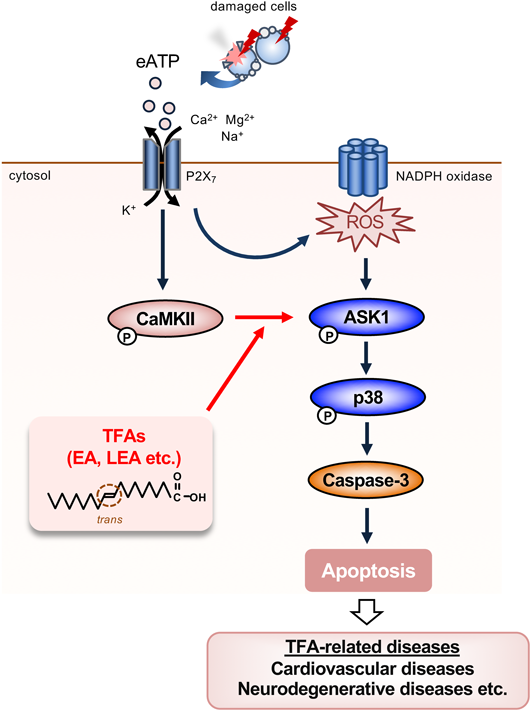
4.1. TFAs Promote eATP-Induced Apoptosis through Enhancing Activation of the ASK1-p38 Mitogen-Activated Protein (MAP) Kinase PathwayeATP is well known to serve as a potent inducer of inflammation and cell death by binding to P2X and P2Y purinergic receptor as a ligand.42) When ATP leaks out from the injured cells, it binds to the P2X purinoceptor 7 (P2X7) that acts as a ligand-gated cation channel, and subsequently induces nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activation downstream of a non-selective cation influx, resulting in ROS generation; ROS directly triggers activation of the stress responsive kinase apoptosis signal-regulating kinase 1 (ASK1), which in turn activates a stress-responsive MAP kinase p38, and ultimately apoptosis, a programmed cell death.43) We found that in the presence of EA, eATP-induced phosphorylation of ASK1 and p38 (a hallmark of their activation) and cell death was substantially enhanced, accompanied with elevation of caspase-3 activation and DNA ladder formation, common markers of apoptosis, in a mouse macrophage cell line RAW264.7.44) Whereas LEA, one of the most common iTFAs, strongly promoted eATP-induced apoptosis as did EA, OA and linoleic acid (LA, C18 : 2 c9 c12) as their respective cis isomers did not. Furthermore, the most typical SFA palmitic acid (PA, C16 : 0) scarcely promoted eATP-induced apoptosis. Importantly, calcium/calmodulin-dependent kinase II (CaMKII), an upstream kinase of ASK1,45) was identified to play a critical role in the pro-apoptotic function of TFAs. EA-mediated enhancement of ASK1/p38 phosphorylation and cell death was suppressed by a CaMKII inhibitor KN-93. Moreover, EA enhanced eATP-driven phosphorylation of CaMKII at Thr-286, which correlates with its activation. These results collectively suggest that TFAs, such as EA and LEA, strongly and specifically enhance eATP-induced activation of the ASK1-p38 MAP kinase pathway in a CaMKII-dependent manner, and thereby promote apoptosis (Fig. 2). P2X7 is highly expressed in macrophages localized at the atherosclerotic lesions, and the ATP-P2X7 signaling activation is implicated in the atherosclerosis progression.46) Therefore, TFAs may exacerbate atherosclerosis through enhancing macrophage apoptosis via the ATP-P2X7 signaling, and further promoting leakage of DAMPs from the atherosclerotic lesions (e.g. ATP) and inflammation (Figs. 2, 3).
The apoptosis signaling pathway via the P2X7-ROS-ASK1-p38 axis was originally identified in macrophages,43) but no report has determined its role in other types of cells until recently. Importantly, we have shown that this apoptosis signaling pathway is also conserved in microglial cell lines, including BV2 and MG6, and EA drives its activation in response to eATP.47) Microglia are derived from hematopoietic myeloid lineage, and serve as the resident macrophages of the central nervous system.48) Upon stress stimuli, including DAMPs, ROS and endotoxins, microglia trigger inflammatory responses and cell death, and consequently promote development of various NDs, such as Parkinson’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis (ALS).48–50) Taken together, TFAs may promote eATP-induced microglial pro-inflammatory signaling and cell death, and consequently aggravate NDs.
4.2. TFAs Promote DNA-Damage Induced Apoptosis via Multiple Signaling PathwaysDNA is damaged by many kinds of stresses, such as UV, ROS, and anti-cancer agents, resulting in generation of various types of DNA lesions.51,52) Since DNA lesions cause mutations and genomic instability that have adverse effects on various diseases including cancer, cells elicit DNA damage responses (DDRs); when the level of DNA damage is low, cells maintain survival by triggering cell cycle arrest, DNA repair and senescence,53) but once DNA is damaged beyond repair, cells undergo programmed cell death, usually apoptosis, which is closely associated with the onset and progression of TFA-related diseases including CVDs54,55) and NDs.56) Our recent study revealed that TFAs, such as EA and LEA, promote apoptosis in response to DNA damage caused by doxorubicin (Dox),57) a DNA topoisomerase II inhibitor inducing double strand breaks (DSBs).57) EA facilitated both mitochondrial ROS (mitoROS) generation and Dox-induced c-jun N-terminal kinase (JNK) activation in a mutually dependent manner, and thereby augmented apoptosis. The pro-apoptotic effect of EA was not affected by knockout of either p53, the crucial transcription factor for DDRs,58,59) or ASK1, a previously identified effector of TFAs,44) but rather blocked by knockdown of SH3 domain-binding protein which preferentially associates with Burton’s tyrosine kinase (Sab, also called as SH3 domain binding protein 5, SH3BP5).60) Sab localizes at the outer membrane of mitochondria, and serves as an adaptor recruiting activated JNK in response to various stresses including DNA damage, consequently inducing mutual enhancement of mitoROS generation and JNK activation.61) Sab knockdown consistently reduced EA-meditated increase in JNK activation and mitoROS generation induced by Dox. These data suggest that TFAs promote feedforward amplification of the mitoROS generation and JNK activation, and ultimately apoptosis (Fig. 4). Since the mitochondrial JNK-Sab-ROS pathway has been reported to play a pro-apoptotic role in diverse types of cells, such as liver cells, neuronal cells and cardiomyocytes,61) this pathway may play a causative role in TFA-related diseases including NASH, NDs and CVDs.
In a most recent study, we found that TFAs, including EA and LEA, also facilitate apoptosis induced by a DNA alkylating agent cisplatin, a common inducer of DNA interstrand crosslinks (ICLs) that blocks RNA transcription and DNA replication, but interestingly, in a different manner from that induced by Dox.62) Although p38/JNK MAP kinases were hyperactivated by EA and accordingly promoted apoptosis in response to both Dox and cisplatin, these pro-apoptotic effects of EA were canceled by knockout of either p53 or ASK1 in the case of cisplatin treatment, but not Dox treatment. We further revealed that EA facilitates cisplatin-induced ROS generation by reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in a fashion dependent on receptor-interacting protein 1 (RIP1), which is known to act as an accessory protein of NADPH oxidase complex downstream of TNF receptor (TNFR) activation63,64); increased ROS in turn enhances the activation of ASK1-p38/JNK MAP kinase pathway, and ultimately p53-dependent apoptosis (Fig. 4). Thus, TFAs promote DNA damage-induced apoptosis at least in two distinct mechanisms, likely according to the types of DNA damage, DSBs and ICLs. One of the most common sources of ICLs is acetaldehyde, which can be generated endogenously in metabolic processes, and also by unhealthy habits like alcohol consumption and cigarette smoking; accumulation of acetaldehyde increases the risk of various diseases such as metabolic syndrome, NDs, and CVDs.65–67) Intriguingly, EA promoted acetaldehyde cell death, which was partly inhibited p53 knockout,62) supporting the notion that TFA consumption may aggravate such diseases, together with unhealthy lifestyles and abnormal acetaldehyde metabolism.
5. CONCLUSION
In a series of our studies, we have demonstrated that in response to stresses, such as eATP and DNA damage, TFAs enhance activation of intracellular signaling pathways that induce inflammation and cell death, which can well explain the etiology of TFA-related diseases (Figs. 2–4). Based on our findings, we propose a novel concept for TFA toxicity in which TFAs serve as an enhancer of stress responses including inflammation and cell death. We believe that our novel concept and the molecular basis for TFA toxicity will contribute to the understandings of the underlying mechanisms of, and prevention and treatment of TFA-related diseases including metabolic syndrome, CVDs and NDs.
Acknowledgments
I would like to acknowledge the valuable advices, supports and contributions to this project of Dr. Takuya Noguchi and Dr. Atsushi Matsuzawa at Lab of Health Chemistry, Graduate School for Pharmaceutical Sciences, Tohoku University. I also thank all members of Lab of Health Chemistry for helpful discussions and cooperations, and my family for supporting me throughout my life. This work was supported by JSPS KAKENHI Grant Numbers JP18K14894 and JP20K07011. This work was also supported by the Japan Foundation for Aging and Health, Sapporo Bioscience Foundation, Grant for Basic Science Research Projects from the Sumitomo Foundation, and Lotte Research Promotion Grant.
Conflict of Interest
The author declares no conflict of interest.
Notes
This review of the author’s work was written by the author upon receiving the 2021 Pharmaceutical Society of Japan Award for Young Scientists.
REFERENCES
- 1) Gebauer SK, Psota TL, Kris-Etherton PM. The diversity of health effects of individual trans fatty acid isomers. Lipids, 42, 787–799 (2007).
- 2) Bendsen NT, Christensen R, Bartels EM, Astrup A. Consumption of industrial and ruminant trans fatty acids and risk of coronary heart disease: a systematic review and meta-analysis of cohort studies. Eur. J. Clin. Nutr., 65, 773–783 (2011).
- 3) Estadella D, da Penha Oller do Nascimento CM, Oyama LM, Ribeiro EB, Damaso AR, de Piano A. Lipotoxicity: effects of dietary saturated and transfatty acids. Mediators Inflamm., 2013, 137579 (2013).
- 4) Mensink RP, Zock PL, Kester AD, Katan MB. Effects of dietary fatty acids and carbohydrates on the ratio of serum total to HDL cholesterol and on serum lipids and apolipoproteins: a meta-analysis of 60 controlled trials. Am. J. Clin. Nutr., 77, 1146–1155 (2003).
- 5) Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Aggarwal N, Schneider J, Wilson RS. Dietary fats and the risk of incident Alzheimer disease. Arch. Neurol., 60, 194–200 (2003).
- 6) Micha R, Mozaffarian D. Trans fatty acids: effects on metabolic syndrome, heart disease and diabetes. Nat. Rev. Endocrinol., 5, 335–344 (2009).
- 7) Johnston PV, Johnson OC, Kummerow FA. Occurrence of trans fatty acids in human tissue. Science, 126, 698–699 (1957).
- 8) Thomas LH, Winter JA, Scott RG. Concentration of transunsaturated fatty acids in the adipose body tissue of decedents dying of ischaemic heart disease compared with controls. J. Epidemiol. Community Health, 37, 22–24 (1983).
- 9) Thomas LH, Winter JA, Scott RG. Concentration of 18 : 1 and 16 : 1 transunsaturated fatty acids in the adipose body tissue of decedents dying of ischaemic heart disease compared with controls: analysis by gas liquid chromatography. J. Epidemiol. Community Health, 37, 16–21 (1983).
- 10) Mensink RP, Katan MB. Effect of dietary trans fatty acids on high-density and low-density lipoprotein cholesterol levels in healthy subjects. N. Engl. J. Med., 323, 439–445 (1990).
- 11) Willett WC, Stampfer MJ, Manson JE, Colditz GA, Speizer FE, Rosner BA, Sampson LA, Hennekens CH. Intake of trans fatty acids and risk of coronary heart disease among women. Lancet, 341, 581–585 (1993).
- 12) Diet, nutrition and the prevention of chronic diseases. World Health Organ. Tech. Rep. Ser., 916, i–viii, 1–149, backcover (2003).
- 13) Restrepo BJ, Rieger M. Trans fat and cardiovascular disease mortality: evidence from bans in restaurants in New York. J. Health Econ., 45, 176–196 (2016).
- 14) Brandt EJ, Myerson R, Perraillon MC, Polonsky TS. Hospital admissions for myocardial infarction and stroke before and after the trans-fatty acid restrictions in New York. JAMA Cardiol., 2, 627–634 (2017).
- 15) Mozaffarian D, Aro A, Willett WC. Health effects of trans-fatty acids: experimental and observational evidence. Eur. J. Clin. Nutr., 63 (Suppl. 2), S5–S21 (2009).
- 16) Machado RM, Nakandakare ER, Quintao EC, Cazita PM, Koike MK, Nunes VS, Ferreira FD, Afonso MS, Bombo RP, Machado-Lima A, Soriano FG, Catanozi S, Lottenberg AM. Omega-6 polyunsaturated fatty acids prevent atherosclerosis development in LDLr-KO mice, in spite of displaying a pro-inflammatory profile similar to trans fatty acids. Atherosclerosis, 224, 66–74 (2012).
- 17) Afonso MS, Lavrador MS, Koike MK, Cintra DE, Ferreira FD, Nunes VS, Castilho G, Gioielli LA, Paula Bombo R, Catanozi S, Caldini EG, Damaceno-Rodrigues NR, Passarelli M, Nakandakare ER, Lottenberg AM. Dietary interesterified fat enriched with palmitic acid induces atherosclerosis by impairing macrophage cholesterol efflux and eliciting inflammation. J. Nutr. Biochem., 32, 91–100 (2016).
- 18) Monguchi T, Hara T, Hasokawa M, Nakajima H, Mori K, Toh R, Irino Y, Ishida T, Hirata KI, Shinohara M. Excessive intake of trans fatty acid accelerates atherosclerosis through promoting inflammation and oxidative stress in a mouse model of hyperlipidemia. J. Cardiol., 70, 121–127 (2017).
- 19) Matthan NR, Welty FK, Barrett PH, Harausz C, Dolnikowski GG, Parks JS, Eckel RH, Schaefer EJ, Lichtenstein AH. Dietary hydrogenated fat increases high-density lipoprotein apoA-I catabolism and decreases low-density lipoprotein apoB-100 catabolism in hypercholesterolemic women. Arterioscler. Thromb. Vasc. Biol., 24, 1092–1097 (2004).
- 20) van Tol A, Zock PL, van Gent T, Scheek LM, Katan MB. Dietary trans fatty acids increase serum cholesterylester transfer protein activity in man. Atherosclerosis, 115, 129–134 (1995).
- 21) Gatto LM, Sullivan DR, Samman S. Postprandial effects of dietary trans fatty acids on apolipoprotein(a) and cholesteryl ester transfer. Am. J. Clin. Nutr., 77, 1119–1124 (2003).
- 22) Vendel Nielsen L, Krogager TP, Young C, Ferreri C, Chatgilialoglu C, Norregaard Jensen O, Enghild JJ. Effects of elaidic acid on lipid metabolism in HepG2 cells, investigated by an integrated approach of lipidomics, transcriptomics and proteomics. PLOS ONE, 8, e74283 (2013).
- 23) Shao F, Ford DA. Elaidic acid increases hepatic lipogenesis by mediating sterol regulatory element binding protein-1c activity in HuH-7 cells. Lipids, 49, 403–413 (2014).
- 24) Oteng AB, Kersten S. Mechanisms of action of trans fatty acids. Adv. Nutr., 11, 697–708 (2020).
- 25) Ginter E, Simko V. New data on harmful effects of trans-fatty acids. Bratisl. Lek Listy, 117, 251–253 (2016).
- 26) Rock KL, Kono H. The inflammatory response to cell death. Annu. Rev. Pathol., 3, 99–126 (2008).
- 27) Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. Int. J. Cell Biol., 2010, 214074 (2010).
- 28) Valenzuela CA, Baker EJ, Miles EA, Calder PC. Eighteencarbon trans fatty acids and inflammation in the context of atherosclerosis. Prog. Lipid Res., 76, 101009 (2019).
- 29) Smit LA, Katan MB, Wanders AJ, Basu S, Brouwer IA. A high intake of trans fatty acids has little effect on markers of inflammation and oxidative stress in humans. J. Nutr., 141, 1673–1678 (2011).
- 30) Da Silva MS, Julien P, Bilodeau JF, Barbier O, Rudkowska I. Trans fatty acids suppress TNF-alpha-induced inflammatory gene expression in endothelial (HUVEC) and hepatocellular carcinoma (HepG2) cells. Lipids, 52, 315–325 (2017).
- 31) Suzuki M, Asai Y, Kagi T, Noguchi T, Yamada M, Hirata Y, Matsuzawa A. TAK1 mediates ROS generation triggered by the specific cephalosporins through noncanonical mechanisms. Int. J. Mol. Sci., 21, 9497 (2020).
- 32) Yokosawa T, Yamada M, Noguchi T, Suzuki S, Hirata Y, Matsuzawa A. Pro-caspase-3 protects cells from polymyxin B-induced cytotoxicity by preventing ROS accumulation. J. Antibiot. (Tokyo), 72, 848–852 (2019).
- 33) Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, Gargiulo G, Testa G, Cacciatore F, Bonaduce D, Abete P. Oxidative stress, aging, and diseases. Clin. Interv. Aging, 13, 757–772 (2018).
- 34) Noguchi T, Suzuki M, Mutoh N, Hirata Y, Tsuchida M, Miyagawa S, Hwang GW, Aoki J, Matsuzawa A. Nuclear-accumulated SQSTM1/p62-based ALIS act as microdomains sensing cellular stresses and triggering oxidative stress-induced parthanatos. Cell Death Dis., 9, 1193 (2018).
- 35) Hirata Y. Reactive oxygen species (ROS) signaling: regulatory mechanisms and pathophysiological roles. Yakugaku Zasshi, 139, 1235–1241 (2019).
- 36) Noguchi T, Sekiguchi Y, Kudoh Y, Naganuma R, Kagi T, Nishidate A, Maeda K, Ishii C, Toyama T, Hirata Y, Hwang GW, Matsuzawa A. Gefitinib initiates sterile inflammation by promoting IL-1beta and HMGB1 release via two distinct mechanisms. Cell Death Dis., 12, 49 (2021).
- 37) Hirata Y, Katagiri K, Nagaoka K, Morishita T, Kudoh Y, Hatta T, Naguro I, Kano K, Udagawa T, Natsume T, Aoki J, Inada T, Noguchi T, Ichijo H, Matsuzawa A. TRIM48 promotes ASK1 activation and cell death through ubiquitination-dependent degradation of the ASK1-negative regulator PRMT1. Cell Reports, 21, 2447–2457 (2017).
- 38) Ma WW, Zhao L, Yuan LH, Yu HL, Wang H, Gong XY, Wei F, Xiao R. Elaidic acid induces cell apoptosis through induction of ROS accumulation and endoplasmic reticulum stress in SHSY5Y cells. Mol. Med. Rep., 16, 9337–9346 (2017).
- 39) Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med., 18, 59–68 (2012).
- 40) Sauvat A, Chen G, Muller K, Tong M, Aprahamian F, Durand S, Cerrato G, Bezu L, Leduc M, Franz J, Rockenfeller P, Sadoshima J, Madeo F, Kepp O, Kroemer G. Trans-fats inhibit autophagy induced by saturated fatty acids. EBioMedicine, 30, 261–272 (2018).
- 41) Sarnyai F, Donko MB, Matyasi J, Gor-Nagy Z, Marczi I, Simon-Szabo L, Zambo V, Somogyi A, Csizmadia T, Low P, Szelenyi P, Kereszturi E, Toth B, Csala M. Cellular toxicity of dietary trans fatty acids and its correlation with ceramide and diglyceride accumulation. Food Chem. Toxicol., 124, 324–335 (2019).
- 42) Faas MM, Saez T, de Vos P. Extracellular ATP and adenosine: the Yin and Yang in immune responses? Mol. Aspects Med., 55, 9–19 (2017).
- 43) Noguchi T, Ishii K, Fukutomi H, Naguro I, Matsuzawa A, Takeda K, Ichijo H. Requirement of reactive oxygen species-dependent activation of ASK1-p38 MAPK pathway for extracellular ATP-induced apoptosis in macrophage. J. Biol. Chem., 283, 7657–7665 (2008).
- 44) Hirata Y, Takahashi M, Kudoh Y, Kano K, Kawana H, Makide K, Shinoda Y, Yabuki Y, Fukunaga K, Aoki J, Noguchi T, Matsuzawa A. trans-Fatty acids promote proinflammatory signaling and cell death by stimulating the apoptosis signal-regulating kinase 1 (ASK1)-p38 pathway. J. Biol. Chem., 292, 8174–8185 (2017).
- 45) Takeda K, Matsuzawa A, Nishitoh H, Tobiume K, Kishida S, Ninomiya-Tsuji J, Matsumoto K, Ichijo H. Involvement of ASK1 in Ca2+-induced p38 MAP kinase activation. EMBO Rep., 5, 161–166 (2004).
- 46) Peng K, Liu L, Wei D, Lv Y, Wang G, Xiong W, Wang X, Altaf A, Wang L, He D, Wang H, Qu P. P2X7R is involved in the progression of atherosclerosis by promoting NLRP3 inflammasome activation. Int. J. Mol. Med., 35, 1179–1188 (2015).
- 47) Hirata Y, Nada Y, Yamada Y, Toyama T, Fukunaga K, Hwang G, Noguchi T, Matsuzawa A. Elaidic acid potentiates extracellular ATP-induced apoptosis via the P2X 7-ROS-ASK1-p38 axis in microglial cell lines. Biol. Pharm. Bull., 43, 1562–1569 (2020).
- 48) Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci., 15, 300–312 (2014).
- 49) Hanisch UK, Kettenmann H. Microglia. active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci., 10, 1387–1394 (2007).
- 50) Streit WJ, Xue QS. Life and death of microglia. J. Neuroimmune Pharmacol., 4, 371–379 (2009).
- 51) Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer, 16, 20–33 (2016).
- 52) Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature, 461, 1071–1078 (2009).
- 53) Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol. Med., 12, 440–450 (2006).
- 54) Mercer J, Mahmoudi M, Bennett M. DNA damage, p53, apoptosis and vascular disease. Mutat. Res., 621, 75–86 (2007).
- 55) Shah NR, Mahmoudi M. The role of DNA damage and repair in atherosclerosis: A review. J. Mol. Cell. Cardiol., 86, 147–157 (2015).
- 56) Madabhushi R, Pan L, Tsai LH. DNA damage and its links to neurodegeneration. Neuron, 83, 266–282 (2014).
- 57) Hirata Y, Inoue A, Suzuki S, Takahashi M, Matsui R, Kono N, Noguchi T, Matsuzawa A. trans-Fatty acids facilitate DNA damage-induced apoptosis through the mitochondrial JNK-Sab-ROS positive feedback loop. Sci. Rep., 10, 2743 (2020).
- 58) Tsuchida M, Yokosawa T, Noguchi T, Shimada T, Yamada M, Sekiguchi Y, Hirata Y, Matsuzawa A. Pro-apoptotic functions of TRAF2 in p53-mediated apoptosis induced by cisplatin. J. Toxicol. Sci., 45, 219–226 (2020).
- 59) Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol., 20, 199–210 (2019).
- 60) Matsushita M, Yamadori T, Kato S, Takemoto Y, Inazawa J, Baba Y, Hashimoto S, Sekine S, Arai S, Kunikata T, Kurimoto M, Kishimoto T, Tsukada S. Identification and characterization of a novel SH3-domain binding protein, Sab, which preferentially associates with Bruton’s tyrosine kinase (BtK). Biochem. Biophys. Res. Commun., 245, 337–343 (1998).
- 61) Win S, Than TA, Kaplowitz N. The regulation of JNK signaling pathways in cell death through the interplay with mitochondrial SAB and upstream post-translational effects. Int. J. Mol. Sci., 19, 3657 (2018).
- 62) Hirata Y, Takahashi M, Yamada Y, Matsui R, Inoue A, Ashida R, Noguchi T, Matsuzawa A. trans-Fatty acids promote p53-dependent apoptosis triggered by cisplatin-induced DNA interstrand crosslinks via the Nox-RIP1-ASK1-MAPK pathway. Sci. Rep., 11, 10350 (2021).
- 63) Kim YS, Morgan MJ, Choksi S, Liu ZG. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell, 26, 675–687 (2007).
- 64) Noguchi T, Tsuchida M, Kogue Y, Spadini C, Hirata Y, Matsuzawa A. Brefeldin A-inhibited guanine nucleotide-exchange factor 1 (BIG1) governs the recruitment of tumor necrosis factor receptor-associated factor 2 (TRAF2) to tumor necrosis factor receptor 1 (TNFR1) signaling complexes. Int. J. Mol. Sci., 17, 1869 (2016).
- 65) Setshedi M, Wands JR, Monte SM. Acetaldehyde adducts in alcoholic liver disease. Oxid. Med. Cell. Longev., 3, 178–185 (2010).
- 66) Zhang Y, Ren J. ALDH2 in alcoholic heart diseases. molecular mechanism and clinical implications. Pharmacol. Ther., 132, 86–95 (2011).
- 67) Chen CH, Ferreira JC, Gross ER, Mochly-Rosen D. Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol. Rev., 94, 1–34 (2014).